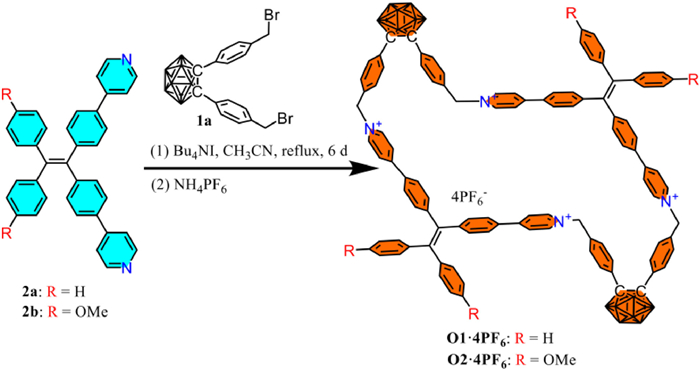
Scheme 1.
Synthesis of cyclophanes O1·4PF6 and O2·4PF6.
Synthesis and properties of tetraphenylethene cationic cyclophanes based on o-carborane skeleton
Rongjian Chen , Jiahui Liu , Caixia Lin , Yuanming Li , Yanhou Geng , Yaofeng Yuan

Supramolecular chemistry research has been closely linked with the development of macrocyclic chemistry. The rational design and efficient synthesis of novel macrocyclic hosts play a crucial role in advancing research in the field of supramolecular chemistry [1–3]. Cationic cyclophanes represent a novel class of macrocyclic hosts capable of solubilization in water or organic solvents through counterion conversion, such as the pioneering research on viologen cyclophanes reported by the Hünig team and the “blue box” discovered by Professor Stoddart's [4–6]. Cationic cyclophanes have been developed and extensively applied in research fields such as mechanically interlocked molecules [4,7–9], host-guest chemistry [10,11], optoelectronic materials [12,13], catalysis [14] and biomedical imaging [15,16]. Tetraphenylethene (TPE) is a typical aggregation-induced emission (AIE) unit with high quantum yields, facile modification and convenient synthesis. In supramolecular chemistry, TPE derivatives are also commonly used as building blocks for constructing many macrocycles or cages [17–19]. Cao et al. groups have developed a series of cationic macrocycles or cages containing TPE, which are capable of recognizing anions or biologically active molecules [20–28].
Carborane is a three-dimensional cage-like structure with a icosahedron shape. It has advantages such as three-dimensional aromaticity, good redox properties, thermal stability, rigid structure and high boron content [29]. Due to the unique structure and properties of carborane, macrocycles based on carborane skeleton have been well studied since the 1990s [30–34]. For example, Jin and coworkers have utilized carborane as the framework structure to develop a series of metal-coordinated macrocycles and cages, which can achieve the separation of light hydrocarbons through interactions such as dihydrogen bonding [35–38]. Carborane embedded necklace-shaped nanohoops with high a solution quantum yield were synthesized by Li and coworkers [39]. Very recently, our group reported carborane-based triangular aromatic alkyne macrocycles, which also demonstrated the AIE effect and can identify nitroaromatic compounds [40].
Although carborane-based macrocycles or cages have been well developed, the water-soluble derivatives are still limited due to the hydrophobic of carborane. To overcome the solubility issue and improve the applicability in aqueous solvents of carborane-based macrocycles, in this study we introduced o-carborane into readily soluble cationic cyclophanes. Moreover, to confer optical properties on the macrocycle, we chosed the TPE unit as functional block to synthesize two novel fluorescent macrocycles O1·4PF6 and O2·4PF6. Notably, O2·4PF6 displayed a near-infrared (NIR) emitting by introducing methoxy groups on the outer side of the TPE unit, while the near-infrared emitting macrocycles without porphyrin structures are rare reported [41]. The cationic cyclophanes O1·4PF6 and O2·4PF6 have Z-shaped hydrophobic cavities and positively charged electrostatic binding sites, which gives them the potential to accommodate electron-rich guests and bind to anions [42]. Nucleotides, with their electron-rich bases and phosphate groups, are ideal guest molecules. Thus, we explored the recognition properties of O1·4PF6 and O2·4PF6 towards nucleotides.
In the synthesis (Scheme 1), we employed template-free and highly diluted reaction conditions. Firstly, 1.0 equiv. of 1,2-bis(4-bromomethylphenyl)-o-carborane (1a) was reacted with 2.0 equiv. of TPE derivative 2a or 2b containing pyridine groups for one day without isolating the intermediate product. Then, an additional equivalent of dibromide 1a was added, and the reaction was continued for five days to obtain the crude product with Br− counterion. Subsequently, the Br− counterion was transformed into PF6− for facilitating the purification and separation of macrocycles, resulting in separation yields of 25% for O1·4PF6 and 37% for O2·4PF6, respectively. Most cyclization experiments typically require a two-step synthesis [41,43,44] or need the introduction of a template for cyclization [45,46]. For some reported one-pot ring closure reactions without the template, the resulting yields were generally below 20% [15,23]. In contrast, our approach does not require a two-step synthesis or using a template, and the yields are higher. The macrocycles O1·4PF6 and O2·4PF6 were successfully characterized using 1H NMR, 13C NMR, HR-MS and single-crystal X-ray diffraction. In the 1H NMR spectra (Fig. 1) of O1·4PF6 and O2·4PF6, it can be found that the chemical shifts of the two macrocycles are similar except for the benzene ring of the TPE units outside the ring, indicating that they have similar cyclic structures. Additionally, compared with the corresponding non-macrocyclic compounds T1·2PF6 and T2·2PF6, the proton chemical shifts of the pyridine and the TPE moieties in the ring (d, e, f, g) are higher than those of the non-cyclized compounds (d′, e′, f′, g′), which could be attributed to the shielding effect after cyclization, since the chemical shifts of the exocyclic benzene ring of TPE unit (i, h) and (i′, h′) are similar. Moreover, for the benzene ring connected to the methylene, the chemical shift of the hydrogen atom (a) linked to the o-carborane is higher than that of hydrogen atom (b) adjacent to the pyridine unit, since the pyridinium cation is a stronger electron-withdrawing group than o-carborane.

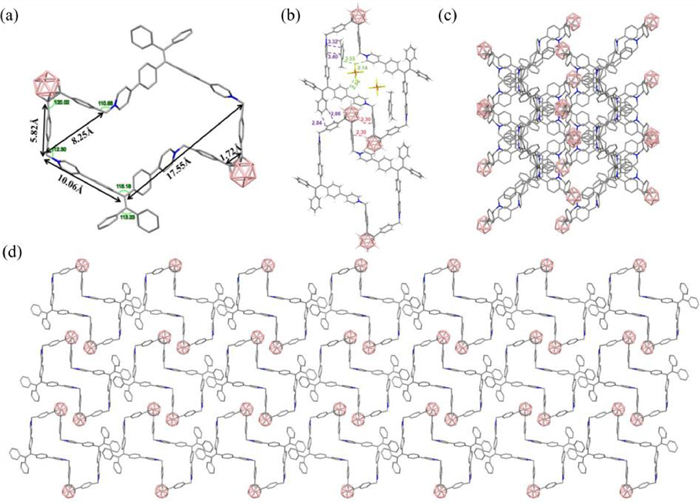
We obtained block-like crystals of O1·4PF6 by evaporating a mixture of xylene and acetonitrile as the solvent (Fig. 2a and Table S3 in Supporting information). O1·4PF6 exhibits a center-symmetric structure, and the cavity of the ring resembles a distorted Z-shape structure. We measured the size of the ring and found that the main cavity of the ring is approximately 10.0 Å × 17.6 Å. The central pyridinium cation is not perfectly parallel, with one cation slightly inward and the other outward. The distance between the two methylene groups in the cyclic compound with an o-carborane framework is approximately 8.3 Å, which is longer than the distance between the two methylene groups in the non-cyclized compound 1a. The side length connecting the o-carborane is 5.8 Å, and the C—C bond length in the o-carborane is 1.72 Å, which is similar to the pre-cyclization state and longer than the pure o-carborane C—C bond length of 1.62 Å [47]. The angle between the benzene ring connected to the o-carborane and the Ccage-Ccage bond is 120.0°. The two benzyl groups are also oriented in the same direction, with angles of 110.6° and 112.3°, respectively, slightly larger than the ideal tetrahedral angle of 109.5°. These results suggest that compound 1a, which contains the o-carborane skeleton, exhibits a certain degree of deformability. Moreover, the two TPE units have different conformations, coexisting in P and M configurations, making the cyclophane internal racemic. In the crystal, we also discovered that the cavity of the ring can accommodate two m-xylene molecules and two counterions. The counterions are present in the central part of the Z-shaped cavity, while the m-xylene solvent molecules are located on both sides of the Z-shaped cavity. There are many weak interactions between the cyclophane and the counterions, solvent molecules and within the cyclophane itself. In addition to π···π, CH···π, CH···F interactions, BH···HC dihydrogen bonding interactions were also found, as shown in Fig. 2b. The stacking of O1·4PF6 crystals demonstrates the formation of tunnel-like structures (Figs. 2c and d). Similarly, O2·4PF6 crystals were obtained by using a volatile acetonitrile-p-xylene mixed solvent. The crystal data is shown in Table S6 (Supporting information). The crystallographic analysis (Fig. S93 in Supporting information) demonstrates that O2·4PF6 shares a cavity structure akin to that of O1·4PF6, with marginally different cavity sizes ascribed to the variance of solvents used. The TPE units inside O2·4PF6 also adopt an internal racemic structure. Introducing methoxy groups on the outer side of the tetraphenylethylene does not significantly affect the molecular cavity structure. We also discovered that O2·4PF6 can accommodate three p-xylene solvent molecules. This indicates that the ring-shaped cavity structure has the potential to accommodate guest molecules.
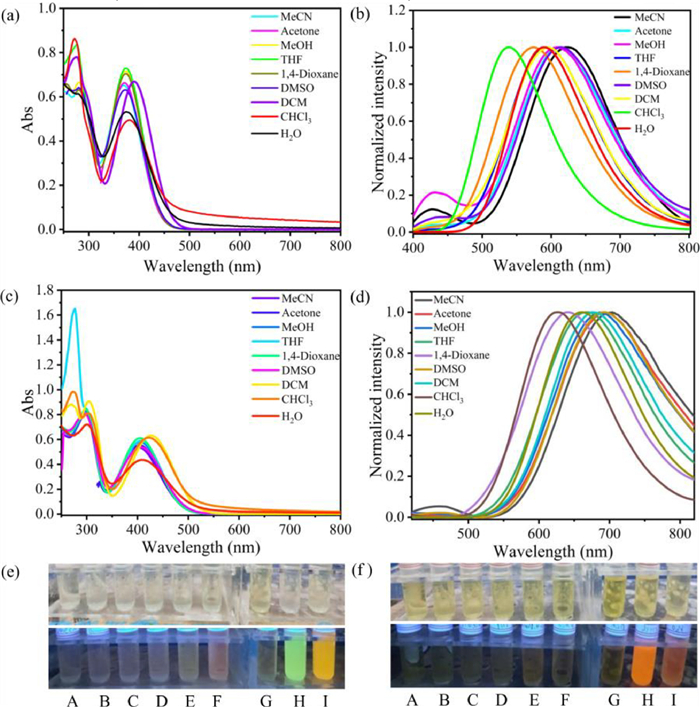
The cyclophanes O1·4PF6 and O2·4PF6 consist of o-carborane and tetraphenylethylene structures, and the C—C bonds of o-carborane can undergo intramolecular vibrational energy dissipation in solution, while the four phenyl rings in tetraphenylethylene can freely rotate in solution, thereby dissipating energy. The vibrational motion of the C—C bonds in o-carborane and the rotation of the phenyl rings in tetraphenylethylene result in the overall molecular motion, leading to weak luminescence in solution. Furthermore, the intramolecular motion may be influenced to different extents in different solvent environments, thus exhibiting different optical properties. We conducted the UV–vis spectroscopy and fluorescence spectroscopy tests of these two cyclophanes in various solvents. The results (Fig. 3, Tables S1 and S4 in Supporting information) demonstrate that these two cyclophanes exhibit varying absorption and emission wavelengths in different solvents. From the UV–vis absorption spectra, it can be observed that the maximum absorption wavelength of O1·4PF6 is around 380 nm, while for O2·4PF6, it is around 405 nm. O2·4PF6 exhibits a more significant redshift compared to O1·4PF6, which is attributed to the introduction of methoxy groups. In the fluorescence emission spectra, the maximum emission peak of O1·4PF6 in acetonitrile solution is at 621 nm, while it undergoes varying degrees of blueshift in other solvent environments. We normalized the fluorescence spectra, which clearly show the degree of blueshift. Among them, the maximum emission peak of O1·4PF6 in water is 590 nm, blueshifted by 31 nm, while in chloroform, it is 539 nm, blueshifted by 82 nm. Under UV irradiation, the fluorescence enhancement behavior of O1·4PF6 in these two solutions can be clearly observed, emitting green light in chloroform and yellow light in water. Absolute quantum yield testing shows that compared to other solvents, O1·4PF6 exhibits higher fluorescence quantum yield behavior in water and chloroform solutions, indicating chloroform and water are poor solvents for O1·4PF6 that can cause its aggregation. Similarly, the maximum emission peak of O2·4PF6 in acetonitrile is at 704 nm, while in water it is at 661 nm, and in chloroform, it is at 627 nm, blueshifted by 43 nm and 77 nm, respectively. Under UV irradiation, O2·4PF6 emits bright red light in water and chloroform, while emitting weaker light in other solvents, indicating that water and chloroform are also their poor solvents. Here, we also found that simple structural modification by introducing methoxy groups can tune the molecular luminescent properties. In acetonitrile solution, O2·4PF6 exhibits a redshift absorption of 33 nm and a fluorescence redshift of 83 nm compared to O1·4PF6. This inspires the development of new macrocycles through structural control. Moreover, both O1·4PF6 and O2·4PF6 exhibit large Stokes shifts in different solvents. The Stokes shift range for O1·4PF6 in different solvents is 159–251 nm, while for O2·4PF6 it is 205–301 nm (Tables S1 and S4). Fluorophores with large Stokes shifts have great applications in the field of bioimaging and sensing [48].
To investigate the difference in optical properties between the cyclized compounds O1·4PF6 and O2·4PF6 and the non-cyclized model compounds T1·2PF6 and T2·2PF6, we also performed photophysical property tests on T1·2PF6 and T2·2PF6 (Table S7, Figs. S94–S97 in Supporting information). In acetonitrile solvent, T1·2PF6 exhibits UV–vis absorption at a wavelength of 365 nm and a maximum emission wavelength of 596 nm, while T2·2PF6 exhibits absorption at a wavelength of 390 nm and emission at a wavelength of 695 nm. Similarly, the introduction of methoxy groups causes a redshift in both the UV–vis and fluorescence spectra of T2·2PF6 compared to T1·2PF6. Moreover, they exhibit stronger fluorescence emission ability in water than in acetonitrile. The cyclized compounds and their corresponding non-cyclized model compounds have similar UV–vis absorption and fluorescence spectra shapes, but the maximum absorption and emission wavelengths are redshifted. This indicates that the main luminescent ability of the cyclized compounds is concentrated in the tetraphenylethene derivatives containing pyridinium, but the cyclized compounds O1·4PF6 and O2·4PF6 have stronger conjugation behavior than the non-cyclized compounds T1·2PF6 and T2·2PF6. The optimized structures and frontier orbital information of molecules 2a, 2b, T12+, T22+, O14+ and O24+ were obtained through theoretical calculations (Table S8 and Figs. S98–S102 in Supporting information). The results show that the molecular orbitals of the HOMO and LUMO energy levels of O14+ and O24+ are mainly distributed in the tetraphenylethene derivatives containing pyridinium that participate in cyclization, while the part with an o-carborane as the framework hardly contributes. This also indicates that the photophysical properties mainly originate from the tetraphenylethene derivatives containing pyridinium.
Since cyclophanes O1·4PF6 and O2·4PF6 contain TPE structural units, and TPE is a typical AIE group, cyclophanes can exhibit an AIE effect. Photophysical tests in different solvents showed that water and chloroform are poor solvents for the cyclized compounds. We tested the fluorescence spectra of O1·4PF6 at different water contents (Figs. 4a and b). As the water content increases, the fluorescence does not change significantly when the water content is below 70%, but a significant enhancement and blue shift of fluorescence occur after exceeding 70%, with the maximum fluorescence intensity reaching 90%, and the maximum fluorescence enhancement reaching 95 times. The intensity slightly decreases at 99%, possibly due to the settling of nano-aggregates. This indicates that O1·4PF6 exhibits good AIE behavior and the aggregation state occurs after a water content of 70%. Similarly, O2·4PF6 (Fig. S63 in Supporting information) also exhibited similar AIE effects in aqueous solution. However, in chloroform (Fig. S30 in Supporting information), aggregation only occurs when the chloroform content exceeds 80%, and obvious green fluorescence appears only when the chloroform content reaches 99%, existing in the form of a suspension. This demonstrates that the aggregation behavior of O1·4PF6 differs between water and chloroform. We believe that chloroform can enter the cavities of the cyclized compounds more effectively than water, displacing the good solvent and further restricting the intramolecular rotational motion. This leads to the non-planarity of the phenyl rings in the tetraphenylethene structure and a decrease in conjugation, resulting in the maximum blue shift. To further demonstrate the correlation between the fluorescence performance and molecular motion, we conducted temperature-dependent fluorescence spectroscopy tests on O1·4PF6 (Fig. S31 in Supporting information). As the temperature decreases, the fluorescence gradually increases, and below the freezing point of acetonitrile, the fluorescence emission ability is significantly stronger than in the solution state. This indicates that lower temperature can cause restriction of intramolecular motions, thereby resulting in fluorescence enhancement. We also investigated the solid-state luminescence behavior of the cyclized compounds (Figs. 4c and d, Table S7 in Supporting information). In the solid state, O1·4PF6 emits intense orange-yellow fluorescence at 596 nm, with a lifetime (τ) of 4.31 ns. In the aqueous phase, the lifetime is slightly longer at 5.24 ns, while in pure acetonitrile solvent, it is only 0.73 ns. This indicates that the aggregated state exhibits better luminescence behavior. However, the emission wavelengths of the cyclized compounds are inconsistent in water, chloroform, and the solid state, which is mainly due to the different conformations they adopt in the aggregated state.

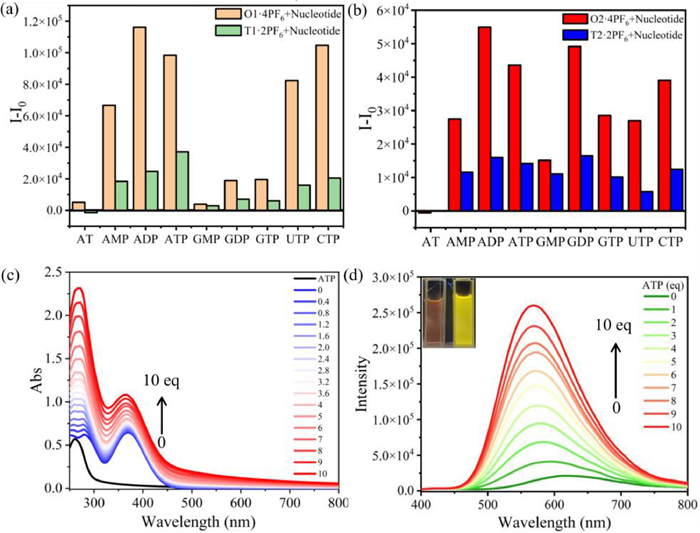
Bio-molecule nucleotides contain phosphate groups, pentose sugars, and nitrogenous bases, forming various types of nucleotides with life information according to different connection methods. Nonetheless, these molecules lack fluorescent groups and are difficult to be detected and identified. Fluorescence detection, characterized by its exceptional sensitivity, is a widely employed analytical technique [49–51]. Our cyclized compounds have cationic groups, cyclic hydrophobic cavities, and fluorescent groups, and we anticipated that these TPE cationic cyclophanes can recognize and detect nucleotides through electrostatic interactions, hydrophobic interactions and other interactions. Nine nucleotides (Scheme S2 in Supporting information) were selected for detection, including AT with no phosphate, AMP and GMP with one phosphate, ADP and GDP with two phosphate groups, and ATP, UTP, CTP and GTP with three phosphate groups. We conducted fluorescence titration experiments by gradually adding aqueous solutions of various nucleotides to acetonitrile solutions of O1·4PF6 and O2·4PF6 (Figs. S41–S49, S75–S83 in Supporting information). We selected to present a column chart of fluorescence enhancement by adding 4.0 equiv. of different nucleotides (Fig. 5). O1·4PF6 and O2·4PF6 show minimal fluorescence response to AT. O1·4PF6 demonstrates significant enhancement towards AMP, ATP, ADP, UTP, CTP and the fluorescence saturation was achieved around 40.0 equiv. of nucleotides. Conversely, it shows weaker enhancement towards GMP, GDP and GTP, which have guanine as the base. This indicates that besides the role of phosphate groups, the variation in bases also plays a significant influence on fluorescence changes. As for O2·4PF6, it shows the strongest recognition towards ADP, followed by GDP, which is different from the behavior of O1·4PF6. This interesting phenomenon can be primarily attributed to photo-induced electron transfer (PET), with O1·4PF6 having a stronger electron transfer with guanine compared to O2·4PF6 [28]. In addition, we conducted UV–vis titration (Fig. 5c, Figs. S32–S40 and S66–S74 in Supporting information) and fluorescence titration (Fig. 5d, Figs. S41–S49 and S75–S83 in Supporting information) experiments by gradually adding 0–10.0 equiv. of nucleotides. We found that, except for AT, both the UV–vis absorption enhancement ability and fluorescence enhancement ability showed a linear relationship with the equivalent amount of added nucleotides. We calculated their detection limits using LOD = 3σ/k (Tables S2 and S5 in Supporting information), and they all exhibited low detection limits except for AT. Furthermore, we compared the recognition abilities (Figs. 5a and b) of O1·4PF6 and O2·4PF6 with the non-cyclized model compounds T1·2PF6 and T2·2PF6. The results showed that the cyclic compounds exhibited stronger recognition abilities toward nucleotides compared to the model compounds. This is primarily due to the cyclic compounds having more positively charged structures, resulting in enhanced recognition abilities.
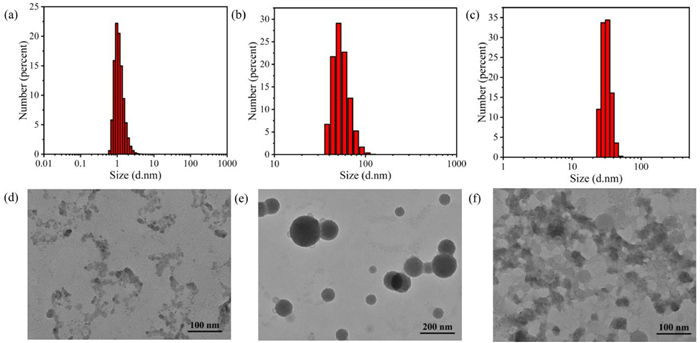
The size and morphology of the monomeric and aggregated forms of O1·4PF6 and O2·4PF6 were studied utilizing transmission electron microscopy (TEM) and dynamic light scattering (DLS) (Fig. 6, Figs. S60–S62, S91 and S92 in Supporting information). DLS analysis showed that O1·4PF6 exhibited a size distribution centered around 1.0 nm in acetonitrile solution, indicating good solubility in acetonitrile. Upon the addition of 10.0 equiv. of ATP, the particle size distribution was concentrated around 60.0 nm. In the aqueous phase, the particle size distribution was concentrated around 30.0 nm, indicating aggregation occurred in both poor solvents and upon the addition of nucleotides. TEM images also revealed that O1·4PF6 in pure acetonitrile solution appeared amorphous, but after the addition of ATP, it self-assembled into solid nanospheres. In water, TEM shows it formed hollow nanospheres of around 30.0 nm or irregular aggregates, which corroborated the results obtained from DLS measurements. The fact that O1·4PF6 exhibited a size of 30.0 nm in water indicated that it possessed a certain degree of water solubility and retained its cation binding site. Nucleotides are typically soluble in water and carry negative charges, so we expected to investigate the binding ability of O1·4PF6 with nucleotides through electrostatic interactions in aqueous solutions. Therefore, we also conducted fluorescence titration experiments of O1·4PF6 with the nucleotides AMP, ADP, ATP, CTP, GTP and UTP in aqueous solutions (Figs. S50–S56 in Supporting information). While AMP and ADP exhibited weak changes, ATP, CTP, UTP and GTP mainly showed fluorescence quenching behavior. It may be due to the aggregation of small nanoparticles into larger ones causing sedimentation. However, among these triphosphate nucleotides, GTP showed the most significant decrease in fluorescence intensity because G was a fluorescence quencher. This is consistent with the weaker fluorescence enhancement observed during GTP titration in acetonitrile, indicating that different bases still play an important role in fluorescence changes. Similarly, we also tested the size of O2·4PF6 aggregates in water. TEM analysis (Fig. S92 in Supporting information) showed that O2·4PF6 formed nanostructures with a size of approximately 60 nm in water. O2·4PF6 also exhibited fluorescence quenching behavior when detecting nucleotides in the aqueous phase (Figs. S84–S90 in Supporting information). However, at 10.0 equiv. of nucleotides, ATP showed the most significant decrease in fluorescence intensity compared to other nucleotides, rather than GTP. This may be due to that O2·4PF6 emits red light, making it difficult for photo-induced electron transfer to occur with GTP.
Due to the helical chirality of TPE, it can undergo P/M rotation. However, the macrocycle O1·4PF6 exhibits an internal racemic state in the crystal (Fig. 2a), lacking chirality. Additionally, circular dichroism (CD) spectroscopy tests conducted in acetonitrile confirmed the absence of chirality. Consequently, we anticipate that introducing a guest molecule will induce chirality in O1·4PF6. Attempts were made to introduce different types of nucleotides into O1·4PF6, and their CD spectra (Figs. S57 and S58 in Supporting information) and circularly polarized luminescence (CPL) spectra (Fig. S59 in Supporting information) were tested. However, no new chiral signals were observed, indicating the failure of the chirality induction strategy. This may be due to the strong electrostatic interactions, which led to the rapid aggregation of O1·4PF6, preventing the change in the helical direction of the TPE moiety. Additionally, the large cavity of the cyclophane may have made it difficult for the chiral structure of the guest to simultaneously induce the helical directions of both TPE units within the cyclophane.
In summary, we synthesized two macrocycles O1·4PF6 and O2·4PF6 efficiently without a template, which contain the o-carborane skeleton and tetraphenylethene cation. This expands the family of supramolecular cyclic compounds containing carborane. The structures of the target compounds were confirmed by single-crystal diffraction analysis, revealing the Z-shaped cavities in both macrocycles. By adjusting the side groups without changing the cavity size, the emission wavelength of the macrocycles can be tuned to achieve near-infrared emission, inspiring the development of novel macrocycles. Both cyclophanes exhibited solid-state luminescence, AIE effect, as well as the macrocycles exhibit multi-color fluorescence properties in different solvents. Research on nucleotide recognition indicates that compared to their model compounds, these two macrocycles exhibit stronger fluorescence enhancement towards nucleotides in acetonitrile, and demonstrate low detection limits for other nucleotides except for AT. In an aqueous environment, nucleotides primarily exhibit fluorescence quenching behavior towards the macrocycles, with GTP showing the most significant quenching effect on O1·4PF6. DLS and TEM studies indicate that O1·4PF6 forms aggregates in water, and its acetonitrile solution can self-assemble with ATP to form solid nanospheres. Furthermore, the excellent fluorescence properties and large Stokes shift of these macrocycles hold promise for future use in cellular imaging applications in the field of biomedicine.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Rongjian Chen: Writing – original draft, Investigation, Formal analysis, Data curation. Jiahui Liu: Methodology, Data curation. Caixia Lin: Writing – review & editing, Project administration, Investigation. Yuanming Li: Writing – review & editing, Methodology. Yanhou Geng: Supervision, Funding acquisition. Yaofeng Yuan: Supervision, Funding acquisition.
This work is supported by the National Natural Science Foundation of China (No. 22071025), the Joint Independent Innovation Fund of Tianjin University and Fuzhou University (No. TF2021-3), the Young and Middle-aged Teacher Education Research Project of Fujian Province (No. JAT210020). The authors thank the Measurement Center of Fuzhou University for their sample testing service. Thank Prof. Zhan-Feng Ju (Fujian Institute of Research on the Structure of Matter) for the assistance with single-crystal testing.
Supplementary material associated with this article can be found, in the online version, at doi:
S.N. Lei, H. Xiao, Y. Zeng, et al., Angew. Chem. Int. Ed. 59 (2020) 10059–10065. doi: 10.1002/anie.201913340
F. Yang, Y. Li, R. Li, et al., Org. Chem. Front. 9 (2022) 2902–2909. doi: 10.1039/D2QO00443G
F. Yang, M. Zhen, S. Wang, et al., Chin. Chem. Lett. 33 (2022) 5088–5091. doi: 10.1016/j.cclet.2022.03.123
X.-Y. Chen, H. Chen, J.Fraser Stoddart, Angew. Chem. Int. Ed. 62 (2023) e202211387. doi: 10.1002/anie.202211387
B. Odell, M.V. Reddington, A.M.Z. Slawin, et al., Angew. Chem. Int. Ed. 27 (1988) 1547–1550. doi: 10.1002/anie.198815471
W. Geuder, S. Hünig, A. Suchy, Angew. Chem. Int. Ed. 22 (1983) 489–490. doi: 10.1002/anie.198304891
Y. Wang, J. Sun, Z. Liu, et al., Angew. Chem. Int. Ed. 55 (2016) 12387–12392. doi: 10.1002/anie.201605454
Y. Lu, D. Liu, Y.J. Lin, et al., J. Am. Chem. Soc. 143 (2021) 12404–12411. doi: 10.1021/jacs.1c06689
A. Garci, A.H.G. David, L.Le Bras, et al., J. Am. Chem. Soc. 144 (2022) 23551–23559. doi: 10.1021/jacs.2c10591
J.C. Barnes, M. Jurícek, N.L. Strutt, et al., J. Am. Chem. Soc. 135 (2013) 183–192. ˇ doi: 10.1021/ja307360n
I. Neira, A. Blanco-Gómez, J.M. Quintela, et al., Acc. Chem. Res. 53 (2020) 2336–2346. doi: 10.1021/acs.accounts.0c00445
H. Chen, S. Hou, Q. Wu, et al., Matter 4 (2021) 3662–3676. doi: 10.1016/j.matt.2021.08.016
Y. Li, Y. Dong, L. Cheng, et al., J. Am. Chem. Soc. 141 (2019) 8412–8415. doi: 10.1021/jacs.9b02617
Y. Li, N. Li, G. Li, et al., J. Am. Chem. Soc. 145 (2023) 9118–9128. doi: 10.1021/jacs.3c00800
X. Dong, X. Dai, G. Li, et al., Adv. Sci. 9 (2022) 2201962. doi: 10.1002/advs.202201962
F. Yang, R. Li, W. Wei, et al., Angew. Chem. Int. Ed. 61 (2022) e202202491. doi: 10.1002/anie.202202491
L. Bian, Y. Liang, Z. Liu, ACS Appl. Nano Mater. 5 (2022) 13940–13958. doi: 10.1021/acsanm.2c01250
J. Zhang, W. Kang, X.D. Xu, Org. Chem. Front. 10 (2023) 6225–6239. doi: 10.1039/D3QO01426F
H.T. Feng, Y.X. Yuan, J.B. Xiong, et al., Chem. Soc. Rev. 47 (2018) 7452–7476. doi: 10.1039/C8CS00444G
L. Wang, Z. Guo, L. Cheng, et al., Dye. Pigm. 216 (2023) 111364. doi: 10.1016/j.dyepig.2023.111364
L. Cheng, H. Zhang, Y. Dong, et al., Chem. Commun. 55 (2019) 2372–2375. doi: 10.1039/C9CC00599D
C. Qin, Y. Li, Q. Li, et al., Chin. Chem. Lett. 32 (2021) 3531–3534. doi: 10.1016/j.cclet.2021.05.006
H. Zhang, L. Cheng, H. Nian, et al., Chem. Commun. 57 (2021) 3135–3138. doi: 10.1039/D1CC00303H
H. Duan, F. Cao, M. Zhang, et al., Chin. Chem. Lett. 33 (2022) 2459–2463. doi: 10.1016/j.cclet.2021.11.010
J.B. Xiong, D.D. Ban, Y.J. Zhou, et al., RSC Adv. 12 (2022) 6876–6880. doi: 10.1039/D2RA00293K
J.H. Wang, J.B. Xiong, X. Zhang, et al., RSC Adv. 5 (2015) 60096–60100. doi: 10.1039/C5RA09721E
S.N. Lei, H. Cong, Chin. Chem. Lett. 33 (2022) 1493–1496. doi: 10.1016/j.cclet.2021.08.068
H. Duan, T. Yang, Q. Li, et al., Chin. Chem. Lett. 35 (2024) 108878. doi: 10.1016/j.cclet.2023.108878
R. Núñez, M. Tarrés, A. Ferrer-Ugalde, et al., Chem. Rev. 116 (2016) 14307–14378. doi: 10.1021/acs.chemrev.6b00198
X. Yang, C.B. Knobler, M.F. Hawthorne, Angew. Chem. Int. Ed. 30 (1991) 1507–1508. doi: 10.1002/anie.199115071
R.N. Grimes, Angew. Chem. Int. Ed. 32 (1993) 1289–1290. doi: 10.1002/anie.199312891
I.T. Chizhevsky, S.E. Johnson, C.B. Knobler, et al., J. Am. Chem. Soc. 115 (1993) 6981–6982. doi: 10.1021/ja00068a069
W. Jiang, I.T. Chizhevsky, M.D. Mortimer, et al., Inorg. Chem. 35 (1996) 5417–5426. doi: 10.1021/ic960354k
C. Viñas, R. Núñez, F. Teixidor, Large molecules containing icosahedral boron clusters designed for potential applications, Boron Science, N.S. Hosmane, CRC Press, New York, 2012, pp. 701–731.
P.F. Cui, X.R. Liu, G.X. Jin, J. Am. Chem. Soc. 145 (2023) 19440–19457. doi: 10.1021/jacs.3c05563
S.T. Guo, P.F. Cui, X.R. Liu, et al., J. Am. Chem. Soc. 144 (2022) 22221–22228. doi: 10.1021/jacs.2c10201
P.F. Cui, X.R. Liu, Y.J. Lin, et al., J. Am. Chem. Soc. 144 (2022) 6558–6565. doi: 10.1021/jacs.2c01668
P.F. Cui, Y.J. Lin, Z.H. Li, et al., J. Am. Chem. Soc. 142 (2020) 8532–8538. doi: 10.1021/jacs.0c03176
M. Zhu, Q. Zhou, H. Cheng, et al., Angew. Chem. Int. Ed. 62 (2023) e202213470. doi: 10.1002/anie.202213470
Q. Qu, M. Fu, C. Lin, et al., Org. Chem. Front. 10 (2023) 3293–3299. doi: 10.1039/D3QO00487B
A.H.G. David, A. Garci, S. Abid, et al., J. Am. Chem. Soc. 145 (2023) 9182–9190. doi: 10.1021/jacs.3c01244
X. Chi, J. Tian, D. Luo, et al., Molecules 26 (2021) 2426. doi: 10.3390/molecules26092426
A. Garci, Y. Beldjoudi, M.S. Kodaimati, et al., J. Am. Chem. Soc. 142 (2020) 7956–7967. doi: 10.1021/jacs.0c02128
A. Garci, J.A. Weber, R.M. Young, et al., Nat. Catal. 5 (2022) 524–533. doi: 10.1038/s41929-022-00799-y
W. Liu, Y. Tan, L.O. Jones, et al., J. Am. Chem. Soc. 143 (2021) 15688–15700. doi: 10.1021/jacs.1c06333
Q.S. Fang, L. Chen, Q.Y. Liu, Chin. Chem. Lett. 28 (2017) 1013–1017. doi: 10.1016/j.cclet.2016.12.002
Y. Sha, Z. Zhou, M. Zhu, et al., Angew. Chem. Int. Ed. 61 (2022) e202203169. doi: 10.1002/anie.202203169
G. Yin, Y. Li, S. Li, et al., Dye. Pigments 198 (2022) 110013. doi: 10.1016/j.dyepig.2021.110013
Y. Yue, F. Huo, F. Cheng, et al., Chem. Soc. Rev. 48 (2019) 4155–4177. doi: 10.1039/C8CS01006D
Q.Y. Cao, L.X. Huang, S. Koo, et al., Sens. Actuator. B: Chem. 368 (2022) 132091. doi: 10.1016/j.snb.2022.132091
B.S. Morozov, A.S. Oshchepkov, I. Klemt, et al., JACS Au 3 (2023) 964–977. doi: 10.1021/jacsau.2c00658

Figure 1 1H NMR spectra (500 MHz, 298 K, CD3CN) recorded for (a) T1·2PF6, R = H; (b) O1·4PF6, R = H; (c) T2·2PF6, R = OMe; (d) O2·4PF6, R = OMe.

Figure 2 The single-crystal X-ray structures of (a) O1·4PF6 and (b) weak intermolecular interactions between molecules. Illustration of the packing of O1·4PF6 along the (c) a-axis and (d) b-axis. Color code: C, gray; N, blue; B, pink. For clarity, all hydrogen atoms, counterions, and solvent molecules are omitted.

Figure 3 (a) UV–vis absorption spectrum and (b) fluorescence emission spectrum of O1·4PF6 (1.0 × 10−5 mol/L) under different solvent conditions, λex = 365 nm. (c) UV–vis absorption spectrum and (d) fluorescence emission spectrum of O2·4PF6 (1.0 × 10−5 mol/L) under different solvent conditions, λex = 400 nm. UV light illuminated fluorescence images of (e) O1·4PF6 and (f) O2·4PF6 in different solvents. A-I are acetonitrile, acetone, methanol, tetrahydrofuran, dioxane, dimethyl sulfoxide, dichloromethane, chloroform, water.

Figure 4 (a) Fluorescence spectra and (b) plot of maximum emission intensity of O1·4PF6 (1.0 × 10−5 mol/L) versus H2O fraction in MeCN–H2O mixture. (c) The solid-state fluorescence spectrum and (d) solid-state fluorescence lifetime of O1·4PF6. (Inset) Fluorescence images of O1·4PF6 versus H2O fraction (0–99%) in MeCN–H2O mixture under UV light (365 nm). λex = 365 nm, Ex/Em slit = 3 nm.

Figure 5 (a) The I-I0 of O1·4PF6 with 4.0 equiv. nucleotides. (b) The I-I0 of O2·4PF6 with 4.0 equiv. nucleotides. (c) UV–vis titration spectra of O1·4PF6 (1.0 × 10−5 mol/L) in MeCN with ATP (0–10.0 equiv.). (d) Fluorescence emission spectra of O1·4PF6 (1.0 × 10−5 mol/L) in MeCN with the ATP (0–10.0 equiv.). (Inset) Fluorescence images of O1·4PF6 (1.0 × 10−5 mol/L) in MeCN (left) and with 10.0 equiv. ATP (right) under UV light (365 nm).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: