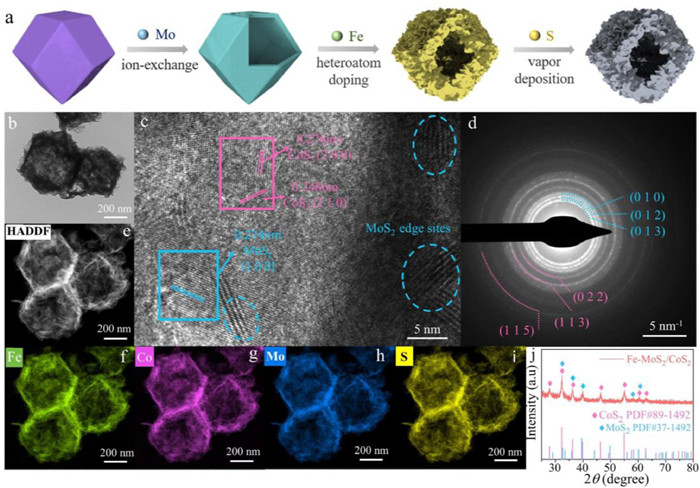
Figure 1.
(a) Synthesis illustration, (b) TEM image, (c) HRTEM image, (d) selected area electron diffraction (SAED) patterns, (e) HAADF-STEM image, (f-i) element mapping images and (j) XRD patterns of Fe-CoS2/MoS2.
Built-in electric field and core-shell structure of the reconstructed sulfide heterojunction accelerated water splitting
Shudi Yu , Jie Li , Jiongting Yin , Wanyu Liang , Yangping Zhang , Tianpeng Liu , Mengyun Hu , Yong Wang , Zhengying Wu , Yuefan Zhang , Yukou Du

The electrolysis of water is considered to be one of the most promising technologies for large-scale hydrogen production, but it requires the efficient electrocatalysts to overcome the high overpotential caused by the slow kinetics of the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER) [1,2]. In low-cost non-noble metal based materials, transition metal sulfides (TMSs) have attracted more attention in the field of electrocatalytic overall water splitting (OWS) due to their special structural and electronic properties [3,4]. It is well known that layered molybdenum disulfide (MoS2) is a platinum-like catalyst to drive HER, but has poor performance for OER [5]. While the sulfides, oxides, and hydroxides of 3d transition metals have consistently demonstrated excellent OER activity. Therefore, the combination of 3d transition metal compounds with MoS2 is expected to fabricate HER and OER bifunctional catalysts. It has been confirmed that the built-in electric field (BEF) at the heterojunction in this compound facilitates electron transfer at the interface and causes a D-band shift, which improves the adsorption/desorption capacity of the catalytic site for the reaction intermediates [6-8]. Meanwhile, 3d transition metal element is a classic electron donor. The introduction of heteroatom dopant such as Fe, Ni or Co not only provides additional catalytic sites, but also activates the inert basal plane of MoS2 through in-plane substitution, which improve its intrinsic catalytic activity [9-11].
Typically, sulfides known as pre-catalysts tend to irreversibly self-reconstruct into metal oxyhydroxides (MOOH) during the OER process [12,13]. Some scholars claim that the hybrid compound of MOOH shell and sulfide core as well as the adsorbed sulfate ions synergistically exhibit more exceed OER properties than the straightforwardly synthesized MOOH [14,15]. While an increasing number of scholars believe that more reconstruct groups lead to higher catalytic activity. The dense active layer seriously impacts the further penetration of the electrolyte and the successful transport of charge and related intermediates, which greatly reduces the quantity and intrinsic activity of the post-OER catalysts [16,17]. The mechanism of the accelerated water oxidation and the structural correlation between the reconstructed MOOH and the pre-catalyst remain unclear. Therefore, it is necessary to identify the real active site of metal sulfides and design feasible pre-catalysts for the development of rational electrocatalysts.
Herein, we successfully synthesized a hollow dodecahedral Fe-doped CoS2/MoS2 heterogeneous catalyst for electrolysis of water by a simple and mild “ion-exchange–heteroatom doping–vapor deposition” method. The resulting Fe-CoS2/MoS2 had an overpotential of 178/375 mV at 10/100 mA/cm2 for OER, and the overpotential of 92 mV at 10 mA/cm2 for HER. Comprehensive analysis showed the difference of energy band between the heterogeneous components spontaneously formed BEF, which accelerated the charge transfer and enhanced the catalytic performance. Meanwhile, trace Fe doping rearranged the local charge and activated the inert basal plane of MoS2, outputting the ideal HER activity. In particular, the synergistic effect of reconstructed “TMS-MOOH” core-shell structure resulted in fortified OER performance. Therefore, the Fe-CoS2/MoS2‖Fe-CoS2/MoS2 couple showed excellent electrocatalytic activity and durability, highlighting the importance of heteroatom doping and BEF for efficient bifunctional electrocatalyst.
The materials and detailed preparation of catalysts can be found in Supporting information.
Fig. 1a illustrated the synthesis procedure of hollow Fe-CoS2/MoS2 polyhedron. Combined with the Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images in Fig. 1b and Figs. S1c-f (Supporting information), it can be seen that Co-MOF and sodium molybdate generated a semi-transparent hollow Co/Mo layered double hydroxide (Co/Mo LDH) by ion exchange and etching. After Fe doping, the leaf-like nanosheets wrapped around the polyhedron surface, presenting the successful fabrication of the hierarchical structure. Subsequently, the final vulcanized catalyst was obtained by typical chemical vapor deposition (CVD). The MOF precursor endowed it a unique nanocage morphology with larger specific surface area, uniform distribution of active sites, sufficient internal space and shorter electron/species mass diffusion path [18-20]. High-resolution TEM (HRTEM) was employed to confirm the presence of sulfide heterostructures (Fig. 1c). Lattice fringes with spacing of 0.248 nm and 0.274 nm correspond to the (210) crystal plane of CoS2 and the (100) crystal plane of MoS2, respectively, consistent with a sequence of polycrystalline rings in the selective electron diffraction (SAED) patterns in Fig. 1d. The slightly different crystal plane spacing compared to the pure material was due to the random substitution of Co and Mo by Fe atoms. Additionally, high-angle annular dark field aberration corrected scanning TEM (HAADF-STEM) images, energy-dispersive X-ray spectroscopy (EDS) result and element mapping images showed uniform distribution and relative content of each element in Fe-CoS2/MoS2 (Figs. 1e-i). In order to highlight the advantages of the catalyst, dopant-free CoS2/MoS2 and Mo-free Fe-CoS2 were also prepared. Their morphology and composition were similar to Fe-CoS2/MoS2 (Figs. S1k-n in Supporting information). Besides, in the X-ray diffraction (XRD) pattern (Fig. 1j), the correlated diffraction peaks were highly consistent with MoS2 (JCPDS No. 37–1492) and CoS2 (JCPDS No. 89–1492), and the characteristic peaks of MOF and LDH precursors (Figs. S2 and S3 in Supporting information) disappeared completely, proving the successful synthesis of sulfide in CVD process. It is worth noting that the low Fe content led to no obvious peaks of FeS2, but the slightly shifted pattern before and after doping validated the lattice distortion caused by Fe (Fig. S4 in Supporting information). Moreover, the Raman spectra in Fig. S5 (Supporting information) displayed two characteristic peaks attributed to MoS2 at 373.86 and 401.18 cm−1 wavenumbers, and a series of peaks belonging to CoS2 at 183.99, 466.55, 489.87, 516.37, 577.03 and 612.84 cm−1, which further revealed that the sample was a multi-component nanocomplex containing CoS2 and MoS2.
As a classical heterogeneous electrocatalytic reaction, the migration of OWS-related intermediates at the solid-liquid-gas three-phase interface involves multiple electron and proton transfers, which means that the performance of the catalyst is connected with the electron coupling on the surface. Therefore, X-ray photoelectron spectroscopy (XPS) was carried out to analyze the electronic structure and surface chemical state of the related catalysts. The survey spectrum in Fig. S6 (Supporting information) depicted that the Fe-CoS2/MoS2 catalyst contained Fe, Co, Mo and S elements, while there was no Fe in CoS2/MoS2 and no Mo in Fe-CoS2. The Co 2p spectrum of Fe-CoS2/MoS2 in Fig. 2a could be well fitted to two different spin–orbit doublets: characteristic peaks at 780.90 and 796.83 eV were assigned to the 2p3/2 and 2p1/2 states of Co3+, while the other peaks at 784.72 and 801.27 eV stemmed from the 2p3/2 and 2p1/2 states of Co2+. Significantly, the peaks shifted towards the lower binding energy compared to Fe-CoS2, suggesting that the strong coupling between the different components at the internal heterojunction resulted in the electrons transfer from MoS2 to CoS2, which regulated the electronic structure and optimized catalytic performance [21].
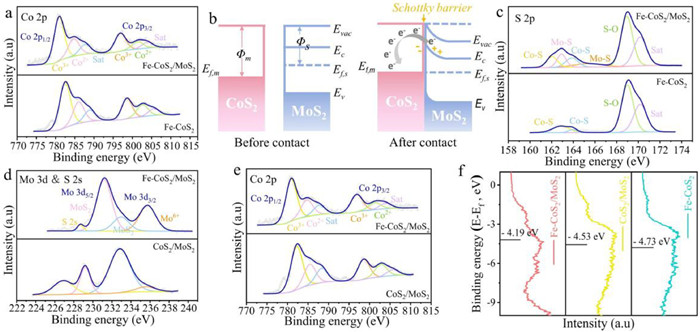
Additionally, the corresponding band structure was ascertained by ultraviolet photoelectron spectroscopy (UPS) (Fig. S7 in Supporting information). The work functions (Φ) of Fe-CoS2 and Fe-CoS2/MoS2 were calculated to be 5.20 and 4.92 eV (Ef = 0 eV), respectively. This difference was attributed to the combination of Co substrate and MoS2. In other words, Φ value of the CoS2 was bigger than that of MoS2, resulting in the electrons transfer in the composite material. Combined with band theory, electron migration behavior of Fe-CoS2/MoS2 was further understood. As a typical semiconductor material, the Fermi level of MoS2 is higher than that of metallic CoS2 (Ef,s > Ef,m) [22]. According to the minimum energy principle, electrons in the semiconductor (MoS2) tended to flow into the metal (CoS2), inducing the formation of BEF and the energy band bending of semiconductor, which formed a Schottky barrier at the contact surface (Fig. 2b and Fig. S8 in Supporting information). The formation of BEF led to different electron-deficient/electron-rich states on the catalyst surface, which were utilized as active adsorption sites to trap reactants and intermediates with opposite charges, ultimately promoted reaction kinetics and selectivity [23]. Besides, in the S 2p section of Fe-CoS2/MoS2 in Fig. 2c, the characteristic peaks from 160 eV to 165 eV could be indexed to the S22− species bridging CoS2 and MoS2, revealing that the Co-S-Mo bond successfully formed heterogenous CoS2/MoS2 interface. And the peaks at 168−171 eV were due to the oxidation by air. While in the corresponding spectrum of Fe-CoS2, only the signal associated with the Co-S bond appeared. The construction of heterojunctions via coupling materials with different energy level structures is an effective path to regulate the intrinsic activity of catalysts [24]. Whether the interfacial contact is tight or not is related to the smooth flow of charge at the heterojunction [25]. In the prepared composite catalyst, the common S atom promoted the close bonding of CoS2 and MoS2, generating a direct charge flow channel between them and accelerating the charge transfer process.
Furthermore, the high resolution XPS spectra of Fe-CoS2/MoS2 and CoS2/MoS2 were compared to investigate the charge modulation of Fe dopant. As shown in Fig. 2d, two distinct peaks at 231.32 and 232.91 eV observed in the Mo 3d XPS spectra of Fe-CoS2/MoS2 were correspond to the 3d5/2 and 3d3/2 states in the MoS2 component, respectively. The signal of 228.72 eV was derived from the S 2s peak and was related to the metal-sulfur (M-S) bond [26]. And the hexavalent Mo(Ⅵ) was oxidized from the air. Compared with the undoped CoS2/MoS2 catalyst, the Mo 3d characteristic peaks moved towards the positive direction, which was opposite to the trend of Co 2p spectra (Fig. 2e). It is clear that Fe dopant promoted Mo acted as an electron donor, thus the positive charge density around it increased, and the migrated electrons were enriched near Co sites. The concentration gradient of the electrons accelerated the formation of BEF, which redistributed the local charge and modulated the chemical environment on the catalyst surface [27,28].
The surface valence band photoelectron spectra (VBS) of catalysts were analyzed to uncover their D-band electron energy (Ed) levels. Fig. 2f displayed that the Ed values of Fe-CoS2/MoS2, CoS2/MoS2 and Fe-CoS2 are −4.19, −4.53 and −4.73 eV, respectively, which implied the BEF and heteroatom Fe effectively tailored the D-band center of Fe-CoS2/MoS2, thus enhancing the adsorption of intermediates and promoting the kinetics of water decomposition process [29,30].
Considering that sulfides usually undergo irreversible structural reconfiguration during OER process to form MOOH as the true catalytically substances, linear sweep voltammetry (LSV) tests with multiple continuous scans of various sulfide catalysts in this work were first performed to verify the effect of their structural reconfiguration on OER electrocatalytic performance [31]. Taking Fe-CoS2/MoS2 as an example, the overpotential obtained in the first three scans initially decreased and then further stabilized, maintaining at 178 mV to reach the current density (j) of 10 mA/cm2, with obvious self-optimization phenomenon (Fig. S9a in Supporting information). The corresponding Tafel slopes also confirmed this trend (Fig. S9b in Supporting information). In addition, Fe-CoS2/MoS2 had the smallest semicircular radius and the lowest charge transfer resistance (Rct) after three electrochemical impedance spectroscopy (EIS) tests (Fig. S9c in Supporting information). Based on the above self-activated OER properties, it can be inferred that the designed sulfide catalysts such as Fe-CoS2/MoS2 underwent structural restructuring under alkaline OER condition and generated stable active substances to drive the oxidation of water.
Electrochemical measurements of self-activated electrocatalysts were carried out to assess their real OER activity. The LSV curves displayed that Fe-CoS2/MoS2 required low overpotential of 178 and 375 mV to achieve the current density of 10 and 100 mA/cm2, respectively, which was much lower than CoS2/MoS2 (206 mV@10 mA/cm2, 425 mV@100 mA/cm2) and Fe-CoS2 (221 mV@10 mA/cm2, 495 mV@100 mA/cm2) (Fig. 3a). Their Tafel curves obtained from LSV data were illustrated in Fig. 3b. The Tafel slope of Fe-CoS2/MoS2 was 81.86 mV/dec, which was 19.12 and 39.76 mV/dec smaller than CoS2/MoS2 and Fe-CoS2, respectively, indicating that the current density increased the fastest when Fe-CoS2/MoS2 driven the catalytic process. Besides, their charge transfer characteristics was evaluated by EIS. As shown in Fig. 3c, the corresponding curve of Fe-CoS2/MoS2 had the smallest semicircle in the Nyquist plots, conforming to the minimum fitted Rct value in Table S1 (Supporting information), which verified its faster reaction kinetics. In addition, the electrochemical double-layer capacitance (Cdl) values (Fig. S10 in Supporting information) converted by the cyclic voltammetry (CV) curves of each sample (Fig. S11 in Supporting information) reflected the relative size of their electrochemically active surface areas (ECSAs). As expected, Fe-CoS2/MoS2 presented the largest Cdl and ECSA values, underscoring that it had the most active sites and the strongest intrinsic activity (Fig. 3d). The above unparalleled OER performance was inseparable from the synergistic effect of multiple metal sites of Fe, Co and Mo and the adjustment of the energy band by BEF. Furthermore, the long-term stability was investigated by chronopotentiometry (CP) at the reference current density of 10 mA/cm2 and the high current density of 100 mA/cm2 (Fig. 3e). The CP curves of Fe-CoS2/MoS2 remained flat during the 60-h test, and the potential increased by only 17.4% and 16.2% of the initial measured value, respectively. This amazing durability could also be demonstrated by the slightly shifted polarization curve after stability testing (Fig. 3f).

Additionally, two other catalysts (Fe-CoS2/MoS2–350 and Fe-CoS2/MoS2–450) with similar crystalline phase were obtained by simply adjusting the vulcanization temperature from 400 ℃ to 350 ℃ and 450 ℃ (Fig. S12 in Supporting information). Judging from their TEM and SEM images in Fig. S13 (Supporting information), the catalyst could still maintain the hollow nanocage morphology when the temperature rose to 450 ℃, but the nanocage surface was slightly damaged, which might lead to the collapse of the internal cavity structure and overlap of the active sites, thus affecting the catalytic performance. Combined with their electrochemical test results in Fig. S14 (Supporting information), it could be concluded that 400 ℃ is the most suitable temperature for vulcanization, so that the catalyst had the smallest overpotential, Tafel slope, Rct and the maximum Cdl/ECSA value. It could be speculated that the higher ECSA of sulfurized catalyst at 400 ℃ was closely related to its electrocatalytic activity. In theory, the reactivity between the sulfur vapor and precursor was promoted as the temperature increased. However, when the vulcanization temperature was too high, the product was more likely to aggregate, collapse or carbonize, which adversely effected the surface area, and might even be accompanied by some side reactions that inhibit the active effect [32]. Moreover, compared with CoS2/MoS2 and Fe-CoS2, the reinforcing OER activity testified that the high efficiency of the heterostructure was less affected by the vulcanization temperature.
The HER properties of the prepared catalysts were also tested in the same three-electrode system. Strikingly, no apparent self-activation phenomenon similar to OER was found in the electrochemical testing. As expected, Fe-CoS2/MoS2 presented an outstanding overpotential of 92 mV@10 mA/cm2, which was 25 mV and 99 mV lower than CoS2/MoS2 (117 mV) and Fe-CoS2 (191 mV), respectively (Fig. 3g). Besides, their HER kinetics were assessed by the Tafel slopes. Obviously, Fe-CoS2/MoS2 exhibited an optimum reaction activity with low Tafel slope of 73.7 mV/dec, which was much lower than 82.06 mV/dec of CoS2/MoS2 and 143.87 mV/dec of Fe-CoS2, demonstrating a faster rate of hydrogen evolution at the cathode (Fig. 3h). As is known to all, MoS2 is a promising platinum-like catalyst for HER [33]. Benefiting from the defects and the reconfiguration of local charge caused by Fe atoms, its inert basal plane was effectively activated, thus greatly increasing the number of exposed active sites [34-36]. Besides, the electrochemical testing process is often accompanied by the generation of abundant sulfur vacancies, which not only exposed more accessible active sites and thus promoted the adsorption of reactants and intermediates, but also effectively modified the electronic structure around the catalytic sites and enhanced the intrinsic activity [37]. It could be speculated that there was a close synergy between the two defects produced by heteroatom dopants and S vacancies, which together drove the HER process [5,38]. Furthermore, at continuous electrolysis for 50 h at current density of 10 mA/cm2, only a negligible voltage increment of Fe-CoS2/MoS2 was observed (Fig. 3i), exhibiting remarkable robustness, which could also be reflected by the almost overlapping polarization curves before and after the CP test (Fig. 3j). Impressively, the polyhedral morphology and crystal structure of Fe-CoS2/MoS2 did not change after HER stability test (Figs. S15a and b in Supporting information), but the HRTEM image displayed discontinuous lattice fringes (Fig. S15c in Supporting information). It was proved that the catalyst maintained the sulfide structure in the long-term HER process, and no surface reconstruction occurred, resulting in no evident self-activation phenomenon in the electrochemical test.
Based on the extraordinary OER and HER bifunctional catalytic activity of Fe-CoS2/MoS2, an overall water splitting device was assembled utilizing it as both cathode and anode in 1.0 mol/L KOH. As presented in Fig. 3k, the Fe-CoS2/MoS2‖Fe-CoS2/MoS2 couple required a low voltage of 1.51 V to drive the current density of 10 mA/cm2, and the Faradaic efficiency (FE) for both O2 and H2 production was close to 100% (Fig. S16 in Supporting information). Moreover, the long-term stability of OWS is a critical parameter to evaluate the practical application of the catalyst. The cell voltage of the electrolyzer negligibly increased by only 30 mV after 53 h-operation at a constant current of 10 mA/cm2, outperforming RuO2‖Pt/C which had a highly fluctuating CP curve (Fig. 3l).
In order to further clarify the vulcanization-induced electrochemical self-reconstruction, multifarious characterizations of post-OER Fe-CoS2/MoS2 were employed to reveal the true catalytic substance. After long-term OER testing, TEM images illustrated the well-retained nanocage structure, which was in great agreement with the catalyst before the reaction (Fig. 4a). Additionally, dense vacancies created by electrochemical etching could be observed in HRTEM images (Fig. 4b), which facilitated the conductivity and full exposure of active adsorption sites [39]. Meanwhile, amorphous defect layer adjacent to sulfide phase was attributed to disordered oxyhydroxide (denoted as Fe-(Co/Mo)OOH), which could provide plentiful active edge sites of CoS2 and MoS2 [40,41]. Moreover, its XRD pattern showed the initial diffraction peaks of sulfides became extremely weak (Fig. 4c), and the unique Raman peak at 501.72 cm−1 was derived from vibration mode of CoOOH (Fig. S17 in Supporting information), indicating that the catalyst was partially converted to amorphous Fe-(Co/Mo)OOH in the long-term OER. It was rational to infer that the leaching of sulfur on the catalyst surface under OER condition promoted the phase transformation into a dense passivation layer of oxyhydroxide with high intrinsic activity, enveloped the internal sulfide, prevented further phase reconstruction, and formed the core-shell structure of “MS-MOOH” (Fig. 4d). XPS measurements were carried out to deeply investigate the changes in the electronic structure after surface reconstruction. In the high-resolution XPS spectra of Co and Mo, the positive shift of the peaks and the increase of high-valence state metals indicated that the metal sites were oxidized during OER process (Figs. 4e and f, Fig. S18 in Supporting information). For S 2p, the strong peaks at 165.93 and 167.09 eV were assigned to the SO42− from pre-oxidation (Fig. 4g) [42]. It has been widely reported that the nconspicuous sulfate ions adsorbed on the catalyst surface in favour of improvement for OER activity and stability [43-45]. The peaks of M-S bond and S22− decreased significantly after the electrolysis, verifying the leaching of S. For O 1s region, three peaks appeared at 530.54, 531.24, and 531.92 eV belonged to metal-oxygen (M-O), hydroxyl oxygen (H—O) and H2O, respectively (Fig. 4h). Compared with the initial material, the peaks corresponding to M-O and hydroxyl oxygen were sharply heightened, further demonstrating the generation of Fe-(Co/Mo)OOH. Therefore, the surface evolution of Fe-CoS2/MoS2 involved in OER is as follows: During the beginning stage of the reaction, the catalyst exists in the form of metal sulfide heterojunction, and the BEF from hybrid sulfide plays a predominant catalytic role. Afterwards, H2O is further oxidized to produce the MOOH shell, which collaborates with the sulfide to facilitate OER and prevent the complete conversion of internal sulfide. In summary, the oxyhydroxide formed a new cooperative coordination structure on the basis of the original sulfide heterojunction, which not only retained the initial internal electric field, but also rearranged the charge at the heterogeneous interface, and ultimately achieved the optimal OER activity.

Furthermore, the valence electron configuration and coupling interplay of Co, Mo, and Fe co-existing at the core-shell interface have been analyzed. In the complex of Fe-CoS2/MoS2 and Fe-(Co/Mo)OOH, Co2+ had the electronic configuration of 3d7 (t2g6 eg1). Its π-symmetric t2g orbitals were fully occupied with relative lower energy, while an unpaired electron was distributed in the eg d-orbital with relative higher energy, interacting with the bridging O2− via the π-donation. Based on the Hund’s rule, Fe3+ had the half-filling 3d5 (t2g3 eg2) electronic structure with the lowest energy. Two unpaired electrons of Mo4+ were distributed in the t2g orbit (Fig. 4i). The spin single electrons of Mo4+, Co2+ and Fe3+ were all bridged with O2− by π-donation, therefore we established the “Mo-O-Co-O-Fe” unit model (Fig. 4j) [46,47]. Evidently, two electrons with the same spin direction in Mo4+ were relatively repelling each other, thus the electron-electron (e−-e−) repulsion between Mo4+ and O2− enhanced the π-donation of O2− to Co2+, resulting in spontaneous transfer of electrons from Mo4+ to Co2+. Meanwhile, owing to the coupling effect of Mo, when the electron cloud density of Co atom increased, there would be weak e−-e− repulsion between Co2+ and O2−, reinforcing the π-donation via O-Fe. Besides, since Fe with 5 high-spin single electrons had the lowest energy, when it was coupled with Co, the electrons of Co tended to continuously transfer to Fe along the energy gradient according to the principle of the lowest energy. The similar redistribution of electrons also occurred in the self- reconstructed CoS2/MoS2 and Fe-CoS2. In summary, the dopant Fe, multi-metallic sites and the reconstructed core-shell structure synergistically regulated the electronic structure of the catalyst and promoted the electrons interaction and transfer. Excitingly, compared with the directly synthesized oxyhydroxide, the as-designed catalyst had lower overpotential and Tafel slope (Fig. S19 in Supporting information), again emphasizing the superior electrocatalytic activity of MS-MOOH complex over pure sulfide and oxyhydroxide.
In conclusion, the bifunctional catalytic activities of Fe-CoS2/MoS2 are attributed to the following aspects: (1) BEF at the heterogeneous interface modified the energy band and electronic structure. (2) The in-situ surface reconstruction in OER generated the MS-MOOH core-shell structure as the true catalytic sites. (3) Defects resulted from heteroatom doping and electrochemical etching promoted the directional transfer of interfacial electrons and improved the utilization rate of basal plane in MoS2. (4) The hollow polyhedral structure was beneficial for the utilization of active sites and electron/mass transport during electrocatalysis. This novel work offers new insights into the design of compound catalysts based on interface engineering and defect engineering.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Shudi Yu: Writing – review & editing, Writing – original draft, Methodology, Investigation. Jie Li: Investigation. Jiongting Yin: Investigation. Wanyu Liang: Investigation. Yangping Zhang:. Tianpeng Liu: Investigation. Mengyun Hu: Investigation. Yong Wang: Supervision, Resources. Zhengying Wu: Supervision, Resources. Yuefan Zhang: Investigation. Yukou Du: Supervision, Resources, Funding acquisition.
This work was supported by National Natural Science Foundation of China (Nos. 52073199 and 52274304).
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Hao, S.F. Hung, W.J. Zeng, et al., J. Am. Chem. Soc. 145 (2023) 23659–23669. doi: 10.1021/jacs.3c07777
S. Zhao, F. Hu, L. Yin, L. Li, S. Peng, Sci. Bull. 68 (2023) 1389–1398. doi: 10.1016/j.scib.2023.06.001
W. Peng, A. Deshmukh, N. Chen, et al., ACS Catal. 12 (2022) 3743–3751. doi: 10.1021/acscatal.2c00328
W. He, R. Zhang, D. Cao, et al., Small 19 (2022) 2205719.
X. Liu, X. Jiang, G. Shao, et al., Small 18 (2022) 2200601. doi: 10.1002/smll.202200601
Y. Yao, J. Wu, Q. Feng, et al., Small 19 (2023) 2302015. doi: 10.1002/smll.202302015
J. Yin, J. Jin, Z. Yin, et al., Nat. Commun. 14 (2023) 1724–1733. doi: 10.1038/s41467-023-37360-9
S. Zhao, Y. Wang, Y. Hao, et al., Adv. Mater. 36 (2024) e2308925. doi: 10.1002/adma.202308925
V.P. Pham, G.Y. Yeom, Adv. Mater. 28 (2016) 9024–9059. doi: 10.1002/adma.201506402
S. Ahmed, X. Ding, N. Bao, et al., Chem. Mater. 29 (2017) 9066–9074. doi: 10.1021/acs.chemmater.7b02593
D. Yu, Y. Hao, S. Han, et al., ACS Nano 17 (2023) 1701–1712. doi: 10.1021/acsnano.2c11939
M. Zheng, K. Guo, W.J. Jiang, et al., Appl. Catal. B 244 (2019) 1004–1012. doi: 10.1016/j.apcatb.2018.12.019
W. Huang, J. Li, X. Liao, et al., Adv. Mater. 34 (2022) 2200270. doi: 10.1002/adma.202200270
N. Yao, G. Wang, H. Jia, et al., Angew. Chem. Int. Ed. 61 (2022) e202117178. doi: 10.1002/anie.202117178
M. Chen, Y. Zhang, R. Wang, et al., J. Energy Chem. 84 (2023) 173–180. doi: 10.1016/j.jechem.2023.05.009
X. Li, J. Zhao, J. Zhou, Q. Wang, Green Chem. 25 (2023) 10684–10692. doi: 10.1039/d3gc03210h
X. Liu, J. Meng, J. Zhu, et al., Adv. Mater. 33 (2021) 2007344. doi: 10.1002/adma.202007344
L. Yu, H. Hu, H.B. Wu, X.W. Lou, Adv. Mater. 29 (2017) 1604563. doi: 10.1002/adma.201604563
M. Yang, C.H. Zhang, N.W. Li, et al., Adv. Sci. 9 (2022) 2105135. doi: 10.1002/advs.202105135
R. Wang, J. Liu, J. Xie, et al., Appl. Catal. B 324 (2023) 122230. doi: 10.1016/j.apcatb.2022.122230
S. Zhao, S.F. Hung, L. Deng, et al., Nat. Commun. 15 (2024) 2728. doi: 10.1038/s41467-024-46750-6
S. He, H. Du, K. Wang, et al., Chem. Commun. 56 (2020) 5548–5551. doi: 10.1039/d0cc01726d
L. Chen, J.T. Ren, Z.Y. Yuan, Adv. Energy Mater. 13 (2023) 2203720. doi: 10.1002/aenm.202203720
X. Wang, X. Wang, J. Huang, et al., Nat. Commun. 12 (2021) 4112–4122. doi: 10.1038/s41467-021-24511-z
J. Jing, J. Yang, W. Li, Z. Wu, Y. Zhu, Adv. Mater. 34 (2021) 2106807.
Y. Li, W. Wang, B. Huang, et al., J. Energy Chem. 57 (2021) 99–108. doi: 10.1016/j.jechem.2020.08.064
S. Ni, H. Qu, H. Xing, et al., Chin. J. Chem. Eng. 41 (2022) 320–328. doi: 10.1016/j.cjche.2021.09.026
S. Ni, H. Qu, Z. Xu, et al., Appl. Catal. B 299 (2021) 120638. doi: 10.1016/j.apcatb.2021.120638
X. Chen, Z. Qiu, H. Xing, et al., Appl. Catal. B 305 (2022) 121030. doi: 10.1016/j.apcatb.2021.121030
D. Li, L. Zhao, J. Wang, C. Yang, Adv. Energy Mater. 13 (2023) 2204057. doi: 10.1002/aenm.202204057
C.X. Zhao, J.N. Liu, C. Wang, et al., Energy Environ. Sci. 15 (2022) 3257–3264. doi: 10.1039/d2ee01036d
S. Yu, D. Liu, C. Wang, et al., J. Colloid Interface Sci. 653 (2023) 1464–1477. doi: 10.1109/tse.2022.3178096
Y. Wu, F. Li, W. Chen, et al., Adv. Mater. 30 (2018) 1803151. doi: 10.1002/adma.201803151
H.J. Liu, S. Zhang, Y.M. Chai, B. Dong, Angew. Chem. Int. Ed. 62 (2023) e202313845. doi: 10.1002/anie.202313845
N. Shaikh, I. Mukhopadhyay, A. Ray, Int. J. Hydrogen Energy 48 (2023) 15944–15955. doi: 10.1016/j.ijhydene.2023.01.098
X. Gao, Y. Zhou, Z. Cheng, et al., Appl. Surf. Sci. 547 (2021) 149113. doi: 10.1016/j.apsusc.2021.149113
Q. Wang, H. Xu, X. Qian, G. He, H. Chen, Appl. Catal. B 322 (2023) 122104. doi: 10.1016/j.apcatb.2022.122104
W. Wu, C. Niu, C. Wei, et al., Angew. Chem. Int. Ed. 58 (2019) 2029–2033. doi: 10.1002/anie.201812475
L. He, N. Wang, M. Xiang, et al., Appl. Catal. B 345 (2024) 123686. doi: 10.1016/j.apcatb.2023.123686
Y. Zhang, Y. Zhang, H. Zhang, et al., Coord. Chem. Rev. 448 (2021) 214147. doi: 10.1016/j.ccr.2021.214147
H. Chen, X. Liang, Y. Liu, et al., Adv. Mater. 32 (2020) 2002435. doi: 10.1002/adma.202002435
Y. Zhang, X. Shen, C. Song, Z. Ji, F.H. Du, J. Mater. Chem. A 11 (2023) 8904–8911. doi: 10.1039/d3ta00635b
H. Liao, T. Luo, P. Tan, et al., Adv. Funct. Mater. 31 (2021) 2102772. doi: 10.1002/adfm.202102772
Y. Shi, W. Du, W. Zhou, et al., Angew. Chem. Int. Ed. 59 (2020) 22470–22474. doi: 10.1002/anie.202011097
S. Yu, N. Zhang, J. Li, et al., ACS Sustain. Chem. Eng. 12 (2024) 4551–4564. doi: 10.1021/acssuschemeng.3c07704
J. Lv, P. Liu, R. Li, et al., Appl. Catal. B 298 (2021) 120587. doi: 10.1016/j.apcatb.2021.120587
J.G. Li, H. Sun, L. Lv, et al., ACS Appl. Mater. Interfaces 11 (2019) 8106–8114. doi: 10.1021/acsami.8b22133

Figure 1 (a) Synthesis illustration, (b) TEM image, (c) HRTEM image, (d) selected area electron diffraction (SAED) patterns, (e) HAADF-STEM image, (f-i) element mapping images and (j) XRD patterns of Fe-CoS2/MoS2.

Figure 2 High resolution XPS spectra of Fe-CoS2/MoS2, CoS2/MoS2 and Fe-CoS2: (a, e) Co 2p, (c) S 2p and (d) Mo 3d. (b) Diagram of energy band and BEF in Fe-CoS2/MoS2. (f) VBS of Fe-CoS2/MoS2, CoS2/MoS2 and Fe-CoS2.

Figure 3 (a, g, k) LSV curves, (b, h) Tafel plots, (c) Nyquist plots, (d) Cdl and ECSA values, (e, i, l) CP curves and (f, j) LSV curves before and after CP test in OER, HER and OWS.

Figure 4 (a) TEM image, (b) HRTEM image, (c) XRD pattern of Fe-CoS2/MoS2 after OER. (d) “MS-MOOH” core-shell structure. XPS spectra for (e) Co 2p, (f) Mo 3d, (g) S 2p, and (h) O 2p of Fe-CoS2/MoS2 before and after OER. (i) The electron configuration of Co, Fe and Mo in the core-shell interface. (j) Electronic coupling in “Mo-O-Co-O-Fe” unit model.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: