Center for Carbon-based Electronics and Key Laboratory for the Physics and Chemistry of Nanodevices, School of Electronics, Peking University, Beijing 100871, China
b.
Spin-X Institute, School of Microelectronics, South China University of Technology, Guangzhou 511442, China
c.
School of Materials and Energy, Guangzhou Key Laboratory of Low-Dimensional Materials and Energy Storage Devices, Guangdong University of Technology, Guangzhou 510006, China
d.
Faculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming 650093 China
e.
i-Lab, CAS Key Laboratory of Nanophotonic Materials and Devices, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou 215123, China
f.
CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, CAS Research/Education Center for Excellence in Molecular Sciences, Beijing National Laboratory for Molecular Sciences (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
g.
State Key Laboratory for Organic Electronics and Information Displays & Jiangsu Key Laboratory for Biosensors, Institute of Advanced Materials, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing University of Posts and Telecommunications, Nanjing 210023, China
h.
Institute of Functional Nano and Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials and Devices, Soochow University, Suzhou 215123, China
i.
Hefei National Research Center for Physical Sciences at the Microscale and Synergetic Innovation Center of Quantum Information and Quantum Physics, University of Science and Technology of China, Hefei 230026, China
j.
Institute of Physics & University of Chinese Academy of Sciences, Beijing 100190, China
k.
State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
l.
College of Chemistry, Beijing Normal University, Beijing 100875, China
m.
Beijing National Laboratory for Molecular Sciences (BNLMS), College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
n.
School of Material and New Energy, South China Normal University, Shanwei 516600, China
o.
International Center for Quantum Materials, Collaborative Innovation Center of Quantum Matter, Interdisciplinary Institute of Light-Element Quantum Materials and Research Center for Light-Element Advanced Materials, School of Physics, Peking University, Beijing 100871, China
p.
Herbert Gleiter Institute of Nanoscience, School of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China
q.
Research Center of Green Catalysis, College of Chemistry, Zhengzhou University, Zhengzhou 450001, China
r.
Key Laboratory for Advanced Materials and Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, State Key Laboratory of Chemical Engineering, School of Chemistry and Molecular Engineering, East China University of Science Technology, Shanghai 200237, China
s.
School of Physics and Information Technology, Shaanxi Normal University, Xi’an 710119, China
t.
Department of Chemistry, Northeast Normal University, Changchun 130024, China
u.
CAS Key Laboratory of Standardization and Measurement for Nanotechnology, National Center for Nanoscience and Technology, Beijing 100190, China
v.
College of Science, Henan Agricultural University, Zhengzhou 450002, China
w.
School of Physics and Wuhan National High Magnetic Field Center, Huazhong University of Science and Technology, Wuhan 430074, China
x.
Department of Physics, School of Physics and Materials Science, Nanchang University, Nanchang 330033, China
y.
State Key Laboratory of Low-Dimensional Quantum Physics, Department of Physics, Tsinghua University, Beijing 100084, China
z.
Donostia International Physics Center, Centro de Fisica de Materiales CFM/MPC, CSIC-UPV/EHU, 20018 San Sebastián, Spain
aa.
Institute for Molecular Science, National Institutes of Natural Sciences, Okazaki, Aichi 444-8585, Japan
ab.
Interdisciplinary Materials Research Center, School of Materials Science and Engineering, Tongji University, Shanghai 201804, China
ac.
Tianjin Key Laboratory of Epigenetics for Organ Development of Preterm Infants, Central Laboratory, Tianjin Fifth Central Hospital, Tianjin 300450, China
ad.
School of Physical Science and Technology, Center for Transformative Science, ShanghaiTech University, Shanghai 201210, China
ae.
School of Chemical Science, University of Chinese Academy of Sciences (UCAS), Beijing 100049, China
af.
National Synchrotron Radiation Laboratory, Department of Chemical Physics and Key Laboratory of Surface and Interface Chemistry and Energy Catalysis of Anhui Higher Education Institutes, University of Science and Technology of China, Hefei 230029, China
ag.
Department of Chemical Sciences National Natural Science Foundation of China, Beijing 100085, China
Received Date:
09 February 2024 Accepted Date:
26 May 2024 Revised Date:
14 May 2024 Available Online:
15 December 2024
Abstract:
Surface chemistry focuses on the investigation of the adsorption, migration, assembly, activation, reaction, and desorption of atoms and molecules at surfaces. Surface chemistry plays the pivotal roles in both fundamental science and applied technology. This review will summarize the recent progresses on surface assembly, synthesis and catalysis investigated mainly by scanning tunneling microscopy and atomic force microscopy. Surface assemblies of water and small biomolecules, construction of Sierpiński triangles and surface chirality are summarized. On-surface synthesis of conjugated carbo- and heterocycles and other kinds of carbon nanostructures are surveyed. Surface model catalysis, including single-atom catalysis and electrochemical catalysis, are discussed at the single-atom level.
Surface chemistry is a fascinating interdisciplinary field that focuses on investigating chemical processes occurring on the surfaces of solid substances. It involves studying the adsorption, migration, assembly, activation, reaction, and desorption of atoms and molecules at surfaces [1-27]. The main objective is to understand the structures and properties of solid surfaces and uncover fundamental principles that govern the physical and chemical changes happening on them [28-31]. Surface chemistry combines knowledge from various disciplines such as chemistry, physics, biology, mathematics, materials, and engineering. It acts as a crucial bridge between and fundamental disciplines and different application areas. Surface chemistry plays a pivotal role and has a profound impact in catalysis, energy, information, and materials science. Over the past century, surface chemistry has experienced rapid development and achieved remarkable accomplishments. Notably, I. Langmuir, an American scientist, was awarded the Nobel Prize in Chemistry in 1932 for his significant contributions to the proposal and study of surface chemistry [32]. Similarly, G. Ertl, a German scientist, received the Nobel Prize in Chemistry in 2007 for his groundbreaking research in solid surface chemistry [33].
Scanning tunneling microscopy (STM) and atomic force microscopy (AFM) as well as other scanning probe microscopies (SPM) are powerful tools for imaging, spectroscopy and manipulation at the single-atom level. This review will summarize the recent progresses on surface assembly, synthesis and catalysis investigated mainly by SPM. First, controlled molecular surface assembly and precise characterization are summed up. New structures of water on surfaces are studied by hydrogen-sensitive scanning probe microscopy. Various assembled structures are prepared by tuning intermolecular interactions, substrates and guests molecules. Fractal Sierpiński triangles are constructed through hydrogen bonds and coordination interactions. The induction, amplification and transfer of surface charity are elucidated. In the second part, recent advances about on-surface synthesis are outlined. Different kinds of surface reactions are introduced, such as dehalogenation, dehydrogenation reactions, surface-supported polymerization reaction, and phenyl migration reactions. The application of these reactions to synthesize conjugated carbo- and heterocycles and other kinds of carbon nanostructures are surveyed. In the third part, we summarize the progresses on the surface model catalysis, including single-atom catalysis, cluster catalysis and surface catalysis. Finally, we recapitulate the basic principle of electrochemical STM and its application on the investigation of adsorption, diffusion, reconstruction and catalytic reaction.
2.
Assembly on surfaces
2.1
Water investigated by hydrogen-sensitive scanning probe microscopy
The interaction of water with solid surfaces is of vital importance to an incredibly wide range of scientific disciplines and applied fields, such as the nucleation and growth of ice, water desalination, heterogeneous catalysis, electrochemistry, wetting and corrosion [34,35]. Over the past several decades, water-solid surfaces/interfaces have been intensively studied by surface-sensitive imaging and spectroscopic techniques [35], including vibrational spectroscopy [35,36] (reflection absorption infrared spectroscopy (RAIRS), high resolution electron energy loss spectroscopy (HREELS)), He scattering [37], low-energy electron diffraction (LEED) [35], X-ray absorption/photoelectron spectroscopy (XAS/XPS) [38] and SPM [39]. Here, we want to introduce the recent advances of qPlus-based noncontact AFM (nc-AFM) and scanning tunneling microscopy/spectroscopy (STM/S), which enable to identify the protons in water in real and energy space. Then we will discuss applications of H-sensitive STM/AFM in probing the structure, dynamics and nuclear quantum effects of interfacial water and ion hydrates. At last, we present an outlook on the further directions and challenges for SPM techniques in studying water-solid interfaces.
2.1.1
H-sensitive scanning probe microscopy and spectroscopy
2.1.1.1
Orbital imaging by STM
Many fundamental and key issues of water-solid interfaces are closely related to the protons in water, such as the structure of H-bonded networks, hydrogen-atom transfer through H-bonds and nuclear quantum effects of water [35,40-49]. As a consequent, identifying the protons in water is indispensable for the accurate characterization of the structure and dynamics of interfacial water. Using STM, frontier orbitals of individual water molecules has been visualized with submolecular resolution [50], which allowed to discriminate the O—H directionality of water monomers and tetramers on Au(111) supported NaCl(001) surface in real space. This is achieved by decoupling water electronically from the Au(111) metal substrate with bilayer insulating NaCl film, then the intrinsic molecular orbitals are preserved. Moreover, the molecular density states (DOS) of water around fermi level could be modified by tuning the tip-water coupling.
2.1.1.2
H-sensitive qPlus-AFM imaging
However, determining the molecular structure through orbital imaging is not straightforward and is usually aided by density functional theory (DFT) calculations. The development of qPlus-based AFM technique (Fig. 1a) presents the capability in identifying the chemical structures of molecules on surfaces with superior resolution [51-54]. The H-bonding skeleton of interfacial water has been directly visualized [55,56] by measuring the short-range Pauli repulsive force [57] with a functionalized tip, which providing the opportunity for seeing the water molecules on various surfaces, especially for the insulating substrates and thick ice. In order to further discern the H atoms of water molecules, the imaging mechanism based on electrostatic interactions was proposed [58], according to the fact that oxygen and hydrogen atoms in water are oppositely charged. DFT calculation revealed that the CO molecule at the tip apex was slightly negatively charged, showing quadrupole-like charge distribution (inset of Fig. 1a). The weak high-order electrostatic force between the quadrupole-like CO-terminated tip and the polar water molecules is more sensitive to the atomic details comparing with the conventional electrostatic force between the monopole tip and water, which presents a featureless attractive background in the AFM images.
Figure 1
Figure 1.
(a) Schematic of the qPlus-AFM setup. The inset shows the calculated charge distribution of the CO at the tip apex. (b) High-resolution AFM images and the atomic models of water dimer (left panel), tetramer (middle panel) and sodium ion hydrates (right panel) on NaCl(001)/Au(111) surface. (c) Left panel: Force curves of individual hydroxyls on the In2O3(111) surface with OH-terminated tip. Right panel: Schematic of the experiment setup. (d) The correlation between H bond length and the force minima. (e) Schematic of the experimental setup and d2I/dV2 spectra of a water monomer on NaCl(001)/Au(111) surface with a Cl-tip (R for rotational, B for bending, and S for stretching). Reproduced with permission [58-62]. Copyright 2018, 2021, Springer Nature; Copyright 2016, American Chemical Society; Copyright 2016, The American Association for the Advancement of Science; Copyright 2018, CC-BY 4.0.
As shown in Fig. 1b, using a CO-terminated tip, the AFM images of water clusters and ion hydrates acquired at large tip heights present submolecular contrast [58,59], in which the crooked depressions (dashed curves) and the bright protrusions (white arrows) steam from the positively charged hydrogen and the negatively charged oxygen, respectively. As a consequence, the position of the H atoms is directly visualized in real space with a nearly nonperturbative manner by measuring the weak high-order electrostatic force, and the structures of H-bonding networks are determined with atomic precision. The H-sensitive AFM imaging method could be applied to a wide range of H-bonding molecular systems.
2.1.1.3
Force curve measurement
Except for high-resolution imaging, force curves enable to quantitatively obtain the height difference of water molecules in the H-bonding network. More recently, the H-bonding strength at the single-bond level could be measured by recording the minima of force (F(z)) between the individual hydroxyl on oxide surface and OH-functionalized tip (Fig. 1c) [60]. It is revealed that the minima of F(z) show a linear correlation with the H-bond length between the tip and the surface OH group (Fig. 1d). The weaker H-bond formed between the tip and surface hydroxyl leads to the stronger OH covalent bond within hydroxyl gets due to the competition for the proton. Consequently, measuring the force minima provides the route for the direct assessment of the acidity of individual surface hydroxyls.
2.1.1.4
Tip-enhanced IETS
In spite of the superior resolution of STM/AFM, they are poor in chemical identification. Inelastic electron tunneling spectroscopy (IETS) based on STM, evented by Ho et al., provides the opportunity for overcoming this limitation [63,64]. STM-IETS enables to probe the vibrational signals of molecules at the single-bond limit, discriminate different isotopes at the single-molecule level and identify the chemical structures with submolecular resolution [63,65,66]. In addition, tip-enhanced IETS has been developed by tuning the IET process from off-resonance to near/on-resonance [67,68]. As shown in Fig. 1e, a Cl-terminated tip was introduced to selectively gate the highest occupied molecular orbitals (HOMO) of water molecule toward EF, which significantly enhances the electron-vibration coupling, resulting in a resonantly enhanced IET process [62]. In such a resonance case, the d2I/dV2 spectrum of a single water molecule shows the prominent vibrational features of rotational (R), bending (B) and stretching (S) modes. In particular, the H-bonding strength could be obtained by tracking the red shift of OH stretching mode, which allows to extract the nuclear quantum effects (NQEs) of H bonds at the single-bond level (see details in Section 2.1.3). Therefore, tip-enhanced IETS opens up a new way for accessing the H degrees of freedom of interfacial water in energy space.
2.1.2
Adsorption, nucleation and reaction of water on solid surfaces
2.1.2.1
Water clusters
With the development of STM and nc-AFM, directly visualizing the atomic structure of interfacial water become possible. The original "bilayer ice" model proposed by TM [69] and Henderson [34] was found to be unsuitable for describing the diversity water structures at the interface. At temperatures below 20 K, the internal structure of the metal-supported water hexamer was identified as the initial cluster on close-packed Cu(111) [40] and Au(111) [70] surface (Figs. 2a and b). Recently, using qPlus-AFM with a CO-terminated tip, Chen et al. [71] unambiguously visualized the common water clusters (Fig. 2c), which are composed of a centrally flat lying hexamer surrounded by three pentagonal rings, on both hydrophilic Pt(111) and hydrophobic Cu(111) surfaces. The common clusters are critical nucleus, which play a key role in the initial stages of ice nucleation. However, they are only kinetically stable since there is not enough thermal energy for free diffuse. As the temperature increased to 120–160 K, water molecules gain sufficient thermal energy and can reassemble into ordered 1D [42,55,72], 2D [44,48,56,73] and amorphous 3D [74] structures or partially dissociated structures on high chemical reactivity metal surfaces [55,75].
Figure 2
Figure 2.
(a) 3D STM image of water hexamer on Au(111) surface. (b) High-resolution STM image and corresponding atomic model of water hexamer on Cu(111) surface. (c) STM, height-dependent AFM, and proposed atomic model of water clusters on Pt(111) and Cu(111) surface. (d) Height-dependent AFM, and proposed atomic model of () R19° monolayer ice on Ni(111) surface. (e) Height-dependent AFM and atomic model of buckled BHI on corrugated Au(110)-1 × 2 surface. (f) Constant-height AFM images and corresponding atomic model of bilayer hexagonal ice on Au(111) surface. (g) Growing process for zigzag (left) and armchair (right) edges. Reproduced with permission [40,56,70,71,73,89]. Copyright 2018, American Chemical Society; Copyright 2007, 2020, Springer Nature; Copyright 2019, 2022, American Physical Society; Copyright 2023, CC-BY 4.0.
The adsorption behavior of water on metal surface is determined by the competition between water-water and water-substrate interactions. On hydrophilic metal surfaces, water often exhibits wetting behavior due to strong interaction with metal surfaces such as Ni, Pd, Pt, Ru. Specifically, the first water wetting layer comprising fused pentagonal, hexagonal and heptagonal rings on close-packed hydrophilic metal surface (Ni(111) [76], Pd(111) [77,78], Pt(111) [44,48,79] and Ru(0001) [48,77,78]), was characterized by STM. Recently, Shiotari et al. [56] directly imaged the hydrogen-bonding structures of the () R19° monolayer ice on Ni(111) surface using qPlus-AFM (Fig. 2d). This work revised the previous DFT-optimized " di-vacancy structure" [76] according to the force-curve measurements. Apart from close-packed metal surface, there is considerable interest in high chemical activity open surfaces and high-index metal surfaces. For example, on Cu(110) [55] and Ni(110) [80] surface, water molecules will partially dissociate, forming 1D or 2D mixed water-hydroxyl structures at higher temperatures (> 150 K). More intriguingly, the first layer on the stepped Cu(511) surface adopts an "ice-like" arrangement [81,82], creating ordered H-up water molecules as donor sites to stabilize the second and continued multilayer ice growth. In fact, the NaCl(001) surface is also regarded as a hydrophilic surface. Chen et al. reported a novel type of 2D ice formed by water tetramers on an insulating NaCl(001) film [83]. Each water tetramer was treated as a single unit and could interconnect into 1D tetramer chains and a 2D wetting layer through a novel bridging mechanism. Interestingly, this tetragonal bilayer ice breaks the Bernal-Fowler-Pauling ice rules and might be a general occurrence on hydrophilic surface with tetragonal symmetry.
2.1.2.3
2D ice on hydrophobic surfaces
On hydrophobic metal surfaces, the adsorption structure of ice is dominated by water-water H-bonding interaction, often resulting in non-wetting behavior, as observed in surfaces like Cu(111) [74] and Ag(111) [40]. However, an intrinsic ice crystal known as bilayer hexagonal ice (BHI) has been reported on hydrophobic metal surface. BHI was firstly predicted to exist in hydrophobic nanopores by Koga et al. using molecular dynamic (MD) simulations [84] and was subsequently verified on graphene [85], graphite [86], and Au(111) [87,88] surfaces. Recently, Ma et al. [73] directly observed the interlocked BHI on Au(111) surface using qPlus-AFM (Fig. 2f). BHI is composed of two complete hexagonal layers of water molecules. In each water layer, half of the water molecules lie flat (parallel to the substrate), while remaining half stand vertical (perpendicular to the substrate), with one O–H oriented either upward or downward (right panel of Fig. 2f). The ultrahigh resolution of nc-AFM allowed researchers to image the delicate edge structures of BHI, enabling the reconstruction of growth processes combined with MD simulations. For zigzag edges (left panel of Fig. 2g), the growth mechanism, named as collective bridging, involves adding water molecules to the periodic array of pentagons, forming a 56, 665-type structures, and ultimately reverting to the original zigzag edge by adding more water molecules. For armchair edges (right panel of Fig. 2g), the growth process starts with the 5756-type armchair edge, then converts to 5656-type member by adding water molecules, finally leading to the reformation of 5756-type armchair edge through a local seeding mechanism. Notably, these two mechanisms were nicely reproduced by MD simulation, thus might be considered as general phenomenon for 2D ice.
So far, most studies of BHI focus on flat hydrophobic surfaces with hexagonal symmetry. To investigate whether BHI can still exist on corrugated surface with non-hexagonal symmetry, Yang et al. conducted experiments by dosing water onto reconstructed corrugated surface (Fig. 2e) [89]. Surprisingly, using nc-AFM with a CO-tip, they directly visualized the formation of hydrophobic BHI on both Au(110)-1 × 2 and Au(100)-5 × 28 surface with periodic 1D reconstruction. Upon depositing water molecules on the Au(110) surface, a monolayer hexagonal ice formed. In this structure, water molecules on the topmost gold rows exhibited a flat lying configuration, while those in the trenches adopted the H-down configuration. With an increase in coverage, the water molecules in first layer rearranged and adjusted H-bonding orientation, resulting in the formation of a buckled BHI. In addition, they also observed the formation of BHI on Au(100)-5 × 28 surface, further suggesting the generality of BHI. Notably, BHI appears to be quite flexible, making minimal adjustments in H-bonding orientation to accommodate different substrates. The flexibility and robustness of BHI suggests the potential for wide-ranging applications as a new type of 2D material in anti-icing, super-lubrication, and emergent quantum matters.
2.1.2.4
Reaction of water on metal oxide surfaces
The interaction between water and metal oxide surfaces has attracted considerable interest, due to the applications in numerous scientific and technological fields [34]. In comparison to metal surfaces, water molecules typically engage in strong interactions with metal oxides, often leading to dissociation [34,36,46,69,90,91]. This dissociation behavior is attributed to the lone pair on the O atom forming a strong dative bond with the coordinatively unsaturated metal cations. Concerning the kinetic and dynamics of interfacial water on metal oxide surfaces, a fundamental challenge lies in identifying the binding site and dissociation mechanism of water molecules. STM and qPlus-AFM, owing to their extremely high resolution, are well-suited for addressing these challenges.
Rutile TiO2(110) is one of the most extensively studied oxide surfaces [69,92-94], primarily due to its crucial role in advancing our understanding of photocatalytic water splitting. Regarding water adsorption on rutile TiO2(110) surfaces, both experiment and theoretical results consistently indicate that water adsorbs preferred on the surface O vacancies, leading to dissociation into a pair of hydroxy groups (OsurfH) [95]. Interestingly, Wang et al. [96] employed hyperthermal water beams with different energy to probe the water dissociation equilibrium on defect-free rutile TiO2(110). The temperature-dependent experiment yields the deprotonation/protonation barrier of 0.36 eV and the molecule water is preferred over the hydroxy pair by 0.035 eV Besides rutile TiO2(110), research on the adsorption of water molecules on anatase TiO2(101) indicate the water molecules remain intact [97,98].
Apart from the single water molecule, Ma et al. investigated the influence of hydrogen bonding network on water splitting at the anatase-TiO2(001)-(1 × 4) surface [49]. Under light irradiation, the ultraviolet photoelectron spectra (UPS) revealed interesting gap states (GS) emerging at 1.6 eV below EF after water desorption (bottom of Fig. 3a), attributed to the water splitting at the terrace sites, as confirmed by XPS results. Moreover, in situ XPS demonstrated the reduction of Ti4+to Ti3+ after water desorption, signifying that the yielded hydroxy groups supplied excess electrons to the surface, leading to the emergency of the GS. More interestingly, DFT calculations showed that, in contrast to 1H2O and 2H2O, the 3H2O configuration could significantly reduce the barrier to 0.08 eV (Fig. 3b). The third water not only connected adjacent water molecules but also formed H-bonds with the surface bridging O atom, ultimately facilitating water splitting. The critical role of the hydrogen bonding network in decreasing the water dissociation barrier and assisting proton transfer might serve as a general paradigm for water splitting on metal oxides.
Figure 3
Figure 3.
(a) Top: STM image of the water-adsorbed anatase-TiO2(001)-(1 × 4) surface (setpoint: 1.2 V, 10 pA). Bottom: The UPS spectra obtained after the water adsorption. (b) Top: Simulated water splitting pathways, considering 1H2O, 2H2O, and 3H2O configurations. Bottom: Calculated potential energy surface along the water splitting pathways. (c) Dissociated water forming a "dynamic ion pair" on the SrO-terminated surface of Sr3Ru2O7. Top: Adsorption geometry of the lowest-energy configuration (left) and consecutive STM images of water monomer motion at 78 K (right). Bottom: The hopping model with DFT, showing the initial, transition, and the final state. (d) Arrhenius plot for the "dynamic ion pair" motion. (e) Top: Height-dependent AFM images of different water coverage (left: 2.5 H2O/unit cell, right: 8 H2O/unit cell). Bottom: O 1s XPS data show that water molecules are partially dissociated. (f) Top view of the minimum-energy structures calculated by DFT. Reproduced with permission [47,49,101]. Copyright 2022, American Chemical Society; Copyright 2015, Springer Nature; Copyright 2018, National Academy of Sciences.
Over the past few years, multiple research groups have reported that partially dissociated water dimers represent the most stable configuration on certain metal oxide surfaces [99,100]. Recently, Halwidl et al. [47] investigated the adsorption of water molecules on the SrO-terminated surface of Srn+1RunO3n+1 (n = 1, 2), which consists of n layers SrRuO3 and separated by two layers of SrO plane (Fig. 3c). DFT calculations indicated that with an increase in lattice constant, a single water molecule on the SrO surface is expected to dissociate, in contrast to the MgO surface [43] where water molecules remain intact. Indeed, using STM, Halwidl et al. [47] directly visualized the water dissociation, where the (OH)ads species is absorbed in the bridge site of Sr-Sr, and the H is transferred to the neighboring O atom, forming a "dynamic ion pair" composed by (OH)ads and OsurfH. Fitting the Arrhenius equation gives the frequency (10 s−1) and activation energy (187 meV) of this "dynamic ion pair" (Fig. 3d), which is in good agreement with the DFT calculations. With increasing coverage, dissociated water dimers came together to form one-dimensional chains, finally establishing partially dissociated H-bonding networks.
Although notable advancements in metal oxides have been achieved with STM in recent time, qPlus-AFM has emerged as a technique boasting superior resolution [101], especially when functionalized with a CO tip. In a recent study, Meier et al. [101] systematically investigated water adsorption and dissociation on () R45°Fe3O4(001) surface using nc-AFM (Fig. 3e). At low water coverage, they identified partially dissociated water dimers through a combination of qPlus-AFM and XPS results. These dimers can serve as anchors for further water molecules desorption, leading to the formation of partially dissociated trimers, finally generate H-bonding networks wetting the entire surface. In addition, the minimum-energy structures of dimer, trimer and H-bonding network were determined combined with DFT calculations (Fig. 3f).
2.1.3
Nuclear quantum effects of interfacial water
2.1.3.1
Physical picture of NQEs
The anomalous macroscopic properties and isotope effects of water largely arise from the quantum nature of protons, that is, nuclear quantum effects (NQEs) of water (Fig. 4a) [102]. Besides over-barrier hopping, the proton could transfer through quantum tunneling when the height and width of the potential barrier is small. Another quantum behavior of proton is quantum fluctuation or zero-point motion (ZPM) because of the Heisenberg uncertainty principle. The anharmonicity of the O—H stretch in the potential energy surface gives rise to the expansion of O—H covalent bond and the enhancement of H-bonding strength. When the zero-point energy dominates over potential barrier, the proton will completely delocalize, forming a symmetric H bond. Extensive theoretical studies have been made to achieve the accurate and efficient description of NQEs of water [61,103]. However, the experimental quantitative characterization and detection of NQEs is still a great challenge, especially at the single H-bond level.
Figure 4
Figure 4.
(a) Left panel: Symmetric double-well potential. Middle panel: Schematic of the harmonic (red) and an-harmonic (blue) zero-point motion of the proton. Right: Model of symmetric H bond. (b) Tunneling current on the chirality interconversion of tetramer on NaCl(001) surface. (c) NQEs on the H-bonding strength. (d) STM image and atomic model of water-hydroxyl cluster on Cu(110) surface with a symmetric H-bond. (e) AFM image (left panel) of Zundel cation and the snapshot (middle panel) from PIMD calculation. Right panel: The probability distribution of proton from PIMD and MD simulations of Zundel-water structures. Reproduced with permission [62,102,104,107,108]. Copyright 2010, American Physical Society; Copyright 2015, Springer Nature; Copyright 2016, 2022, The American Association for the Advancement of Science; Copyright 2017, Elsevier.
2.1.3.2
Concerted proton tunneling and NQEs of H bond strength
Using a Cl-terminated tip with STM, Meng et al. directly visualized the concerted many-body proton tunneling in a water tetramer on NaCl(001)/Au(111) surface [104]. This is realized by recording the tunneling current to monitor the reversible interconversion of the different H-bonding chirality states of water tetramer (Fig. 4b). The proton transfer dynamics was proved to be irrelevant to the bias, tunneling current and the temperature (5–15 K). In addition, the concerted tunneling manner of four protons was further confirmed by isotope substitution experiments, in which the H2O and D2O mixed tetramer was constructed and discerned by vibrational features using tip-enhanced IETS [105]. Moreover, the vibrational spectroscopy of isolated HOD molecules enables to extracted the H-bonding strength of hydrogen and deuterium, respectively. Consequently, Guo et al. achieved a quantitative assessment of the NQEs on the strength of a single H-bond, revealing that the quantum component of H-bond could be as large as 14% of the bond strength (Fig. 4c) [62]. This work further unraveled that the anharmonic quantum motion of hydrogen nuclei weaken the weak hydrogen bonds and strengthen the relatively strong ones, hinting that symmetric H-bond could emerge at strong H-bonding system.
2.1.3.3
Visualizing symmetric H bond
Kumagai et al. reported the formation of symmetric H-bond in a water-hydroxyl cluster on Cu(110) surface (Fig. 4d) [106]. Very recently, Tian et al. directly visualized the completely delocalization of protons in monolayer water on metal surfaces [107], using H-sensitive nc-AFM technique in a noninvasive manner (Fig. 4e). This is achieved by doping hydrogen atoms on the water layer on Au(111) surface, resulting in the formation of Zundel-type hydronium (H5O2+). The AFM image of Zundel cation exhibits a dimer-like feature (Fig. 4e), in which the bright protrusions are attributed to the oxygen atoms in water and the depression at the center of dimer denotes the shared H+. Path integral molecular dynamics (PIMD) simulations based on DFT calculations confirm the totally delocalization of the extra proton between two water molecule and the simulated AFM image of the PIMS snapshot agrees well with the experimental AFM image (Fig. 4e). More interestingly, the H-bonding network of the Zundel-water overlayer is considerably compressed comparing with the water H-bonding network on Au(111), suggesting the stronger H-bonding strength in the Zundel-water layer which consists of symmetric H bonds. This work opens up a new route for generating symmetric H bond in light-elements systems stabilized by NQEs.
2.1.4
Structure and dynamics of hydrates
2.1.4.1
Molecular hydration
Hydrations are relevant to a wide range of environmental, chemical, biophysical and even technological processes. STM/AFM has been extensively applied to investigate the hydration process of molecules, including small adsorbates [59], organic molecules and biomolecules [109-111]. Cai et al. reported the water dimer induced the local chiral inversion of the adenine molecules, resulting in a mismatched H-bonding superstructure [111]. Xu et al. investigated the solvation of DNA bases and revealed the catalytic role of water molecule in the regulation of the corresponding H-bonding network [109]. They have further observed the water induced chiral separation of RNA bases on Au(111) surface [110]. These works significantly advance our understanding on the crucial role of water in the biomolecular systems.
2.1.4.2
Structure and proton transfer dynamics of hydrated protons
Water-ion interactions attract intensive interests in recent years because of the essential function in electrochemistry, biological ion channels, water desalination and aqueous batteries [112-116]. Using H-sensitive qPlus-AFM technique, Tian et al. directly visualized the Eigen- and Zundel-type hydrated protons within the H-bonding network of water on various metal surfaces (Au(111), Pt(111), Cu(111), Ru(0001)) [107]. They found that the Eigen cations (H9O4+) self-assemble into monolayer structures with local order, while the Zundel cations form long-range ordered structures with 3 × 3 periodicity due to nuclear quantum effects (NQEs) (Figs. 5a and b). Two Eigen cations can combine into one Zundel cation with simultaneous proton transfer from the water layer to the metal surface, corresponding to a new type of Volmer-like step in hydrogen evolution reaction (HER). In addition, it was revealed that the Zundel configuration is preferred on Pt(111) over the Eigen due to the smaller lattice constant and stronger water-surface interaction than Au(111). Such a preference is absent on Au(111), which sheds new light on different reaction kinetics of these two surfaces. Those results are expected to be general on noble metal surfaces and may shed new light on various electrode processes in acidic aqueous environments.
Figure 5
Figure 5.
High-resolution AFM images and the schematic models of (a) Eigen-water and (b) Zundel-water overlayers on Au(111) surface. (c) High-resolution AFM images and the schematic models of the Na+·nD2O, n = 2,3,5 cluster on NaCl(001)/Au(111) surface. (d) Tip-induced diffusion dynamics of Na+ hydrates. Top: Schematic diagram of the Au-mediated inelastic electron excitation of the Na+ hydrates with the STM tip on NaCl(001). Inset of Top: Current-bias relationship of Na+·3D2O with a Cl− tip. Bottom: Comparison of Veff for Na+·nD2O (n = 1–5) at d × the lattice constant. Reproduced with permission [59,107]. Copyright 2022, The American Association for the Advancement of Science; Copyright 2018, Springer Nature.
2.1.4.3
Atomic structure and transport dynamics of alkali ion hydrates
Based on the H-sensitive imaging method, Jiang's group has systematically investigated the hydration structure and transport dynamics of alkali ions (Figs. 5c-d) [59]. Using controlled tip manipulation, sodium ion hydrates with different hydration number (Na+·nD2O, n = 1–5) was constructed on NaCl(001) surface and the atomic geometries were identified unambiguously with the freedom of hydrogen precision (Fig. 5c). They found the pronounced transport capability of Na+·3D2O hydrates due to the symmetry mismatch between the hydrates and the surface lattice. More recently, they observed the nanoscale one-dimensional close packing of interfacial alkali ions at charged surfaces, which was driven by water-mediated attraction. Those results have clear implications for nanofluidic technology and shed new lights on how to control the ion transport in confined environments such as the nanochannels by surface/interface engineering and decoration.
With the development of STM and qPlus-AFM, superior high resolution has been achieved, which enables to access to the H degree of freedom both in real space and energy space. Using H-sensitive imaging and spectroscopic techniques, the following major breakthroughs in the field of water science have been obtained. (1) Atomic imaging of the edge structure and growth of the intrinsic 2D bilayer hexagonal ice [73]. (2) Visualizing the structure and transport dynamics of ion hydrates [59]. (3) Quantitative assessment of the NQEs of interfacial water at single-bond level [62,104,107]. These works not only substantially enrich our knowledges about interfacial water, but also provide the unprecedented opportunity to explore more challenging issues in this field.
Firstly, qPlus-AFM with functionalized tip offers the possibility in answering the below questions about the nucleation and growth of ice. How different surfaces influence the growth of ice? How many layers of water are required to achieve the transition from 2D to 3D ice? What is the structure of the ice surface? How to identify the structure of amorphous ice and the crystallization process? Secondly, the role of water and NQEs in heterogeneous catalysis, molecule/ion solvation, electrochemistry deserves in-depth exploration. Finally, emerging techniques such as combing SPM with advanced sample preparation techniques (such as electrospray ion beam deposition, aqueous freezing), chemical-sensitive spectroscopic methods and ultrafast technologies need to be developed and utilized to study more complex and realistic systems with both high spatial and time resolution, for example, biomolecules, liquid-solid interfaces and water molecules under confinement.
2.2
Assembly of small biomolecules
Among others, biological systems themselves are complex yet fascinating self-assembly systems [117-121]. For instance, DNA, a self-replicating material present in nearly all living organisms that carries generic information, is constructed primarily by the self-assembly of complementary base pairs through hydrogen bonding. Understanding the underlying mechanisms of molecular self-assembly and the contributing interactions, including the synergism and competition within these hierarchical interactions, at the submolecular or even atomic scale is of paramount importance to a wide range of scientific fields, including but not limited to chemistry, biology, and physics [122,123]. In this regard, SPM provides a valuable and versatile toolbox not only to visualize the morphologies and skeletons of individual molecules in real space, but also to discriminate the hierarchy of intermolecular interactions involved and determine the associated structural growing or transformation scenario. At the same time, atomically clean and flat surfaces serve as ideal two-dimensional playgrounds for the target molecules, with their mystery in the self-assembling processes to be unraveled.
Precise regulation of molecular assembly processes and structures is the key to the fabrication and functionalization of low-dimensional nanostructures. Nevertheless, the hierarchy of multi-level interactions and their interacting rules remain elusive in the assembled structures. Direct detection and regulation of these interactions in real space is an immediate means to unravel these mysteries. It has been reported that the reversible transformations between fractals and periodic structures can be achieved by introducing gas molecules (CO and CO2) [124], making it possible for precise regulation of molecular assembly processes and structures. As mentioned above, DNA is one of the most important fine molecular self-assembly systems in nature, and the bases and their derivatives play a key role in the precise replication process of DNA, making them exciting natural systems. By the introduction of metal [125-128], halogen [129,130], salt [131,132], and water [109,110,133-135] as regulating factors, Xu and coworkers have finely regulated the intermolecular non-covalent interactions and assembled structures and revealed the synergistic and competitive mechanisms of multi-level interactions within the assembled motifs.
Taking the interactions between bases and water as an example, Xu and co-workers have elucidated the mechanisms of water-induced molecular tautomerization [133] and realized water-induced structural transformation of molecular nanostructures [136] on surfaces. Based on the introduction of the alkali metal salt NaCl, the precise identification, controllable separation, and interconversion of two guanine (G) tautomers (the canonical G/9H and the non-canonical G/7H forms) were realized [131] on Au(111) earlier, where the competition in intermolecular interactions between the two G tautomers and Na and within the two G tautomers themselves was the key. On this basis, water molecules were then controllably exposed to the G-precovered Au(111) sample at a pressure of ~10−5 mbar under ultra-high vacuum conditions, and interestingly, the formation of a rare G tautomer, G/(3H, 7H), was induced and visualized in real space [133]. Further theoretical calculations elucidated that multiple H2O molecules could construct "water bridges" via hydrogen bonds, suspended above the G molecular plane, and assist in the intramolecular proton transfer processes in a domino fashion, thus reducing the reaction barrier of tautomerization. In addition, the regulating role of water on the self-assembled structures on surfaces was also explored. By exposing the self-assembled nanostructure of adenine (A) molecules to an increasing amount of water, ranging from 10−6, 10−5, to 3 × 10−5 mbar, the gradual structural transformation was experimentally captured, showing a dynamic hydration process [136]. Besides, such a regulating effect of water molecules was driven by the formation of a more stable hydrogen-bonded structure with the involvement of water molecules. To further reveal the interplay between biomolecules and water, two other base molecules, i.e., cytosine (C) [109] and uracil (U) [110], were further explored under the influence of water molecules, as shown in Figs. 6a-k and l-o, respectively.
Figure 6
Figure 6.
STM images showing the structural transformation of cytosines from (a, b) disordered networks with T-junctions to (c, d) one-dimensional cytosine chains on Au(111) after water exposure. Nc-AFM images of the typical (e), T-junctions (f) and one-dimensional chains superimposed with the corresponding structural models (g, h). (i-k) Representative zoomed-in images of molecular dynamics simulations of the transformation of cytosine chains, where red, blue, and green dots represent weak, water-involved, and strong hydrogen bonds, respectively. Reproduced with permission [109]. Copyright 2020, American Chemical Society.
Upon deposition of C molecules onto Au(111), disordered molecular networks were formed in a "glassy state" (Fig. 6a), with the existence of a large number of T-junctions as typically indicated by red arrows (Fig. 6b). Interestingly, after exposing such a C-precovered sample to water atmosphere at a pressure of ~10−5 mbar, an obvious phase transition was observed, with the formation of one-dimensional extended C chains (Fig. 6c). Notably, all the T-junctions were broken up into parallel chains, while the morphology of each molecule appeared to be similar, and water molecules were found to be absent from the molecular structure (Fig. 6d). With the assistance of high-resolution nc-AFM images, the molecular skeletons were clearly resolved, indicating the involvement of various hydrogen-bonded C dimers. The strong and weak hydrogen-bonded dimer configurations (i.e., dimers 1–8 and 9–14) were highlighted with hydrogen bonds depicted in green and red dotted lines, respectively. Before exposure to the water atmosphere, the typical T-junctions (Figs. 6e and f) were composed of several weak hydrogen-bonded C dimers, e.g., dimers 9 and 13, while stronger dimers 1–8 were generally involved in the molecular chain segments. After water exposure, strong C dimer configurations were well preserved (Figs. 6g and h), yet, weaker ones disappeared. DFT calculations further rationalized that the competition in the strength of hydrogen bonds within the hydrated C structures (C–H2O clusters) and within the pure C dimers played a significant role in driving the phase transition. The weaker hydrogen-bonded configurations were thus disturbed by water, resulting in the selective scissoring of T-junctions. Such a water-induced evolution scenario was further verified by the molecular dynamics (MD) simulations, showing the growth processes of C chains both with and without the participation of water molecules. In the absence of water molecules, T-junctions with weak hydrogen bonds were formed as indicated by the red arrow (Fig. 6i). On the contrary, in the presence of water molecules, water-involved hydrogen bonds were formed (indicated by the blue arrows in Fig. 6j), followed by the re-formation of strong hydrogen bonds within C chains after squeezing off water (indicated by the green arrows in Fig. 6k). Consequently, the competition of various hydrogen bonds and their evolution were unambiguously revealed based on the C + H2O prototypical system.
Similarly, based on the interactions between U molecules and water, the structural transformation from a racemic disordered U structure to homochiral U + H2O structures with chiral separation was obtained on Au(111). It is worth noting that in this case water molecules were trapped in the five-membered rings of homochiral U molecules, which appeared as dim dots in the STM image. Such a water-driven self-assembly process was found to be attributed to the preferential binding of water to specific sites of U molecules. It thus provides single-molecule insights into the role that water molecules may play in generating chirality in vivo.
In the above cases, the interactions between water and biomolecules were extensively explored, with competitive intermolecular hydrogen bonding occurring between water and biomolecules and within biomolecules themselves. Due to the relatively weak water-biomolecule interactions, water molecules generally incorporate into the molecular nanostructures and desorb from the hydrated structures in a global manner, while less attention has been paid to the local intermolecular interactions between water and biomolecules. Precise STM manipulation, which allows direct disturbance of individual molecules on surfaces [127,137-146], holds great promise for capturing and detecting dynamic processes in a local manner in real space, leading to the experimental discrimination and evaluation of various intermolecular interactions.
Recently, by applying the hydration of thymine (T) molecules as a model system, Xu and co-workers further explored the local dehydration process of the water-involved T structure in situ by STM lateral manipulations (Fig. 7a) [135]. In the water-involved T structure, a H2O molecule was located at the center of four T dimers (including two green dimers and two blue ones with R and L chirality, respectively), while two OH···O hydrogen bonds were formed between the water molecule and the adjacent T molecules stemming from green dimers. Note that different from the cases of bases C [109] and U [110] in the water atmosphere, where water molecules desorbed globally before desorption of the target biomolecules, the water-involved T structure survived before 350 K without water desorption or perturbation of the hydrogen-bonded configurations. It thus indicated relatively strong intermolecular interactions between water and T molecules in this case, which may allow the influence of water on the intermolecular interactions to be directly regulated and captured by locally removing individual water molecules. Subsequently, successive STM lateral manipulations were performed on individual water molecules as shown in Figs. 7b-e, i and j. When only one water molecule was removed leaving a single vacancy within the structure, the two adjacent green T dimers remained unchanged (Figs. 7b and c). Interestingly, once two water molecules were removed and absent on both sides of a green T dimer, the dimer simultaneously underwent a structural transformation to form the blue dimer with a local chiral inversion (Figs. 7d and e). In addition, a naturally existing situation (Fig. 7f) with characteristically the same structural arrangement as that shown in Fig. 7e was explored in detail by LT-STM and nc-AFM imaging, verifying the formation of a blue dimer with the hydrogen-bonding configuration preserved when both ends were devoid of water molecules (Figs. 7g and h). This also implied the green-to-blue dimer transformation (with chiral inversion) after removal of both water molecules (Figs. 7d and e). Furthermore, the generality of such a chiral inversion phenomenon was also demonstrated by removing all the water molecules on both sides of three consecutive green T dimers, resulting in the R-to-L chiral inversion for all of the three T dimers (Figs. 7i and j). In this way, a local dehydration process was experimentally triggered and detected, revealing the influence of local hydrogen bonding, and the chiral inversion was theoretically verified to be thermodynamically driven. Such a strategy is not only significant for understanding the fundamental mechanisms in biological self-assembly processes especially those driven by water in vivo, but is also extendable to other molecular self-assembly systems to detect and identify different types of non-covalent interactions.
Figure 7
Figure 7.
(a) Schematic illustration showing the local dehydration of water-involved T structure by removing the target water molecules via in-situ STM manipulation, leading to the local chiral inversion of certain T dimers. (b-e, i, j) Sequential STM manipulations on the water molecules (highlighted in blue solid circles) recorded in the same regions. (f) STM images showing the naturally existing water vacancies (depicted by white dotted circles) in the water-involved T structure. (g, h) Magnified STM and nc-AFM images confirming the formation of a blue T dimer in the absence of water molecules on both sides (white dotted circles). Reproduced with permission [135]. Copyright 2022, American Chemical Society.
Controlling molecular self-assemblies and nanostructures at surfaces is a crucial interdisciplinary realm within surface science. In-depth exploration of this captivating field could significantly contribute to the understanding of fundamental principles governing intermolecular interactions and unveil the potential applications of surface structures at the molecular level. The programmed self-assembly of well-designed molecular monomers facilitates the formation of desired nanostructures. Fractal structures are prevalent observed in nature with morphological feature, such as snowflakes, coastlines and leaves, characterized by its self-similarity and non-integer dimensional filling of space [147]. Fractal structures have attracted great attention because of its great significance in science, engineering, mathematics, and aesthetics. As a kind of representative fractal, Sierpiński triangles (STs) were proposed by the Polish mathematician Wacław Sierpiński in 1915 and then theoretically predicated via Monte Carlo simulations [148,149]. The simulation results demonstrated that STs exhibit distinct optical and electronic properties owing to their unique aperiodic yet ordered structures [150-152].
The earliest molecular-scale STs structures were experimentally synthesized by self-assembled DNA tiles [153,154]. Recently, many attempts have been made to generate defect-free STs on surfaces via various self-assembly strategies. Wu's group and Wang's group first reported a whole series of defect-free molecular fractals on metal surface by using specific 120° backbone as the precursor and synergistic halogen and hydrogen bonds as the driving forces [155]. This research paves the way for the preparation of various fractal structures. So far, STs have been fabricated on diverse surfaces via a range of intermolecular forces, encompassing electrostatic interaction, hydrogen bonding, halogen bonding, coordination interaction and covalent bonding [117,156-167].
Assembling STs into high order or large-scale two-dimensional crystal structures is imperative to explore the potential application of STs. But in fact, STs with varying sizes and orientations are dispersed irregularly on the surface due to the random formation of nucleation centers, hindering the dynamic formation of two-dimensional crystal structures. Wang's group has devoted significant efforts to exploring the growth mechanism of STs. By incorporating appropriate external constraints to guide the fractal units towards ordered arrays, they successfully achieved one-dimensional (1D) double chains of STs through the utilization of the templating effect of reconstructed Au(100) surface [158]. The diffusion anisotropy along different surface direction, resulting from high corrugation of the surface reconstruction, suppresses the formation of larger STs-n while promoting elongation of the STs-n along the [011] direction.
The recent work of Wang et al. have successfully fabricated 2D crystals composed of STs as building blocks by employing V-shaped 3-bis(4-pyridyl)-benzene (BPyB) molecule and Fe atoms on Au(111) [167]. Through the deposition of BPyB molecules and Fe atoms onto Au(111), followed by annealing, around 380 K, a series of equilateral STs structures are formed at low coverage, as shown in Figs. 8a-c. The fundamental structural unit of STs consists of three BPyB molecules and one Fe atom through metal-organic coordination (Fig. 8a). In Fig. 8c it illustrates the self-similarity of STs denoted as ST-n, where the order n ranges from 0 to 5. In Fig. 8b, an ST-5 is shown with a total count of 366 molecules and 243 Fe atoms, representing the highest order observed experimentally thus far. Previously, ST-5 were only obtained by combining co-assembly methods and the templating effect of reconstructed Au(100)-(hex) [160]. At high molecular coverages, STs with the order from 0 to 2 are assembled into well-defined 2D crystals on Au(111), as depicted in Figs. 8d-g. These networks are designated as ST-(n + n). By gradually reducing the coverage from values such as 0.80, 0.75, and down to 0.57 ML respectively, the networks of ST-(0′+1), ST-(1 + 1), ST-(1 + 2), and ST-(2 + 2) were accordingly obtained. For instance, upon closer examination and corresponding structural models for homotactic ST-(1 + 2) structures, it was revealed that one ST-1 connected with three vertex positions occupied by adjacent ST-2 triangles. The large size of 2D crystals suggests their thermodynamic stability on the surface due to favorable molecule-substrate interactions facilitated by both small sizes for BPyB molecules and appropriate molecular diffusion capabilities on Au(111). This enables error-correction mechanisms within nodal motifs during the formation process of STs.
Figure 8
Figure 8.
(a) Model depicting a coordination node formed via coordination interaction between three BPyB and one Fe atom. (b) STM image of a fifth-order STs on Au(111). (c) A series of BPyB-Fe-ST-n structures, ranging from 0 to 5, along with their respective models. (d-g) STM images and accompanying models of 2D crystals composed of ST-(n + n). Reproduced with permission [167]. Copyright 2020, American Chemical Society.
On the other hand, the size and symmetry of the fractal structures were match with the surface lattice of Au(111), thereby eliminating structural errors and enhancing the stability of large-scale STs crystals. K-map analysis confirmed that the degree of matching between the Fe-BPyB-Fe structure and Au(111) lattice along 451 direction was maximized, which was consistent with the observed experimental ST-(n + n) fractal assembly structure on the substrate. Furthermore, DFT calculations revealed that all Fe atoms in the fractal structure preferentially adsorbed at energetically favorable hollow sites along 451 direction, ensuring stable existence of two-dimensional STs crystal structures. Both K-map analysis and theoretical calculations underscored that efficient molecular diffusion and excellent lattice matching between molecules and substrates played pivotal roles in constructing two-dimensional STs crystals. These findings not only provided insights into preparing fractal crystal structures with diverse bonding modes but also laid a foundation for exploring their properties and potential applications on a larger scale in future studies.
In order to explore the functional advantages of fractals in plants, fungi, and animals, etc., it is necessary to construct biomolecular fractals. The stochastic peptide and protein fractals have been previously designed [168]. However, the structures of these disordered fractals are quite intricate, making it extremely challenging to study their growth mechanisms at the molecular level. From this, Wang et al. generated a series of STs with global organizational chirality through the co-assembly of L-tryptophan and 1,3-bi(4-pyridyl)benzene molecules on Ag(111) [120], as shown in Fig. 9, which can serve as a model system for biological fractals. By depositing BPyB and L-Trp molecules on Ag(111) at a ratio where L-Trp exceeded BPyB by more than 2:1, they were able to form a series of the STs. The multiple STs were denoted as L-TrpBPyB-ST-n (n = 0, 1, 2, 3), where n is the order of STs. Each L-Trp-BPyB-ST-n consisted of 3n+1L-Trp and 1/2 × 3n+1 BPyB molecules. For instance, an L-Trp-BPyB-ST-3 contained 42 BPyB molecules and 81 L-Trp molecules. In Fig. 9b, each V-shaped bright protrusion at a 120° angle represented a BPyB molecule while the dim trimers among them corresponded to L-Trp molecules. According to DFT calculations on the structure of an L-Trp-BPyB-ST-0, L-Trp molecules adopt a zwitterionic state and formed a trimer stabilized by cyclic hydrogen bonds between oxygen atoms from carboxylate groups and hydrogen atoms of neighboring tryptophan ammonium groups. Additionally, the interaction between L-Trp and BPyB molecules involved hydrogen bonding between the hydrogen atoms (N—H) of indole in L-Trp and the nitrogen atoms in BPyB. Previous studies have demonstrated that STs formed by achiral molecules expressed local organizational chirality on surfaces as a result of asymmetric surface adsorption, leading to the random dispersion of two mirrored images with equal probability [155]. In this study, it was observed that the coadsorption of chiral and achiral compounds on substrates gives rise to the emergence of globally organized chirality. The L-Trp molecules maintain their inherent chirality on Ag(111) due to specific enantioselective hydrogen bonding interactions with BPyB molecules, resulting in higher-order L-Trp-BPyB with consistent handedness. The organizational chirality of these STs originated from the asymmetric intermolecular interaction between L-Trp and BPyB molecules, leading to globally homochiral STs where all structures possessed the same handedness. Furthermore, it was discovered that by switching from L-tryptophan to D-tryptophan, the chirality of STs can be easily altered. This successful construction of homochiral STs through a bottom-up approach provides insights into understanding complex biomolecular hierarchical processes at the single-molecule level.
Figure 9
Figure 9.
(a) Approach towards creating biomolecular STs with global organizational chirality. STs formation is possible when 120° V-shaped organic molecules interact with metal atoms on surfaces. If chiral biomolecules imitate metal atoms in forming nodes with these V-shaped ligands, biomolecular ST formation would occur. It is expected that all resulting STs would have consistent handedness. (b) L-Trp-BPyB-ST-n prepared via a co-assembly method with n ranging from 0 to 3. Reproduced with permission [120]. Copyright 2021, American Chemical Society.
As previously mentioned, the utilization of specific assembly techniques can lead to the creation of ordered yet aperiodic structures, such as fractals and quasicrystals. Understanding the growth mechanisms of these structures is crucial for their controlled production on surfaces. Therefore, it becomes imperative to study the structural transitions. However, achieving reversible transformations between crystalline and fractal structures remains a challenging task. Wang et al. made an interesting discovery that STs underwent a change into 1D chains with the assistance of CO and CO2 [124]. On Au(111), 4,4′′-dicyano-1,1′:3′,1′′-terphenyl (C3PC) molecules and Fe atoms formed STs through three-fold coordination interactions (Fig. 10a). Subsequently, gas molecules (CO and CO2) were introduced to modify the assembly structure on the surface covered by STs. Upon dosing CO molecule onto the sample with a pressure of 5.0 × 10−6 for 120 s (600 Langmuir (L)) at room temperature, regular long chains approximately aligned along < 211 > directions of Au(111) were obtained (Fig. 10b). The protrusions in zoom-in STM image clearly revealed themselves in the long chains and were found to be preferentially located at iron atoms, attributed as axially bonded CO molecules. Applying a voltage pulse of 2.0 V allowed desorption of CO from Fe atom. Spin-polarized DFT calculations were conducted to investigate the mechanism underlying the transformation from STs to chains. It was revealed that CO molecules play a crucial role in facilitating this transformation. Initially, CO preferentially bound to Fe and altered 3-fold Fe(C3PC)3 to 4-fold Fe(CO)x(C3PC)4-x structures. Additionally, CO replaced one C3PC ligand from coordinated structures, allowing freely diffused C3PC molecules to interact with Fe(CO)x(C3PC)4-x and form the final product FeCO(C3PC)4, which serves as the building block for chains on Au(111). Furthermore, CO stabilized released Fe atoms from STs by forming Fex(CO)y clusters like Fe(CO)4. The stable coordination motifs of Fe(C3PC)3 change into those containing axially bonded CO molecule in Fe(C3PC)4 during this process, leading to one-dimensional chain formation. This phenomenon can be attributed to the openness of the fractal structure (Hausdorff dimension 1.59), providing ample space for ligand rearrangements. The interaction energy between Fe(CPC)4 and CO was calculated as 0.71 eV Upon annealing samples, 1D chains reverted back into STs. Additionally, it was observed that structural transition could also be induced by CO2 molecules on Au(111) (Fig. 10c). Axially bonded CO2 exhibited a binding energy of 0.16 eV in Fe(CPC)4CO2 but easily desorbed from its corresponding iron atom. Hence, CO2 did not exist within chains themselves. It was concluded that the structural transformation was induced by co-assembly of CO and catassembly of CO2, respectively. The catassembly theory was first proposed by Prof. Tian's group of Xiamen University [169,170]. Herein, the intermediate process of catassembly was visually observed for the first time through the variable temperature experiment, providing further clarification on the mechanism of catassembly and new insights into exploring growth patterns for more unique structures in future.
Figure 10
Figure 10.
(a) C3PC molecules and Fe atoms formed STs on Au(111) through three-fold coordination interactions. (b) Upon dosing CO molecule on the STs/Au(111) with the pressure of 5.0 × 10−6 torr for 120 s at room temperature, stable coordination motifs changed from Fe(C3PC)3 to Fe(C3PC)4 with an axially bonded CO molecule, transforming STs into one-dimensional chains where CO coassembled with Fe(C3PC)3 units. (c) Subsequently, dosing CO2 of 650 L on the sample induced structural transformation from STs to chains via a molecular catassembly process. Reproduced with permission [124]. Copyright 2023, CC-BY 4.0.
Exploring chirality in supramolecular assembly represents a captivating frontier within the realm of surface chemistry [171-173]. Investigating the fundamental scientific principles governing chiral generation and transfer at the molecular level holds the potential to enhance our comprehension of many pivotal processes, including the origins of chirality in biological systems and enantioselective heterogeneous catalysis [174,175]. The meticulous construction of chiral nanostructures on surfaces offers new possibilities for pioneering the development of novel chiral functional materials and their subsequent applications [176-179].
When a chiral molecule is adsorbed on the surface, the internal asymmetry of the molecule is usually preserved and reflected in its adsorption configuration. If the stereocenter in the molecule can interact with the substrate, a chiral adsorption configuration is generated [180,181]. In addition to the intrinsic chiral molecules, some achiral molecules may also adopt chiral adsorption conformations when they are adsorbed on surface since they cannot rotate freely in space due to the limitation from 2D surface [178,182]. They are called prochiral molecules. Furthermore, the expressions of chirality on 2D surface are analyzed in different levels: chirality induced by molecular packing or the orientation of molecular arrangement relative to the underlying substrate. Symmetrical achiral molecules can also form chiral motifs on a surface due to asymmetry of relative positions of molecules in the monolayer. However, for the racemic chiral molecules or the achiral molecules, a remarkable feature of the surface assembly is that the two mirror-imaged chiral conformations formed by them always exist at the same time and in an equal proportion. Chirality is limited to a local area of the surface and is a local chirality. The whole surface is still racemic. How to achieve a homochiral surface composed of a single chiral structure and to elucidate the mechanism in chiral induction, transmission and amplification are the key challenges in surface chirality research [183,184].
2.4.1
Chiral recognition and 2D crystallization on solid surfaces
Ever since Louis Pasteur's groundbreaking separation of sodium ammonium tartrate enantiomers in 1848, chiral recognition during crystallization is predominant in understanding the origin of homochirality in nature. When compared to the crystallization process occurring in 3D space, the constrained spatial environment on a 2D surface assembly offers an exceptional platform to gain deeper insights into the mechanism and rules of chiral recognition for mixed enantiomers during crystallization (Fig. 11a) [110].
Figure 11
Figure 11.
(a) Illustration for the heterochiral and homochiral recognition. (b) 2D crystallization of racemic mixtures on surface. (c) Br···Br contacts mediated chiral recognition in the self-assembled structure of achiral molecules [191]. (d) Water-induced chiral separation via hydrogen bonding interactions on an Au(111) Surface [110]. Reproduced with permission [110,191]. Copyright 2021, American Chemical Society; Copyright 2022, CC-BY 4.0.
As the racemic mixtures of enantiomers are adsorbed onto a solid surface, they can crystallize into one of the three distinct packing structures: a 2D conglomerate, 2D racemate, or a 2D random solid solution, as shown in Fig. 11b [185]. Mirror-imaged enantiopure assemblies constitute the 2D conglomerate structures, where the enantiomers separate in different domains. However, since the two enantiopure assemblies always emerge on the surface in an equal coverage, the whole surface is still racemic [186]. On the contrary, the 2D racemate are composed of ordered unit cells containing equal proportion of the two adsorbed enantiomers [187]. As for the 2D random solid solution, it is referred to as the disordered crystallization in which both enantiomers are distributed on surface randomly. In this situation, the supramolecular organization is heterochiral with no long-range ordered periodicity. To date, a detailed understanding of what governs enantiomers to crystalize into stable racemates or conglomerates is still elusive. By using STM, the coexistence of 2D racemate and conglomerate crystals for mixed heptahelicene enantiomers are observed on Ag(100). The transition from homochiral nucleates to larger racemic zigzag motifs can be controlled by increasing surface coverage, despite the extended homochiral structure possesses a higher stability [188]. Moreover, a minute difference in molecular structure can also affect the 2D crystallization process. Maeda and coworkers have investigated the 2D chirality of enantiomers of two photochromic diarylethenes on solid surface. The mixed enantiomers of one of the chiral diarylethenes separate spontaneously upon adsorption on surface, while the other diarylethene enantiomers arrange into a racemate crystal in which enantiomeric monomers coexist in an ordered domain [189].
Except for chiral enantiomers, achiral molecules can also form enantiomeric motifs on 2D surface [190]. In this case, chiral recognition process is like that in the monolayer of mixtures of racemic enantiomers. Wan et al. present the evolution of Br···Br contacts in the enantioselective homochiral and heterochiral recognition during supramolecular 2D crystallization (Fig. 11c) [191]. Type Ⅰ Br···Br contacts corresponds to homochiral recognition and dominate on Ag(100) surface at low coverage. When the self-assembled structure is mediated only by Type Ⅰ contacts, 2D conglomerates are formed. In contrast, type Ⅱ Br···Br contacts is responsible for heterochiral recognition and prevail on Ag(100) surface at high coverage, leading to the generation of racemates. Moreover, the matching degree of the molecular aggregate and the substrate plays a vital role in the formation of type Ⅰ Br···Br contacts. On Ag(111) surface which perfectly matches with the 2D conglomerate, type Ⅰ Br···Br contacts occur during the 2D crystallization and generate homochiral domains exclusively.
Additionally, the introduction of foreign species can also induce the 2D crystallization of the two mirror-imaged molecules to interconvert between the conglomerate, racemate, and random solid solution. The biologically relevant molecule uracil (U), one of the RNA bases, is chosen by Xu and co-workers to self-assemble into random structures through hydrogen-bonds on Au(111) surface (Fig. 11d) [110]. After water exposure, the random structure is transformed to a homochiral water-involved lang-range ordered U + H2O structure, leading to 2D conglomerate where chiral separation occurs. Moreover, interestingly, the water-induced homochiral ordered U + H2O structure can also be converted into the ordered racemate by removal of water and the interconversion process is reversable. The origin of chiral separation is supposed to be the preferential hydrogen binding between water and the O1 sites of U molecules, which results in the generation of the energetically favored homochiral (U−H2O−U)2 cluster. Then, the clusters grow into chains, five-membered ring chains and finally into a homochiral 2D conglomerate.
2.4.2
Chiral induction and amplification during molecular assembly on solid surfaces
Despite extensive exploration and research on chiral recognition, predicting whether racemates will undergo chiral segregation or co-crystallization during surface assembly remains a challenging task. The 2D chiral crystallization still lacks controllability, which is supposed to be the research basis for their applications in numerous fields, including asymmetric catalysis, chiral drug synthesis, and chemical sensing. The precise design and regulation of supramolecular chirality represent crucial steps toward establishing a globally homochiral surface from mixed enantiomers. Comprehensive insights into 2D chiral assembly processes, such as chiral induction and amplification, have been thoroughly investigated from various aspects, as summarized in Fig. 12a.
Figure 12
Figure 12.
(a) Schematic diagram of chiral control on 2D surface. (b-d) Co-adsorber induced homochirality at the liquid/solid interface [192]. (e-g) Biasing enantiomorph formation through geometric confinement [193]. Reproduced with permission [192,193]. Copyright 2013, CC-BY-NC—ND; Copyright 2018, American Chemical Society.
For achiral building blocks, a common way to regulate supramolecular chirality is to introduce other chiral species into the achiral assembly system. Although the chemical structure of the newly introduced chiral species maybe very different from that of the achiral building block, it is possible to play a crucial role in the chiral control of the assembly process through molecular interactions. Therefore, the selection of appropriate chiral auxiliary is of great significance for controlling supramolecular chirality. Through hydrogen bonding interactions, chiral dibenzoyl tartaric acid (D/L)-TA was applied to induce globally homochirality in monolayer consisting of an achiral oligo(p-phenylene vinylene) derivative (AOPV3) (Fig. 12b). The remaining hydrogen bond sites in the AOPV3 assembly structure can just enantioselectively match the carboxyl groups in (D/L)-TA. STM experiments result show that when D-TA is introduced, most of the surface is clockwise (CW) assembly structure, and correspondingly, when L-TA is introduced, most of the surface is constituted by counterclockwise (CCW) motif. Furthermore, the global organizational chirality is non-volatile even the chiral auxiliary is removed with a volatile solvent, revealing an obvious memory effect. Using this method, homochiral surface with one preferred handedness constituted by achiral molecules is achieved on 2D surface.
In addition to chiral auxiliaries, the selection of solvent in the assembly system is also crucial [194]. Chiral solvent can affect the adsorption and desorption balance of the solute at the liquid-solid interface, and determine the final chiral characteristics of the supramolecular structure. It has been found that in some supramolecular assembly process, although the solvent is not adsorbed on the surface and does not directly participate in the surface assembly, the solvent molecule, as a chiral seed, can transmit its own chiral information to the supramolecular structure and induce a clear preference to one enantiomorph with only one handedness [195]. For example, by assembling achiral alkoxylated dehydrobenzo[12]annulene (DBA) derivative at the interface of enantiopure chiral solvent and achiral graphite substrate, homochiral networks are fabricated. Based on host-guest interactions, hierarchical chiral assembly can be realized in the multicomponent supramolecular structures, when coronene and isophthalic acid are trapped into the nanowells formed by DBA derivative. Moreover, the structural "size matching" is supposed to be critical in the efficiency of chiral communication. The smaller the difference in the length of the alkoxy chain of the chiral solvent and the DBA, the stronger is the solvent-induced chirality, that is, a less amount of chiral solvent is required to realize 100% chiral induction.
Compared with the methods using chiral solvents or chiral auxiliary, there is usually a more clear and stable interaction mediated by non-covalent bonds between the building block molecules and the co-adsorptive species, which is not only conducive to effective transmission of chiral information, but also provides convenience for studying the mechanism of chiral control at the molecular level [196,197]. In addition, unlike chiral solvents or chiral additives which are always in solution phase, co-adsorbed molecules are directly adsorbed on solid surfaces and participate in supramolecular self-assembly [198,199]. The surface-limited effect and interaction with other adsorbed molecules contribute to a greater steric hindrance and a more efficient enantioselectivity in chiral induction. Wan's group has successfully used small chiral co-adsorbers with simple structure to realize highly efficient chiral regulation in the supramolecular assembly of achiral molecules. As can be seen from Fig. 11, 2D honeycomb networks with overall left-handedness or overall right-handedness are constructed at the liquid/solid interface [192]. The achiral 5-(benzyloxy)isophthalic acid (BIC) derivative and the co-adsorbed octanol can form two mirror-imaged chiral domains through hydrogen bonding and van der Waals interactions. When a chiral center is introduced into the octanol, due to the spatial limitation from the substrate and the specific directivity of intermolecular hydrogen bond, the stereochemical information in octanol can be communicated into the structural unit composed of BIC and co-adsorbed octanol molecules stereoselectively, then further transferred to the entire honeycomb networks (Fig. 12b). If pure R-2-octanol is used as a chiral co-adsorber, only the CW networks can be observed on the surface (Fig. 12c), while if S-2-octanol is used, all of the domains are composed of CCW motifs (Fig. 12d). The co-adsorbed chiral octanol not only acts as chiral seed, but also serves as the building block of the supramolecular assembly.
Compared with the method of introducing chiral information through covalent bonds, using chiral co-adsorbers based on non-covalent interactions to achieve chiral control is simple and convenient, avoiding complicated chiral synthesis. At the same time, since chiral information is introduced through more flexible and controllable non-covalent interactions, precise regulation of chiral control processes can be achieved by adjusting the weak interaction between chiral co-adsorbers and achiral molecules [200]. Changing the position of the stereogenic center relative to the stereocontrolling moiety (the hydroxy end group) in chiral co-adsorber revealing an odd–even effect for the chiral induction in the BIC assembly system, due to the alternation of steric hindrance. Moreover, when the stereogenic center is located closer to the stereocontrolling moiety, the stereochemical information can be communicated to the 2D organizations more reliably [201]. This result provides further mechanistic understanding in the communication of stereochemical information during 2D crystallization at molecular level.
Molecular assembly is the result of the synergistic interactions between molecules and their neighboring molecules, the substrate, and the environment. Beyond the molecular system, geometric confinement and external field may also have a powerful impact on the induction of chirality [202-204]. By introducing chiral environment, the control of surface supramolecular chirality can be realized. For instance, De Feyter et al. have used well-defined nanocorral to achieve geometric confinement and further trigger a chiral bias in the assembly of prochiral molecules at the liquid/HOPG interface, as illustrated in Figs. 12e-g [193]. The HOPG surface is first modified with covalently bounded 3,5-bis-tert-butylphenyl molecules densely, then nanoshaving is performed to create the nanocorrals with specific size, shape and orientation, in the presence of prochiral ISA-OC18 in solution. When the nanocorral boundary is not parallel with the mirror plane of HOPG substrate, a chiral nanocorral is generated. Intriguingly, in this situation, the chiral adsorbed conformation of ISA-OC18 on the exposed graphite surface inside nanocorrals is enantioselective, and the enantioselectivity relies on the nanocorral shape and the orientation relative to the underlying graphite substrate. Statistical analysis reveals that the L chiral square nanocorrals prefer to accommodate left-handed domains of ISA-OC18 (Fig. 12f), while a majority of square nanocorrals with R chirality is fulfilled with right-handed domains (Fig. 12g). In addition, with the increase of the size of nanocorral, the chiral bias will disappear. The chiral control by geometric confinement is supposed to arise from the enantiomeric matching between the nanocorrals and the nucleus formed at the early stage in molecular assembly. Only the molecules that matches the chirality of the nanocorral is stable in the perspective of kinetics and can successively aggregate inside the nanocorral. In contrast, the molecules with conflicting chirality are less stable and will dissolve.
External field is considered as a simple and efficient tool to modulate the supramolecular assembly and is an attractive candidate to induce homochirality [205]. Utilizing external magnetic field, Berg and co-workers realized the chiral control in the monolayer of achiral 4-cyano-4′-octylbiphenyl (8CB) molecules without chiral inputs [206]. When deposited on the graphite, 8CB molecules self-assemble into two enantiomorphs containing enantiopure left- or right-handed configurations in equal quantities and the whole surface is racemic. When a liquid crystal solvent and a magnetic field are applied, the enantiomeric excess of the resulting surface is altered. The supramolecular chirality of the molecular domains is directed by the orientation of the external magnetic field. The left-handed configurations are preferred when the magnetic field rotates −19° from the < 1100 > direction. On the other hand, the rotation of the magnetic field is +19° from the < 1100 > direction triggers a preferential formation of the right-handed domains. Consequently, by this method, the chirality of supramolecular structure is regulated conveniently.
2.4.2.2
Chiral amplification
In general, statistical analysis in the monolayer shows that the mirror-imaged enantiomorphs formed by achiral molecules always exist on surface in equal proportions. To break the symmetry of the crystallization process, a typical approach is to introduce a small number of sergeants (chiral molecules) into a large amount of soldiers (achiral molecules) to direct the achiral soldiers to arrange in only one enantiomeric manner and assemble into a homochiral monolayer. The chirality of the introduced small amount of sergeant molecules determines the preferred chiral characteristic of the entire assembly structure. This method is known as the sergeants and soldiers principle, as illustrated in Fig. 13a. The first report about this principle on 2D surface was the introduction of a small number of chiral tartaric acid molecules (TA) into the achiral succinic acid (SU) monolayer. The introduction of only 2 mol% of chiral TA was enough to control a large number of achiral SU molecules to break the symmetry of the entire surface and form a globally homochiral assembly structure [207]. One possible mechanism in the chirality control is the substrate-mediated process of chirality recognition. Through the selective interaction between TA and the underlying substrate, chiral TA can only be adsorbed on the surface of copper single crystal in a specific chiral adsorption conformation. Thus, the chiral information in TA can be further transmitted to the surrounding achiral molecules through the hydrogen bonding interactions with SU molecules. Furthermore, the correlation of the efficiency of chiral amplification based on sergeants and soldiers principle with the proportion of enantioselective interaction in the total intermolecular interactions has been evaluated by Wan's group (Fig. 13b) [200]. Although the sergeants and soldiers principle can always operate in different organizations, the chiral amplification efficiency is different from each other. The efficiency of chiral amplification increases from the dimer structure, the composite tetramer structure to the trimer structure. Intriguingly, molecular mechanistic simulations reveal that the strengthened trend of chiral amplification is quite in agreement with the increasing proportion of hydrogen bonding interactions in the series of structures, since the stereochemical information in chiral alcohol is always transferred to the achiral BIC molecules through the directional hydrogen bonds. The stronger the enantioselective hydrogen bonds formed between chiral and achiral molecules, the higher the long-range transmission efficiency of chirality.
Figure 13
Figure 13.
(a) Schematic diagram of the sergeants and soldiers principle. (b) Manifesting the chiral amplification process controlled by sergeants-and-soldiers principle in the co-adsorber induced homochiral assemblies at the liquid/solid interface [200]. (c) Schematic diagram of the majority rules. (d) Concentration–modulated supramolecular chirality following the majority rules [212]. Reproduced with permission [200,212]. Copyright 2016, Royal Society of Chemistry. Copyright 2023, CC-BY-NC 4.0.
The molecular self-assembly on surface is usually a stable or metastable process which is relatively stable and close to the state of thermodynamic equilibrium. The concentration of molecules in the system and the assembly temperature has a great influence on the thermodynamic equilibrium of the surface self-assembly process. By changing the temperature or adjusting the molecular concentration, the thermodynamic equilibrium of the original assembly process can be destroyed, to achieve controllable regulation of the nanostructure on surface and affect the supramolecular chirality. De Feyter et al. have combined the temperature control and concentration variation in the operation of sergeants and soldiers principle to unravel two discrete chiral amplification pathways at the liquid/solid interface [208]. At lower solute concentration, the handedness of the enantiomer in majority is amplified after an annealing protocol, furnishing a surface exclusively covered with one enantiomorph preferred by the majority enantiomer. When the annealing protocol is performed at a higher solute concentration, the majority chirality can be entirely reversed. The contrasting results are attributed to the noticeable amplification pathway conducted by the enantioselective host–guest interaction during crystal growth.
Although the chiral domain regions formed by mixed enantiomers always exist simultaneously, studies have found that in some chiral systems, once there is a certain difference in the relative content of two chiral enantiomers, that is, a small excess of one enantiomer is generated, global homochirality with a single handedness will be triggered on the surface. This phenomenon is known as the majority-rules principle, which is also called the chiral nonlinear amplification phenomenon (Fig. 13c) [209,210]. Raval et al. successfully achieved nonlinear symmetry breaking in supramolecular assembly structures by introducing a small enantiomeric excess (ee) in tartaric acid (TA) enantiomer mixtures [211]. R, R-TA and S, S-TA assemble into mirror-imaged chiral structures on surface, respectively. As the content of R, R-TA in solution increases, the chiral domain region corresponding to R, R-TA on surface is enlarged substantially. A slight change in ee value in solution can induce a huge change in supramolecular chirality on the surface. That is, supramolecular chirality can be easily regulated by adjusting the relative content of chiral tartaric acid molecules. As long as the ee value R, R-TA reaches 0.2, the whole surface chirality is biased to homochirality. The calculation method of the monomer ee value in the experiment is as follows: ee = (R - S)/(R + S), where R and S represent the amount of two enantiomers, respectively. Similar chiral amplification effect based on the majority principle has also been found in other supramolecular system, such as the heptahelicene assembly [209], and the global chirality is adjustable.
In addition, the majority rules principle also works in the assembly of achiral molecules where the supramolecular chirality is triggered by chiral co-adsorber. Wan' group has systematically studied the chiral amplification phenomenon in the assembly of 5-(benzyloxy)-isophthalic acid derivatives at the liquid/solid interface [213]. If one of the mirror-imaged co-adsorbers is slightly excessive (ee ≥ 5.2%) in solution phase, then only its preferred chiral assembly structure can evolve on the surface [192]. The enantiomorph with the opposite handedness completely disappears and a globally homochiral surface is generated. The interaction between achiral building block and chiral co-adsorber is enantioselective due to the specific directivity of intermolecular hydrogen bonds, which is beneficial for highly efficient amplification of chirality. Furthermore, it is found that molecular concentration can also affect the efficiency of chiral amplification. Li and co-workers reported that the reduction of molecular concentration in solution contributes to the alternation of 2D crystallization from heterochiral to homochiral and facilitates a more efficient amplification of chirality (Fig. 13d) [212]. To formulate the concentration-dependent chiral amplification phenomenon and illustrate the inherent quantitative correlation between the enantiomers and the induced supramolecular chirality, a 2D cooperative equilibrium model based on Langmuir-type adsorption is proposed. It verifies that a subtle change in the enantiomer concentration can make a significant difference in the expression of chirality at the supramolecular level.
2.4.3
Chiral transfer
2.4.3.1
Chiral transfer from the underlying molecular assembly to the upper layer
Besides the chiral transfer within the self-assembled monolayer, the stereochemical information can also be transferred from 2D to 3D systems. Enantioselective interlayer interactions are prerequisite to precisely control the chirality in the overlayers. Wu and co-workers have revealed the chiral transfer from self-assembled structure of achiral molecules to its top-sitting molecules, giving rise to chiral molecules with electronic chirality rather than conformational chirality, as depicted in Fig. 14a [214]. The achiral titanyl phthalocyanine (TiOPc) molecules assemble on Au(111) surface with their ending oxygen atoms pointing away from the surface, and two enantiomorphic domains involving periodic chiral void are formed. When additional TiOPc molecule are applied onto the assembly, the top-sitting TiOPc molecule locates vertically above the chiral void and becomes chiral, indicating an efficient transmission of chirality between the underlying and the top-sitting TiOPc molecules. It is supposed that the chirality of the top-sitting TiOPc is induced by a charge transfer from its indole rings to the oxygen atoms of TiOPc molecules constituting the chiral void in the bottom layer, which leads to the chirality of the molecular orbitals. Tobe and co-workers have reported the stereocontrolled epitaxial growth of self-assembled bilayers, in which the building blocks within the monolayer are stabilized by van der Waals interactions while additional hydrogen bonds mediate the interactions between layers in the longitudinal direction [215]. Since the chiral information in the monolayer is introduced by hydroxy substituents in molecule which contributes to the formation of hydrogen bonds, the upper assembly needs to adapt a stereospecific manner relative to the first layer and the epitaxial growth of the overlayers is enantioselective (Fig. 14b). Moreover, the hydroxy substituents in the top and bottom layers point in opposite directions, allowing for the formation of hydrogen bonds. Consequently, within the same crystalline domain, though the constituents are the same enantiomers, the supramolecular chirality of the top and bottom layers are opposite.
Figure 14
Figure 14.
(a) The transmission of chirality from the self-assembly of achiral TiOPc to the TiOPc molecule situated on the top of chiral void [214]. (b) STM image and the suggested model for the multilayer growth, with the first and second layers exhibiting opposite interlocking patterns [215]. (c) Characterization of the structures, computational models, and proposed reaction mechanism for the transformation products P1, P2, and P3 resulting from the annealing of (P)-DBH on Ag(111) in the temperature range of 520–670 K [217]. (d) Transferring two-dimensional chirality from self-assembled structures to covalently bonded products [220]. Reproduced with permission [214,215,217,220]. Copyright 2016, 2019, 2020, American Chemical Society; Copyright 2016, Springer Nature.
2.4.3.2
Chiral transfer of molecular assembly in on-surface reactions
An efficient control over the chiral transfer in on-surface reactions remains significant interest in surface chemistry, since it can result in enantioselective formation of chiral products. And it offers a deeper understanding of how surface interactions influence the stereochemical outcome of chemical reactions [216]. Stetsovych et al. have demonstrated a global symmetry breaking in the 2D crystallization of prochiral molecules on the solid achiral surface through a stereocontrolled chemical method (Fig. 14c) [217]. When enantiopure helical (P)-DBH precursor is deposited on Ag(111) surface, most of the surface is covered with homochiral trimers which appear as equilateral triangles. After annealing at 520 K, the intrinsic chiral helical molecules entirely disappear. Instead, two prochiral coronene derivatives are synthesized. Notably, the handedness distribution of the newly formed prochiral molecules is enantioselective, generating a preferential formation of one enantiomer over the other, due to the chiral transfer from the enantiopure helicene molecules to the prochiral adsorbates in on-surface reaction. Further increasing the annealing temperature to 620 K results in a surface with racemic adsorbates. In addition to the intrinsic molecular chirality, the organizational chirality of achiral molecules can also be preserved in the products of on-surface reaction [218,219]. Zhang et al. have reported the chiral transfer from self-assembled structures to covalently bonded products through on-surface aryl−aryl coupling reaction on Au (111) (Fig. 14d) [220]. The initial organizational chirality of the self-assembled monolayer on surface determines the final enantiomorphs of the newly formed oligo-p-phenylenes. As the domains of the precursor are homochiral, after annealing, homochiral islands of produced oligomers can be obtained. In contrast, mixed chiral geometry of the precursor in the assembly leads to the generation of racemic lamellae product. The alkyl chains in precursor play a vital role in the chiral transfer process during the on-surface reaction.
Later, researchers found the chirality of the final products can also be regulated by chiral intermediate states. Yang et al. successfully transfer the chiral information on the self-assembly structure to the C—C bond coupling products on Ag(100), assisted by the metal-organic hybrid tetramer intermediate states (Fig. 15) [219]. Tetramer intermediate states are formed by annealing the surface at 490 K. The tetramers are of chirality and found to self-assemble into enantiomeric islands. More importantly, the chiral expressions of the newly formed dimer products after annealing at higher temperature are fully determined by the chirality of the tetramer intermediate states. The detailed reaction pathways are rationalized by both DFT calculations and synchrotron-based XPS experiments. On the other hand, direct heating the sample to the C—H bond activation temperature resulted in various dimer products on the surface, showing poor reaction selectivity. Thus, by adjusting the self-assembled structures, the chiral transfer in on-surface reaction can be controlled and a covalently bonded products with a specific handedness are generated.
Figure 15
Figure 15.
(a-d) Transferring of the chiral information from the tetramer intermediate states to the dimer reaction products on Ag(100) [219]. Reproduced with permission [219]. Copyright 2019, American Chemical Society.
Intermolecular interactions within self-assembled nanostructures and the influence of local molecular environments on the individual molecules were also explored at the single-molecule level. In this context, molecular orientation is crucial for self-assembly and fabrication of advanced functional organic nanodevices, e.g., organic light-emitting diodes, and can also be regarded as the reflection of molecular intermolecular interactions from local molecular environments. Traditional spectroscopic methods are versatile in distinguishing between horizontal and vertical orientations, but are less accessible in characterizing molecular orientations in the plane, while SPM is particularly powerful in visualizing specific molecular orientations on surfaces in real space and also in directly controlling molecular orientations at the single-molecule level by manipulations. A large number of previous reports focused on the direct control of orientation changes of isolated molecules in a free space by SPM manipulations [137,139,142,221,222]. More recently, molecular orientation selectivity has been elucidated in a constrained situation under the influence of intermolecular interactions arising from local molecular environments (Fig. 16) [223], which are more general cases in realistic applications.
Figure 16
Figure 16.
(a) Schematic illustration showing the orientation selectivity of a Na-TPyP molecule in a "Klotski Puzzle", which was realized by STM manipulations. (b) Two possible pathways for the orientation change of a Na-TPyP molecule, i.e., molecular rotation and conformational change. (c-n) Sequential STM images showing the orientation selectivity of the target sliding-block molecules after STM manipulations (as indicated by red arrows) in "Klotski Puzzles" containing (c-f) a single-cavity, (g-j) a 2 × 2 cavity, and (k-n) a 3 × 3 cavity, respectively. The apparent long axes of the molecules are indicated by blue dashed lines. Reproduced with permission [223]. Copyright 2023, American Chemical Society.
Based on the electrostatic interactions between tetrapyridyl-porphyrin-based molecules (Na-TPyP) and Na (derived from NaCl) [224,225], a close-packed self-assembled chessboard structure was constructed (where Na atoms were located at the center of four pyridyl groups and were invisible in most cases) with the coexistence of some vacancies (Fig. 16a). Na-TPyP molecules with longitudinal and transverse orientations were found to alternate orthogonally. To reveal the interconnection between orientation selectivity and local molecular environments, various types of molecular "Klotski puzzles" were artificially constructed by collecting dispersed vacancies via STM lateral manipulations, including those with two-dimensional cavities, one-dimensional channels, and some more complicated patterns. When the target sliding-block molecules (A, B, and C) were manipulated to the adjacent vacancies from all directions, orientation changes occurred to maintain the orthogonally alternate orientations with respect to their neighbors, thus fitting the local environments in which the target molecules were constrained on three sides (Figs. 16c-f). To systematically explore molecular orientation selectivity in different local environments, the target molecules (D and E) were positioned and manipulated in 2 × 2 and 3 × 3 cavities without any spatial constraints (Figs. 16g-j and k-n), respectively. After moving a single-molecule distance, continuous orientation changes were observed (Figs. 16g-j), whereas after moving a double-molecule distance, the orientations at the destinations appeared to be the same as before (Figs. 16k-n). In both cases, the adjacent constraint was reduced to two-sided, and the eventual orientations of the target molecules always critically fitted their local molecular environments after manipulations, with the interactions with neighboring molecules at the destinations being the key to the orientation selectivity. In addition, such a rule of orientation selectivity was evidenced to be valid as long as at least one side of a target molecule interacted with an adjacent molecule. In the absence of any constraints, no orientation selectivity was achieved, and two molecular orientations interconverted randomly during scanning at ~300 K. Moreover, two possible pathways for the orientation change, i.e., molecular rotation and conformational change, were revealed based on STM observations of maker molecules and DFT calculations (Fig. 16b). Thus, this study further demonstrated the delicate interplay between molecular orientations and intermolecular interactions in molecular self-assembly at the single-molecule level.
Additionally, self-assembly of molecules refers to formation of an orderly arranged monolayer as a result of molecules interacting with each other as well as the substrate, which has been widely used as the interlayer in nano energy devices, such as solar cells [226] and lithium batteries [227]. The self-assembled monolayer (SAM) can be tuned by selecting different anchoring groups, spacer groups and terminal groups of molecules as shown in Fig. 17a. The anchoring group is responsible for the binding between the molecule and the substrate, which provides a driving force of molecular self-assembly. The choice of anchoring group is also important for surface state passivation as well as work function adjustment of the substrate. The spacer group determines not only the steric hindrance of molecule arrangement, but also the decay rate of mass and charge transport. The terminal group is related to hydrophobic and hydrophilic properties, which affects the interaction with the overlayer and adsorbent. In this section, we will take the perovskite solar cells (PSCs) as an example to present the benefits of SAM on device performance.
Figure 17
Figure 17.
(a) Schematic illustration of the effects of anchoring groups, spacer groups, and terminal groups on the properties of self-assembled molecule monolayer (SAM) [226]. (b) The optimization of interfacial band bending of perovskite solar cells with a fullerene derivative SAM. [232]. Reproduced with permission [226,232]. Copyright 2019, CC-BY 4.0; Copyright 2020, John Wiley and sons.
Solution-processed PSCs are promising solar energy harvesting technologies owning to their high-efficiency and low-cost [228-230]. The PSCs are composed of perovskite photoactive layer sandwiched between an ITO/FTO transparent bottom electrode and a metal top electrode. The device performance has been improved significantly by incorporating interlayers between the perovskite and two electrodes. Recently, the further insertion of SAM between the interlayer and the perovskite becomes a popular strategy to realize even more superior device performance, which is explained by various mechanisms, such as interfacial defect passivation, interfacial energy band structure reconfiguration, perovskite morphology optimization [226,231].
Metal oxides, such as n-type ZnO, TiOx, SnOx or p-type NiOx., MoOx, are widely adopted interlayers between the ITO/FTO transparent bottom electrode and the perovskite according to the polarity of the device. However, a high density of trap states on these metal oxide interlayer will not only exacerbate charge carrier recombination, but also cause Fermi level pinning [233-235]. Zuo et al. [233] reported that 3-aminopropionic acid (C3) can be self-assembled on ZnO because of interaction between carboxyl anchoring group of C3 and hydroxy group on the surface of ZnO. The work function of C3-SAM coated ZnO was ~0.65 eV lower than that of ZnO as measured by UPS to facilitate selective electron collection, indicating that C3-SAM formed a strong interfacial dipole with its positive end pointing upwards and the negative end pointing toward the ZnO. Also, the alkaline terminal group of C3-SAM can promote a heterogeneous nucleation of perovskite to realize a low density of trap states as well as a smooth morphology. As a result, the PSC with C3-SAM showed remarkably increasing in power conversion efficiency (PCE). Li et al. [235] reported that P=O anchoring group (4-(3,11-dimethoxy-7H-dibenzo[c,g]carbazol-7-yl)butyl)phosphonic acid (MeO-4PADBC) can binds with hydroxy group of NiOx to provide a driving force of self-assembly of MeO-4PADBC as well as passivate trap states on NiOx. The interfacial energy level alignment between NiOx and perovskite was optimized with the MeO-4PADBC SAM, which facilitated hole extraction and minimizes the voltage loss, leading to a high certified PCE of 25.6% on a masked area of 0.0414 cm2. In addition, the PSC with MeO-4PADBC exhibited a higher thermal stability, which retained 90% of the initial PCE after running for 1200 h at 65 ℃.
The n-type [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) or p-type 2,2′,7,7′-tetrakis(N, N-di-p-methoxyphenylamine)−9,9′-spirobi-fluorene (Spiro-OMeTAD) is the popular interlayer between the perovskite and the top metal electrode depending on the polarity of PSCs. However, the perovskite usually exhibits a high density of trap states on the surface, which is challenging to be efficiently passivated with either PCBM or Spiro-OMeTAD [232,236,237]. Zhang et al. [232] inserted an iodide ionized fullerene derivative (PCBB-3N-3I) between the perovskite and the PCBM, which realized a superior PCE (Fig. 17b). The anchoring group Ⅰ- of PCBB-3N-3I can passivate the uncoordinated Pb2+ of perovskite via electrostatic interaction. Also, the electrostatic interaction promoted the orderly arrangement of PCBB-3N-3I. In addition, the spacer group tris(dimethylamine) at the pendant group of PCBB-3N-3I regulated the steric hindrance between each other to further improve the order degree. As a result, the PCBB-3N-3I SAM exhibited a high dipole moment and increased the interfacial band bending as supported by cross-sectional scanning Kelvin probe microscopy, which facilitated selective electron collection. In contrary, the fullerene derivative before ionization (PCBB-3N) exhibited random orientation, which inhibited the electron collection and even caused an inferior device performance. Lu et al. [236] inserted a series of para-substituted benzenethiol SAM between the perovskite and the Spiro-OMeTAD. The –SH group of benzenethiol SAM acted as the anchoring group towards the uncoordinated Pb2+ on the perovskite, which realized efficient trap passivation. While the para-substitution, such as –CN, –NO2, –SCH3, –OCH3, in the aromatic moiety allowed for tuning the dipole moment of benzenethiol SAM. Among them, the HS-Ph-CN SAM-based PSCs exhibited the highest PCE, which was attributed to the optimized dipole direction and interfacial band bending, leading to selective hole collection.
Organic functional materials have attracted great attention recently since they exhibit a variety of unique physical properties. Studying these materials is vital for both foundational research in chemistry and physics and for the development of future technologies. Compared to inorganic materials, organic materials have the added advantages of low cost, easy fabrication and mechanical flexibility. Many organic materials and devices have shown great value in various fields such as energy, materials, and information due to their excellent performance. Examples of these are organic light-emitting diodes, organic solar cells, and organic field-effect transistors.
Over the past decades, on-surface self-assembly and synthesis of low-dimensional organic functional materials in ultra-high vacuum (UHV) have advanced significantly [238-241]. These materials include organic single molecules, graphene nanoribbons (GNRs), metal-organic frameworks (MOFs), and covalent organic frameworks (COFs). Notably, recent theoretical predictions indicate that organic functional materials have a number of exotic electronic properties, such as high conductivity [242], superconductivity [243], half-metallicity [244], ferromagnetism [245], quantum spin liquid [246], and topologically non-trivial band structures [247,248]. However, most of these research focuses on noble metal substrates such as gold, silver, and copper [238,240,241]. This severely restricts the study of electronic properties of organic functional materials.
On one hand, organic materials often interact strongly with these metal substrates, which can perturb their intrinsic properties. This makes an in-depth study in these systems challenging. To explore the predicted exotic electronic properties, synthesizing these materials on inert surfaces, such as graphene, other van der Waals layered materials, and bulk insulators, is highly desired [249,250]. On the other hand, if organic functional materials could be synthesized on special functional material substrates (like superconductors), the intrinsic properties of organic functional materials could be further expanded on these functional material substrates, leading to richer physical properties. For instance, magnetic impurities on superconductors can lead to Yu-Shiba-Rusinov (YSR) bound states [251-253]. This method can confirm elusive magnetic properties in organic materials, like the magnetism found in the zigzag edges of graphene [254]. Moreover, topologically non-trivial YSR energy bands (i.e., topological superconductivity) can give rise to Majorana fermions [255], which have great application potential in topological quantum computing [256].
Accordingly, by combining SPM imaging, manipulations, and DFT calculations, various interaction modes of regulating factors such as metals, halogens, salts, and water with organic molecules have been directly observed and revealed at the submolecular level, and a variety of means to finely regulate the assembled structures (including ratio, temperature, doping, etc.) have been proposed. Therefore, the fundamental properties of multi-level interactions, such as coordination, hydrogen bonding, and electrostatic ionic interaction, and the rules governing these interactions have been elucidated, providing the basis for further precise construction of stronger intermolecular interactions (e.g., carbon-carbon covalent bonds).
3.
Surface synthesis
Apart from molecular self-assembly dominated by non-covalent interactions, the programmable construction of functionalized low-dimensional nanostructures via the formation of more robust covalent bonds (e.g., C–C bonds) by well-controlled on-surface reactions has long been a goal pursued by surface scientists. Compared to the non-covalent intermolecular interactions as discussed above, the covalent connections are featured by efficient electron transport and high thermal stability and are thereby promising for the application in the construction of discrete one-dimensional nano-chains and single-layered two-dimensional nano-sheets. Guided by this ultimate goal, a large number of studies have been concentrating on monitoring the stepwise reaction evolution processes of molecular precursors and intermediates involved in on-surface reactions toward products [257-259], which help to elucidate the influencing factors of the reactions at the single-molecule level, and then to selectively regulate the reaction pathways and products to precisely synthesize low-dimensional covalent nanostructures. Moreover, by utilizing specific non-covalent intermolecular interactions between molecular precursors or intermediates and regulating factors (e.g., metal atoms), on-surface reaction pathways could be selectively regulated, and directional conversions of reactions could be realized. In such a way, the difference between the on-surface reactions and solution reactions has been gradually clarified.
Precise construction of covalent connections between various molecular components is of great challenge, but essential for the synthesis of low-dimensional carbon-based nanostructures, and is also the basis for building molecular electronic devices. The key lies in understanding the activation and reaction mechanisms of various functional groups during on-surface molecular reactions. Thus, starting from the most common reactions in organic synthesis, C–H and C–Br activation, several pioneering works have explored the on-surface construction of intermolecular C–C covalent bonds, probed the reaction intermediates and products in real space, revealed the evolution mechanisms of the reactive intermediate states, and elucidated the on-surface reaction pathways and mechanisms of C–H and C–Br activation and C–C coupling processes. Accordingly, the covalent linkages of C–C single, double, and triple bonds have been precisely constructed, leading to the atomically precise fabrication of novel carbon-based nanostructures on surfaces.
3.1
CnullH bond activation
The pioneering work of surface-supported polymerization reaction of inert regular alkane was reported by Zhong et al. [260]. The dehydrogenation reactions of sp3 C—H groups were observed to occur at a mild temperature (420 K) on a reconstructed Au(110) surface (Fig. 18a). Accompanied by the polymerization reaction, the Au(110) surface undergoes a structural evolution from (1 × 2) to (1 × 3). Owing to the orientational constraint of the reactant molecules in these one-dimensional reconstructed channels, the reaction takes place with high selectivities (at terminal CH3 or penultimate CH2). The exposed metal surfaces were proposed to play a significant role in triggering such alkane polymerization, which is hardly to achieve in solution. In order to explore the active sites for alkane polymerization on Au(110), control experiments are performed by the same group [261]. The reconstructed Au(110) transforms from (1 × 2) to (1 × 3) by either introducing branched methylidene groups into the aliphatic chain or low-energy electron irradiation prior to the polymerization reaction of alkanes. Systematic STM observations investigate that alkane chains adsorbed on (1 × 3)-Au(110) are more reactive than that on (1 × 2)-Au(110). Such structure-reactivity relationships rely on that the presence of an extra row of gold atoms in the groove of (1 × 3)-Au(110). The exposed Au atoms with lower coordination number make are highly active, enabling dehydrogenation coupling to occur at mild temperatures. In fact, metal atoms with lower coordination can also be found along the step edges. Zhang et al. successfully achieved the coupling of linear alkanes along the step edges of copper surfaces at modulated temperatures [262]. The alkane coupling exclusively occurred at terminal methyl, exhibiting excellent reaction selectivity. Moreover, since direct n-alkane coupling is ascribed to the low coordination number of the copper atoms, such reactions take place on different facets of copper surfaces. Recently, Li et al. reported the direct transformation of n-alkane into all-trans conjugated polyene on Cu(110) through a cascade process of alkane dehydrogenation under surface constraints [263]. DFT calculations reveal that the terminal methyl dehydrogenation is the rate-determining step. As shown in Fig. 18b, once the terminal C—H bond is activated, the dehydrogenated carbon radicals tend to continuously have the hydrogen atoms detached, eventually forming polyene products. In addition to STM/AFM investigations, the reaction products are systematically evidenced by various surface technologies, such as ARPES, IRRAS and TPD. Recently, Hao et al. further demonstrated that the presence of hydroxyl groups can significantly promote the transformation of C(sp3)-H to C(sp2)-H of n-alkanol on Cu(110) [264]. DFT calculations confirm the entire reactions are triggered with the assistance of C=O groups, by both the electron-withdrawing induction and the enhanced molecule-substrate interactions. Afterward, they systemactically investigated the detailed defects during the formation of polyacetylene on Cu(110) [265]. Three distinct linkages are identified using STM and nc-AFM. Recently, concentrating on the C(sp3)-H activation, Tang and colleagues successfully achieved the transformation of non-conjugated poly(o-naphthylene vinylidene) toward conjugated poly(o-naphthylene vinylene) on Au(111) [266]. This transformation occurs through dehydrogenation upon mild annealing, as depicted in Fig. 18c.
Figure 18
Figure 18.
(a) Dehydrogenative polymerization of linear alkane on Au(110) [260]. (b) Direct transformation of n-alkane into all-trans conjugated polyene via cascade dehydrogenation on Cu(110) [263]. (c) Transformation of poly(o-naphthylene vinylidene) into its conjugated derivative poly(o-naphthylene vinylene) via dehydrogenation reaction [266]. Reproduced with permission [260,263,266]. Copyright 2011, American Association for the Advancement of Science; Copyright 2021, 2022, CC-BY 4.0.
A major challenge of on-surface synthesis is how to efficiently steer reaction pathways and improve the yield/quality of products. The molecular adsorption geometry on metal surfaces can influence the reaction behavior significantly. For instance, Zhong et al. reported the different cyclodehydrogenation pathways on surfaces as compared to those in solution [267]. Atomically flat surfaces tend to facilitate the formation of planar benzo-fused perihexacenes, while nonplanar double [7]helicenes are synthesized through solution synthesis. The selective aromatic C—H bond activation is one of the most challenging in organic chemistry. The difficulties arise not only from the high C—H bond dissociation enthalpies, but also the existence of multiple equivalent/quasi-equivalent reaction sites in organic molecules. By using hydroxyl as the directing group, Li et al. realized the regioselective aromatic C—H bonds activations on both Au(111) and Ag(111) surfaces, as shown in Fig. 19a [268]. More importantly, they observed the surface controlled mono-selective ortho C—H activation on Ag(111), and di-selective ortho C—H bond activation on Au(111). This phenomenon gives a first controllable mono/di selective C—H bonds activation on metal surfaces. By combining temperature dependent XPS measurements and DFT calculations, they found the novel phenomenon arises from the binding strength difference between metal substrates and oxygen atoms of phenol derivatives, leading to different competition between dehydrogenation and deoxygenation reactions. Based on this, mono-selective ortho C—H bond activation of phenol was also demonstrated on Cu(111) by Deimel et al. [269]. Achieving precise synthesis of nanostructures with unsaturated carbon skeletons is an urgent issue to address. However, the high chemical activity of unsaturated carbon-carbon bond functional groups results in poor reaction selectivities. For example, linear acetylenic couplings are far from satisfactory, due to the producing of enyne or cyclotrimerization byproducts. Recently, Li et al. reported the synthesis of well-aligned nitrogen-doped one-dimensional graphdiyne (GDY) nanowires with selective on-surface acetylenic homocoupling reactions on Au(111) (Fig. 19b) [270]. The replacement of benzene with pyridine moieties prohibits the cyclotrimerization reaction, and facilitates the linear coupling to produce well-aligned N-doped graphdiyne nanowires. DFT calculations elucidate that the pyridinic nitrogen modification substantially differentiates the coupling motifs at the initial C—C coupling, which is decisive for the formation of graphdiyne.
Figure 19
Figure 19.
The selectivity of reaction pathways. (a) Mono- and di-selective ortho C—H bond activation of phenol derivatives on Au(111) and Ag(111), respectively [268]. (b) Homocoupling reaction of polarized terminal alkynes (TAs) on Au(111) [270]. Reproduced with permission [268,270]. Copyright 2016, 2023, American Chemical Society.
Additionally, the C–C coupling reaction of terminal alkynes on Ag(111) is taken as an example, which was one of the most important potential reactions for the construction of novel carbon nanomaterials with unsaturated carbon skeletons (e.g., graph(di)yne), while the detailed carbon skeletons in the sp2/sp hybridized state were unclear. In light of this aspect, Zhang et al. applied the coupling reaction of terminal alkynes (4,4′-diethynyl-1,1′-biphenyl, abbreviated as DEBP) as a model system (Fig. 20a) [271], and combined STM imaging, STS characterization, and tip-enhanced Raman spectroscopy (TERS) techniques to directly probe the characteristic electronic features and local vibrational properties of the unsaturated carbon skeletons generated via the coupling reaction in real space (Fig. 20b). Based on the characteristic STM topographies, enyne (in the majority) and cumulene connections were tentatively identified as indicated by red and blue arrows in the STM image in Fig. 20a. Subsequently, a more precise determination of the detailed chemical arrangements was realized by means of STM-TERS measurements (Fig. 20b). Charge-density-distribution maps showed that the enyne connection consisted of sp-sp alkynyl and sp2-sp2 alkenyl groups at the linkage, while sp2-sp-sp-sp2 carbon skeleton existed in the cumulene connection (left panel of Fig. 20c), which would lead to different CC stretching modes of sp-carbon (right panel of Fig. 20c), serving as Raman-active tags in the silent region. The TERS data further confirmed the appearance of two different CC stretching modes, i.e., 2090 cm−1 and 2107 cm−1 at the two sites (the blue and red dots), respectively (Fig. 20d), demonstrating the respective chemical arrangement of cumulene and enyne. Such a remarkable sensitivity makes STM-TERS a promising spectroscopic approach to provide chemical information (including but not limited to the determination of bond orders), complementing the well-established STM-AFM imaging technique, which provides molecular skeletons and morphologies with little chemical insight. Thus, the chemical structures of the covalently generated carbon skeletons of the reaction products were finely identified and determined at a single-chemical-bond level, leading to the elucidation of the nondehydrogenative direct C–C coupling reaction (i.e., addition reaction) mechanism of terminal alkynes on Ag(111). Such a study further demonstrates the great potential of combining spectroscopic and topographic information as a novel methodology for chemical identification and dertermination in the field of surface chemistry.
Figure 20
Figure 20.
(a) Schematic illustration showing the selection between C–C coupling reaction and C–H activation of terminal alkynes on Ag(111) under different reaction conditions. (b) STM-TERS measurement on the two newly generated sp-sp2-carbon skeletons (i.e., enyne and cumulene). (c) Charge-density-distribution of enyne and cumulene on Ag(111) (left), and the corresponding CC stretching modes of sp-carbon involved (right). (d) STM-TERS spectra collected at the positions marked in the inset [271]. (e) STM image showing the self-assembled structure of DEBP on Ag(111). (f) Formation of long C–C coupled chains upon annealing at ~370 K. (g–i) Gradual formation of organometallic dimers and chains after O2 dosage. (j) Energy diagram of DFT-calculated reaction pathways of C–H activation on Ag(111) without and with the assistance of oxygen species. (k) Charge-density-difference map of IS2. (l) Structural models involved in the C–H activation pathway in the presence of atomic oxygen [278]. Reproduced with permission [271,278]. Copyright 2021, 2022, American Chemical Society.
On the other hand, the high reactivity of terminal alkynyl groups provides the opportunity to steer the reaction pathways by applying different reaction conditions. Given the importance of selective regulation of chemical reactions in chemistry, catalytic strategies have been extensively developed, including the introduction of gas reactants to surfaces to collide with the preabsorbed molecular species [272-277]. Using the same molecular system, oxygen molecules were dosed to the DEBP-precovered Ag(111) sample held at ~300 K (Fig. 20e) [278]. It brought about the gradual formation of extended organometallic chains (Figs. 20g-i) with high efficiency, which were closely packed together into islands (Fig. 20i) and were distinct from the dispersed C–C coupled chains obtained without the participation of O2 (Fig. 20f), indicating the occurrence of C–H activation at room temperature (~300 K). Moreover, both molecular O2 and atomic O were experimentally found to efficiently lead to C–H activation under mild conditions via an associative and dissociative mechanism, respectively. DFT calculations on the reaction pathways further revealed that the reaction barrier of C–H activation on Ag(111) was drastically reduced due to the facilitation of both oxygen species (Figs. 20j and l), and their attraction to the terminal alkynyl groups was the key to the reduction and the regulation of reaction pathways (Fig. 20k). As a result, the selective regulation of the on-surface reaction pathways of terminal alkynes from C–C coupling to C–H activation was successfully realized by introducing O2, and the catalytic performance and reaction mechanisms of oxygen species in the C–H activation processes were elucidated. This catalytic strategy of introducing gas molecules into molecule-surface systems inspires the selective regulation of chemical reactions by utilizing multi-level non-covalent interactions, and would pave the way for understanding many key interfacial chemical processes. It would also be an inspiration for improving or altering product selectivity and reaction rates for the construction of functional nanostructures with high efficiency and yield.
Based on the revelation of on-surface reaction mechanisms and the development of on-surface reaction strategies, researchers in the field of surface chemistry are further developing the on-surface synthesis methods [157,279]. A series of novel carbon-based nanostructures are then precisely synthesized [258] with high efficiency and selectivity including those in various dimensions, ranging from zero-dimension to two-dimension. Here we would like to take some typical one-dimensional carbon-based nanostructures as examples, such as polyacetylenes and various organometallic metal-carbynes (Fig. 21). Aiming at precise synthesis of one-dimensional carbon-based nano-chains, Xu and co-workers have made a lot of attempts and have successfully synthesized cis- and trans-polyacetylene chains [280] and a variety of metalated carbynes containing different metal atoms (e.g., Cu, Ag, Au) [281-284] as well as different structural periodicities of carbon-carbon triple-bonds (e.g., acetylenic, diacetylenic, and triacetylenic metal-carbynes) [285].
Figure 21
Figure 21.
(a) Scheme of on-surface synthesis of cis-, trans-polyacetylenes, and organometallic Cu-carbynes on Cu(110) from C2H2. (b, c) Nc-AFM and STM images of cis- and trans-polyacetylenes on Cu(110). (d) STS characterization of an individual trans-polyacetylene on Cu (black), at the Cu/oxide interface (blue), and on the oxide (red), respectively. (e) Nc-AFM and STM images, DFT model, and STM simulation of an individual Cu-carbyne on Cu(110). (f) On-surface synthesis and STM/nc-AFM characterization of close-packed Cu-carbyne ribbons from CBr4via an elimination reaction. (g-i) STM/nc-AFM/STS characterization of diacetylenic Au-carbynes on Au(111) synthesized from C4Br4. (j, k) Characterization of triacetylenic Ag-carbynes on Ag(111) synthesized from C6Br6. Reproduced with permission [280-284]. Copyright 2019, Springer Nature; Copyright 2016, 2020, 2022, 2023, American Chemical Society.
Polyacetylene is the earliest discovered conducting polymer, and its conducting mechanisms have been controversial, among which the soliton theory is one of the conducting mechanisms proposed by theorists. Trans-polyacetylene is one of the simplest one-dimensional model systems for studying solitons, and direct experimental observation of solitons in trans-polyacetylene had been a great challenge. For the precise preparation of trans-polyacetylene, the researchers steered the reaction pathways of acetylene (C2H2) on a Cu(110) surface and induced the selective activation of alkynyl groups (Fig. 21a) [280]. By keeping the substrate at room temperature, the carbon-carbon bond was preferentially activated upon the adsorption of acetylene molecules and then coupled to form cis-polyacetylene (Fig. 21b). Subsequently, by modulating the thermodynamics of the reaction, the cis-trans isomerization reaction of polyacetylene was induced, and the surface confinement effect was exploited to finally precisely prepare the monodisperse oriented-grown trans-polyacetylene (Fig. 21c), providing the possibility of fine characterization of its structure and precise measurement of its electronic properties. Spectroscopic measurements revealed that the polyacetylene adsorbed on the metal substrate exhibited metallicity. In order to observe the possible formation of solitons, part of a trans-polyacetylene chain was decoupled from the metal substrate by Cu oxide intercalation, and the decoupled polyacetylene fragment was found to be semiconducting (with a measured bandgap of 2.4 eV), leading to the construction of a heterojunction consisting of a metallic and a semiconducting phase (Fig. 21d). In addition, a soliton-like interface state was presented at the heterojunction and directly observed in real space. This work provides an important support for the in-depth study of the intrinsic physical effects of soliton.
As for the synthesis of metal-carbynes, various molecular precursors, typically like C2H2, CBr4, C4Br4, and C6Br6, have been applied according to the potential C–H and C–Br activation reactions as established previously. For example, acetylene (C2H2) was chosen again and deposited on a Cu(110) surface, which was held at ~450 K instead, and the terminal alkynyl group selectively underwent C–H activation on the surface. By the subsequent metal-organic coupling, one-dimensional metalated carbyne chains were fabricated with [–C≡C–Cu–] as the structural unit (Fig. 21e) [281]. Over the past three years, Xu and co-workers further designed and modified the precursors to expand the family of metal-carbynes and further elucidate their structure-property relationship. Interestingly, based on the CBr4 molecule and surface-assisted elimination reactions on Cu(110), Cu-carbyne ribbons were prepared instead of isolated Cu-carbyne chains, featuring a metal-to-metal alignment between adjacent chains due to the face-to-face π−π stacking between C≡C bonds (Fig. 21f) [284]. Similarly, the elimination reaction of C2Br6 molecules on Ag(110) also resulted in the construction of Ag-carbyne ribbons with a periodic [–C≡C–Ag–] unit. Notably, a width-dependent bandgap modulation was theoretically revealed in both cases, with the bandgap decreasing as the ribbon width increased, exhibiting an alternative strategy for bandgap engineering.
Besides, by modifying two bromine atoms at each end of the cumulene backbone to synthesize the C4Br4 (Br2C═C=C═CBr2) molecule, they triggered the complete debromination on Au(111) by thermal treatment and produced another type of metal-carbyne (Au-carbyne) composed of diacetylenic carbon periodicity, i.e., [–C≡C–C≡C–Au–] (Figs. 21g and h) [282]. STS results exhibited its semiconducting feature Fig. 21i). Furthermore, by exploiting the halogen elimination reaction, a hexabromobenzene (C6Br6) molecule was selected and deposited on Ag(111). Upon complete debromination, the C6 rings experienced a ring-opening reaction and transformed into the polyynic C6 chains, followed by metal addition to form triacetylenic Ag-carbynes, i.e., with a structural unit of [–C≡C–C≡C–C≡C–Ag–] (Fig. 21j) [283]. Meanwhile, a series of one-dimensional metal-carbyne chains containing different metals (Ag and Cu) and periodicities were fabricated on Ag(110) and Cu(110) using C2I4 and C4Br4 as precursors, respectively, and the transformation between acetylenic and diacetylenic organometallic polyyne chains were also experimentally observed [285]. It was also theoretically predicted that their bandgaps would decrease with the increasing number of acetylenic units, while for the same carbon unit, the bandgap was metal dependent in the order of Ag−carbyne > Cu−carbyne > Au−carbyne. Thus, these studies show that in addition to the ribbon width, the metal element and carbon structural periodicity are also significant for bandgap modulation, laying the groundwork for the precise synthesis and measurement of intrinsic metal-free carbyne chains.
Recently, another two kinds of all-carbon allotrope, the cyclo[n]carbons (Cn, n = 10, 14) consisting of two-coordinated sp-hybridized atoms, has been synthesized and characterized, revealing the structure which was open to debate before [286]. The fully halogenated naphthalene (octachloronaphthalene, C10Cl8) and anthracene (decachloroanthracene, C14Cl10) are used as molecular precursors, with the aim of generating C10 and C14 on the surface through tip-induced dehalogenation and retro-Bergman reactions (upper panels in Figs. 22a and b). Further analysis of the AFM images of C10 (lower panels in Fig. 22a) reveals that the C10 structure is not a perfect circle. This pentagon-like shape appearing in the AFM images is probably related to the theoretically predicted intrinsic D5h symmetry of C10. A similar strategy using tip manipulation to induce complete dehalogenation of the precursor results in the formation of C14 accompanied by two-step retro-Bergman ring opening. AFM images (lower panels in Fig. 22b) show that C14 also exhibits a cumulenic feature, which is different from the polyynic C18 and C16 with characteristic bright features [287,288], despite a BLA of 0.05 Å calculated for C14. Such a small BLA cannot be distinguished experimentally by AFM imaging, and AFM image simulations of C14 suggest that, in the case of 0 < BLA < 0.09 Å, structures can be assigned as cumulene-like; when BLA ≥ 0.09 Å, structures can be identified as polyynic.
Figure 22
Figure 22.
Surface synthesis strategies and chemical structure characterization of C10 and C14. (a) The upper panel shows the reaction scheme for the formation of C10. The lower panels show the AFM image, Laplace-filtered AFM image, and atomic structure overlapped on the image of C10, respectively. (b) The upper panel shows the reaction scheme for the formation of C14. The lower panels show the AFM image, Laplace-filtered AFM image, and atomic structure overlapped on the image of C14, respectively. Reproduced with permission [286]. Copyright 2023, Springer Nature.
Based on the reaction mechanisms and regulation rules revealed in these studies, a large number of novel carbon-based nanostructures have been synthesized with atomic precision and in a controllable manner on surfaces by designing molecular precursors containing specific C–H or C–Br bonded functional groups. These recent exciting advances clearly develop and establish new methodologies for surface-confined precise synthesis of novel molecular nanostructures that are intriguing as potential molecular electronics and functional nanomaterials.
3.3
Formation and breaking of non-carbon-carbon bonds
Investigating the formation and breaking of non-carbon-carbon bonds is an intriguing topic in organic chemistry. Amide linkages are the most basic chemical connections in polypeptides and proteins. Recently, Yang et al. demonstrated the formation of amide bonds between carboxyl and amine species through direct dehydration condensation reaction under ultrahigh vacuum conditions, as shown in Fig. 23a [289]. Such amidation reactions could take place on Au(111), but were inhibited on Ag(111) and Cu(111) surfaces. This can be attributed to the lower dehydrogenation and decarboxylation reaction barrier of the carboxyl group on Ag(111) and Cu(111), resulting in the formation of other byproducts. Imines are important products in synthetic chemistry, which can be formed through the Schiff-base reaction between an aldehyde and an amine. The on-surface synthesis of imines was first performed by Weigelt and co-workers on Au(111) under UHV [290]. Afterwards, Gong et al. systematically studied the influence of the stoichiometric ratio of precursor molecules to the reaction products [291]. The underlying mechanisms that lead to the diverse surface patterns are investigated by Monte Carlo simulations. On the other hand, breaking of the chemical bonds is essential to chemical science and chemical industry. As depicted in Fig. 23b, the cleavage of C—O bonds via thermally excitation was reported by Yang et al. leading to the conversion of alkoxybenzene-containing ethers into alcohols on various metal surfaces [292]. They demonstrated that the dealkylation process could be finely controlled by the annealing parameters. Moreover, detailed reaction pathways were elucidated using density functional theory calculations. The entire reaction is initiated by the C—H bond activation of the propyl group, which differs significantly from their homogeneous counterparts in solution.
Figure 23
Figure 23.
Formation and breaking of non-carbon-carbon chemical bonds. (a) Formation of amide bonds between carboxyl and amine species on Au(111) [289]. (b) The formation of phenols through the catalytic dealkylation of ethers on metal surfaces [292]. Reproduced with permission [289,292]. Copyright 2016, 2021, John Wiley and sons.
3.4
Several strategies for synthesis of acetylenic scaffoldings
The blooming on-surface reactions has irritated researcher to focused on more sophisticated carbon nanomaterials, especially that hard to be synthesized in solution chemistry. Recently, on-surface synthesis of acetylenic scaffoldings containing sp1-hybridized carbons has recently received tremendous attentions due to their fascinating electronic and mechanical properties [293]. To the best of our knowledge, up to five distinct on-surface reactions has been applied into explore the feasibility of synthesis of acetylene moieties on surfaces [294-299]. The dehydrogenation coupling of terminal alkyne, named as Glaser coupling, has been more frequently employed for create of diyne moieties (Fig. 24a) [294-296]. Unfortunately, byproducts like enynes and trimerization are often observed, and the yield are relatively lower [294]. To overcome such shortcomings and improve the selectivity of Glaser coupling, different approaches have been focused to decrease the side products, like attaching sterically demanding groups next to the alkyne moiety, selection of a high miller index surface or introducing gasses [278,294,296,300,301]. Besides that, researchers have also attempted to search for other potential on-surface reactions with the aim of direct synthesis of acetylene moieties. In 2018, Kawai et al. display another strategy by introducing a precursor molecule with protection group of trimethylsilyl (TMS) on Cu(111) (Fig. 24b) [298]. With post annealing at 400 K, there undergoes a desilylation followed by C—C homocoupling, leading to the formation of anthracene oligomers linked by diacetylene moieties. Later, Zhu's group then exploited Sonogashira coupling for synthesis of graphyne nanowires with the controlled regulation of high temperature, low coverage and low evaporation rate on Ag(111) (left panel in Fig. 24c), providing the first illustration on the cross coupling applied in graphynes or graphdiynes [302]. By invoking TMS-enthynyl and chlorophenyl groups as a model system, Kawai's group also achieved Sonogashira coupling with chemoselectivity through stepwise silylene tethering and elimination of the ME3Si group on Ag(111) (right panel in Fig. 24c), achieving the synthesis of butadiyne segments [303]. Very recently, Kong et al. introduced alkynyl carboxylic acids (3,3′-(naphthalene-2,6-diyl)dipropiolic acid, NDDA) as a reactive compounds, they demonstrated that decarboxylative coupling are triggered after deprotonation, and finally poly(diynes) were selectively synthesized on Ag(111) (Fig. 24d) [299]. In comparison to the traditional Glaser coupling, decarboxylative coupling displaying here show higher efficiency and higher selectivity at a low reaction temperature. Apart from the above strategies, the synthesis of acetylenic scaffoldings can also be achieved by dehalogenative coupling [304-306]. In addition, Xu's group have realized the controlled synthesis of a series of graphyne and graphdiyne motif by virtue of muti-dehalogenation strategy [297,307,308]. For example, via triggering dehalogenative homocoupling of terminal alkynyl (sp1-carbon atom) bromine on Au(111), they have successfully constructed dimer structures, 1D nanowires and 2D networks with acetylenic scaffoldings [297]. More interestingly, the multi-dehalogenation reaction of tribromomethyl (sp3-carbon atom)-substituted compounds results in the direct formation of C—C triple bonds, and finally the synthesis of graphyne-like nanowires [308]. As mentioned above, the unexpected stepwise dehalogenative coupling reaction of tribromomethyl (sp3-carbon atom) and aryl bromide (sp-carbon atom) groups on Au(111) and Ag(110) provide another avenue for buildup of graphyne-like nanowires. In 2022, Kawai et al. performed the synthesis of multi-block co-oligomers by defluorinative coupling, extending the range of dehalogenation strategy [309].
Figure 24
Figure 24.
(a) Illustration of the first example of Glaser coupling on Ag(111) [295]. (b) Illustration of the synthesis of anthracene oligomers linkered by diacetylene moieties by desilylation followed by C—C homocoupling on Cu(111) [298]. (c) Left panel: illustration of the synthesis of graphyne nanowires by sonogashira coupling anthracene oligomers linked by diacetylene moieties by desilylation followed by C—C homocoupling. Right panel: Illustration of the synthesis of bitadiyne sigments by stepwise silylene tethering and elimination of the ME3Si group on Ag(111) [302,303]. (d) Illustration of the synthesis of poly(diynes) by decarboxylative coupling after deprotonation on Ag(111) [299]. Reproduced with permission [295,298,299,302,303]. Copyright 2012, Springer Nature; Copyright 2018, American Chemical Society; Copyright 2021, 2023, John Wiley and sons.
3.5
Competition and collaboration of dehalogenation and dehydrogenation reactions
Based on the above discussion, both dehalogenation and dehydrogenation reactions on surfaces show huge potentials on advanced fabrication of sophisticated carbon-based nanostructures and nanomaterials with atomic precise. Also, there are number of other on-surface reactions has been exploited for the create of novel robust nanostructures among the most active areas of surface science community, such as Bergman cyclization, decarboxylative polymerization, azide-alkyne click reactions, imine formation [53,216,289,290,310-316]. Deep understanding the advantages/disadvantages of distinct on-surface reactions and their competition/collaboration will help for the rational selection of certain on-surface reactions and facilitate to build up more complicated covalent bonded nanostructures in a finely controlled manner. Take typical dehalogenation and dehydrogenation as an example, the advantages of dehalogenation reactions include: (1) The dehalogenative C—C coupling are able to yield higher selectivity in comparison to the dehydrogenative one. (2) Also, it has been proven to be feasible for multi-dehalogenation reaction at one carbon atom and generation of more than one carbon radicals aiming to the direct synthesis of alkyne and alkene moieties besides the alkane ones, while it seems much more difficult to trigger multi-dehydrogenation reaction. (3) It is easy to control for specific reaction site for dehalogenation reaction via decorated by halogen atoms. But for pure aryl, hydrogen atom attached to certain carbon atoms are preferred to be activated (for example, H atoms attached to meta carbon sites are preferred to dissociate for 4Ph molecule [317]). These advantages of dehalogenation reactions shown above should be attributed to the lower energy barrier of C-X cleavage [318]. Despite that, it should be noted that the dehalogenation reaction will produce halogen atoms as byproducts which may hamper the fine synthesis of products in desired qualities [319].
It is noteworthy to point out that, the different reaction pathway between dehalogenation and dehydrogenation reaction may trigger fully distinct products. As reported by Xu's group, dehydrogenative homocoupling of alkenes (VBP) on Cu(110) surface induced the formation of trans-diene, yet, when the alkenes precursors are replace by alkenyl halide ones (BVBP, where one halogen atom replace one of the terminal hydrogen atom attached to alkene part of VBP), dehalogenation homocoupling occurred and a cis-diene are unexpectedly synthesized instead [320]. Such precise synthesis of stereoselective dienes should be caused by the significantly distinct reaction pathway, Concretely, trans-dienes are realized by C—C coupling between VBP precursors followed by a dehydrogenation process and no organometallic intermediates are observed, while cis-diene products directly result from the demetallation of the most stable cis-form organometallic intermediates after dehalogenation reaction. This finding presented here shed light on the selection of specific on-surface reactions with the aim of delicate synthesis of covalent nanostructures in atomic precision.
Apart from studies focused on certain on-surface chemical reaction, it is of great interest to design precursors with multiple active sites to explore the competition of different on-surface chemical reactions with the aim of developing precisely robust nanostructures or nanomaterials in a controlled manner. As an example, H. Kong et al. select DN molecules with multiple active sites as a model system to investigate the competition between dehalogenation and dehydrogenation/dehydration reactions (Fig. 25a) [321]. On Au(111), it is found that intramolecular dehydrogenation or dehydration firstly occur followed by C—C coupling, results in the synthesis of polymeric chains. Interestingly, when deposited DN molecules on the more active Ag(111) surface, unpredictable C—H coupling are achieved after dehalogenation. This situation can be rationalized by the simultaneous dehalogenation reaction and dehydration, thus the hydrogen atom produced during dehydration can directly transfer to the as-formed carbon radicals generated by dehalogenation reaction.
Figure 25
Figure 25.
(a) Illustration of competition between dehalogenation coupling and dehydrogenation/dehydration reaction of DN molecules on Au(111) and Ag(111) [321]. (b) illustration of synthesis of carbon nanoribbon consisting of period 4–5–6–8 membered rings on Au(111) formed by dehalogenative homocoupling followed by face-to-face dehydrogenation coupling of DIF molecules [324]. (c) Illustration of stepwise dehalogenative homocoupling reaction of precursor molecules where function group of C(sp3)–Br are activated firstly followed by the cleavage of C(sp2)–Br bond [308]. Reproduced with permission [308,321,324]. Copyright 2017, 2023, American Chemical Society; Copyright 2020, Royal Society of Chemistry.
Based on the basic understanding on competition of on-surface reactions, collaboration of hierarchical on-surface reactions undoubtedly will improve their application in more complex covalent nanostructures. As a typical example, the on-surface synthesis of atomic precise graphene nanoribbons is actually achieved by an intermolecular dehalogenative homocoupling with subsequent intramolecular dehydrogenation cyclization reactions [322]. Following the same strategy, Xu's group has illustrated the formation of polyphenyl chains by stepwise reaction of dehalogenative and dehydrogenative coupling of 4-bromobiphenyl (shortened as BBP) molecule on Cu(110) [323]. More interestingly, by selecting 2,7-diiodo-9H-fluorene (DIF) molecules composed of both C-I and -CH2- function group, they have synthesized a more complex carbon nanoribbon consisting of period 4–5–6–8 membered rings on Au(111) (Fig. 25b) with a semiconducting bandgap of 1.4 eV by the dehalogenative homocoupling followed by face-to-face dehydrogenation coupling [324]. As displayed above, most of the on-surface hierarchical synthesis involve remarkably different reactions on surfaces, Xu's group recently pioneered to introduce two types of dehalogenative homocoupling reactions ((i.e., C(sp3)–Br and C(sp2)–Br) hybridized carbon atoms by design and synthesis a precursor molecule of 1-bromo-4-(tribromomethyl)benzene, BTBMB) on Au(111) and Ag(110) surface (Fig. 25c) [308]. They interestingly found that stepwise dehalogenative homocoupling reaction have been realized by delicately controlling the substrate temperature, that is, C-Br activation within tribromomethyl groups followed by C-Br activation of aryl bromide groups, and finally a graphyne nanowire are synthesis.
3.6
Conjugated carbo- and heterocycles
Conjugated carbo- and heterocycles are the basic structural units of organic functional materials. Their sizes and types of covalent linkages have a significant impact on the properties and function of organic functional materials. Therefore, the development of efficient ring formation reactions to precisely construct them is of great importance for both fundamental science and application purposes in the fields of nanoelectronic devices, photonics, and quantum science. On-surface synthesis (OSS) [325], as a bottom-up synthetic method, has been proven to be a powerful tool for atomically precise fabrication of low-dimensional carbon-based nanomaterials with diverse conjugated carbo- and heterocycles over the past 15 years [326-328]. This method relies on covalent coupling reactions of rationally designed precursor molecules that occur on solid substrates such as metal or metal oxide surfaces under ultra-high-vacuum conditions [241,329], and the formation of the conjugated carbo- and heterocycles mainly involves a stepwise process containing the Ullmann coupling and subsequent cyclodehydrogenation [330,331]. However, cyclodehydrogenation involving C—H bond activation reaction requires a high annealing temperature, leading to poor selectivity and poor tolerance of functional groups and limiting the utility.
If the precursor molecules can directly undergo the ring formation reactions on surfaces, the limitations of high temperature-induced cyclodehydrogenation are avoided. However, to achieve highly selective ring formation reactions on surfaces, the following challenges need to be addressed: (1) Inhibiting the formation of linear chains, (2) accelerating the ring formation reaction, (3) improving the selectivity of the ring formation reactions to form target n-membered conjugated rings (Fig. 26). In this part, we review the recent advances in the OSS field and focus on the development and synthetic applications of on-surface conjugated ring formation reactions, mainly including the synthesis of benzenoid and nonbenzenoid conjugated carbo- and heterocycles.
Figure 26
Figure 26.
Selective formation of conjugated carbon- and heterocycles on surfaces.
3.6.1
Formation of benzenoid conjugated rings on surfaces
The six-membered conjugated carbo- and heterocycles are the most common structural units in organic functional materials. Diels–Alder (DA) reaction, is one of the most popular reactions that form six-membered rings in synthetic chemistry, but the spatial restrictions of DA reaction hampered their direct application on surfaces. Until recently, Castro‐Esteban et al. achieved the highly selective on-surface intramolecular DA reaction by using a cyclic strained triyne [332]. This precursor can facilitate the on-surface hexadehydro-Diels–Alder (HDDA) reaction to give the aryne intermediate, which further undergoes the intramolecular cycloisomerization to benzenoid nanographene (Fig. 27a). In 2013, Sun et al. reported the first on-surface Bergman cyclization by using 1,6-di-2-naphthylhex-3-ene-1,5-diynes as precursors to form the phenyl diradical intermediates, which undergo the covalent polymerization to produce one-dimensional polyphenylene chains (Fig. 27b) [312].
Figure 27
Figure 27.
Formation of benzenoid conjugated rings on surfaces. (a) On-surface hexadehydro-Diels–Alder reaction [332]. (b) On-surface Bergman cyclization [312]. (c) On-surface cyclotrimerization of terminal alkynes [333]. (d) On-surface dehydration cyclotrimerization of acetyls [334]. (e) On-surface formation of pyrazine via dehydration cyclocondensation of o-diamine and o-diketone [335]. (f) On-surface formation of triazine via a special dehydration reaction [336]. Reproduced with permission [312,332-336]. Copyright 2021, CC-BY-NC—ND; Copyright 2013, 2015, 2016 American Chemical Society; Copyright 2014, Royal Society of Chemistry; Copyright 2022, CC-BY 4.0.
In addition to on-surface intramolecular cyclization, [2 + 2 + 2] cyclotrimerization of alkynes has also been used to directly synthesize the benzene rings. In an earlier study, Liu et al. reported on-surface cyclotrimerization reactions of alkynes on Au(111) using 1,3,5-tris(4-ethynylphenyl)benzene (TEB) as a key building block, resulting in a two-dimensional covalently bonded polyphenylene nanostructures network (Fig. 27c) [333]. Cycloaddition of alkenes and alkynes is common, but the ring formation reaction of alkanes is rarely involved. In the pioneering work of Fasel et al. the phenylene rings were selectively formed via a formal [3 + 3] dehydrogenation cycloaromatization of isopropyl substituents on Au(111) and Au(110) surfaces [337]. On the anisotropic Au(110) surface, the diffusion of precursors was constrained in one-dimension, avoiding cross-linking competitive reactions, which improved the selectivity of the [3 + 3] cycloaromatization.
The condensation reaction is a kind of classical covalent bond formation reaction in organic chemistry and has been used in the construction of conjugated carbo- and heterocycles. For example, Yang et al. developed the first dehydration cyclotrimerization of acetyl to form a phenyl ring on Ag(111) (Fig. 27d) [334]. Jiang et al. achieved the first in-situ formation of pyrazine on Ag(111) via the dehydration cyclocondensation of o-diamine and o-diketone (Fig. 27e) [335]. More recently, Cheng et al. successfully constructed a triazine ring on Au(111) through a novel dehydration reaction of a special precursor and nitrile (Fig. 27f) [336]. Considering that the desorption temperature of different precursors is different, adjusting the proportion of two reactants can improve the abundance of a dehydration reaction up to 60%.
3.6.2
Formation of nonbenzenoid conjugated rings on surfaces
Nonbenzenoid conjugated structures are common as topological defects in carbon-based nanomaterials because they can tune the electronic properties [338-340]. The five-membered conjugated rings are the most common nonbenzenoid structures and their formation mainly involves the [3 + 2] cycloaddition. For example, the azide−alkyne [3 + 2] cycloaddition on Au(111) or Cu(111) has been developed to form the triazole [310,311]. Subsequently, Palma et al. reported a head-to-tail [3 + 2] cycloaddition of phenanthridinium/azomethine moiety and cyano to form the azaullazine on different substrates (Fig. 28a) [341].
Figure 28
Figure 28.
Formation of nonbenzenoid conjugated rings on surfaces. (a) On-surface [3 + 2] cycloaddition [341]. (b) Steric hindrance-directed [2 + 2] dehalogenative cycloaddition [342]. (c) On-surface [1 + 1 + 1 + 1] cyclotetramerization of terminal alkynes [343]. (d) On-surface [1 + 1 + 1] cycloaddition of isocyanides [316]. Reproduced with permission [316,341-343]. Copyright 2021, American Chemical Society; Copyright 2022, John Wiley and sons; Copyright 2018, 2020, CC-BY 4.0.
The four-membered conjugated ring has the unique characteristics of anti-aromaticity [257,344-348]. The most straightforward way to synthesize a four-membered conjugated ring on surfaces is through the dehalogenative [2 + 2] cycloaddition of ortho-dihalogenated arenes. In the pioneering work of Fasel and Meunier et al. the cycloaddition reactions of ortho-dihalotetracenes on an Ag(111) substrate gave multiple products, including tetracene dimers, trimers, and tetramers [344]. To improve the selectivity of dehalogenative [2 + 2] cycloaddition, Grill reported a highly selective [2 + 2] cycloaddition by selecting Au(110) as a substrate. Recently, Li et al. developed a highly selective [2 + 2] cycloaddition on Au(111) to achieve the first synthesis of ladder phenylenes, where methyl-derived steric hindrance controls the selectivity of the dehalogenative [2 + 2] cycloaddition (Fig. 28b) [342]. Chi et al. reported a highly selective dehalogenative [2 + 2] cycloaddition induced by metal-organic intermediates on Ag(111) [349]. Notably, four-membered conjugated rings have also been synthesized by the [1 + 1 + 1 + 1] cyclotetramerization of phenylacetylene on Cu(100) (Fig. 28c) [343].
Three-membered conjugated carbocycles are the smallest ring structure with a strong strain, thus its synthesis is a great challenge. In 2022, Li et al. developed the first [1 + 1 + 1] cycloaddition reaction with high selectivity and achieved the synthesis of aza[3]radialenes on the Ag(111) surface (Fig. 28d) [316]. They selected 1,4-dichloro-2-isocyanobenzene (DCICB) as the precursor, in which chlorine substituents-derived steric hindrance on Ag(111) ensured the reaction selectivity and orientational order of product.
3.6.3
Phenyl migration reaction
As one of the rearrangement reactions, radical aryl migration is fascinating to the chemical community owing to its potential applications in radical chemistry and organic synthesis [350,351]. However, the mechanism of radical aryl migration has been long debated. Ruan and coworkers reported a case of the formation of various polycyclic aromatic hydrocarbons via a phenyl migration reaction on metal substrates [352]. The precursor, 1,4-dimethyl-2,3,5,6-tetraphenyl benzene (DMTPB), undergoes a phenyl migration and cascaded cyclodehydrogenations after thermal treatment. As illustrated in Fig. 29, annealing the self-assembled DMTPB clusters at elevated temperature, conventional C–C couplings between phenyl groups in the early stage of the reaction are surprisingly absent (no-go products N1 and N2). Instead, four benzene rings migrate to the para-methyl positions, forming Thiele's hydrocarbon, which further transforms into fully conjugated polycyclic aromatic hydrocarbons A1, A2, A3, A4 and A5 via intramolecular cyclodehydrogenation reactions. Various intermediates and products derived from the cascaded cyclodehydrogenations process were directly captured by STM/nc-AFM.
Figure 29
Figure 29.
On-surface generation of polycyclic aromatic hydrocarbons A1, A2, A3, A4, and A5 via a phenyl group migration reaction. Reproduced with permission [352]. Copyright 2023, CC-BY 4.0.
3.6.4
Selective cyclodehydrogenation on metal surfaces
Selective surface synthesis is the key to realize high-quality target nanostructures with a minimum of possible side products [353-356]. Various strategies have been used to tune selectivity of reactants on surfaces, such as substrate templating effects [357], kinetic and dynamic effects [358]. However, the selectivity mechanism during the cyclodehydrogenation reaction induced by molecule tautomerism has rarely been explored. Recently, Zhang and coworkers demonstrated the regioselectivity of 2H-diphenyl-porphyrin (H2-DPP) in cyclodehydrogenation on metal substrates, the reaction pathway is shown in Fig. 30a [359]. H2-DPP monomer forms two configurations (anti- and syn-) via a dehydrogenation coupling, among which the yield of anti-configuration exceeds 90% (Fig. 30b). They found that the regioselectivity of H2-DPP was derived from the reaction energy barrier during the dehydrogenation coupling of different tautomers. Once the hydrogen atoms of the porphyrin macrocycle are replaced by a metal atom (such as Fe), the yield of both two planar products will become nearly close (Fig. 30c).
Figure 30
Figure 30.
(a) The reaction pathway of M-DPP (M = 2H, Cu, Fe) monomers on metal surfaces. (b) High regioselectivity of H2-DPP in cyclodehydrogenation reaction. (c) No selectivity of Fe-DPP in cyclodehydrogenation reaction. Reproduced with permission [359]. Copyright 2023, American Chemical Society.
3.7
Nonbenzenoid hydrocarbons at surfaces: towards graphene isomers
Since the discovery of the first two-dimensional (2D) allotrope of carbon, i.e., graphene, the 2D allotropes of other group-Ⅳ, Ⅴ and Ⅵ elements such as borophene [360,361], phosphorene [362-364], germanene [365,366], silicone [367], and stanine [368] have also been reported in the past two decades. They exhibit strongly different properties and functionalities due to their different composite elements. Moreover, elements such as phosphorous and stannum were reported to have more than one type of 2D allotropes prepared with different methods. For instance, black [362] and blue [364] phosphorenes were prepared by exfoliation of black phosphor and molecular beam epitaxy (MBE) growth, respectively. Buckled and ultra-flat stanenes were prepared by the MBE growth on Bi2Te3(111) [369] and Cu(111) [368] surface, respectively. However, for the carbon element, it is still of great challenge to prepare any other 2D allotropes besides graphene, i.e., the isomers of graphene.
This challenge is caused by the fact that graphene represents the thermodynamically most stable tiling of sp2 carbon atom in 2D space. The growth of other less stable nonbenzenoid isomers therefore requires a more sophisticated kinetic control. Up to date, the well-developed MBE and chemical vapor deposition (CVD) growth of graphene typically employs high substrate temperatures that allow reversible C—C bonds formation. This results in the yielding of the most stable graphene [370]. In contrast, the growth of isomers of graphene requires irreversible C—C bonds formation, which traps the carbon structures in a local minimum. Therefore, the most promising technique for the growth of isomers of graphene is the organic synthetic methods employing C—C coupling reactions. Its additional advantage is the big reservoir of nonbenzenoid hydrocarbons, which act as building blocks to achieve the nonhexagonal tiling of carbon atoms beforehand. Since the isomers of graphene could be reckoned as hydrogen-free 2D conjugated polymers with nonbenzenoid hydrocarbon repeating units. In view of this, the route to isomers of graphene is to achieve 2D polymerization of nonbenzenoid hydrocarbons through C—C coupling/annulation.
The goal of this part is to show the art of designing isomers of graphene from the perspective of synthetic feasibility on the basis of the chemistry of nonbenzenoid hydrocarbons and on-surface synthesis. As a start, we will classify the predicted isomers of graphene regarding their chemical structures. Subsequently, we show the current developments of the on-surface reactions for the synthesis of nonbenzenoid carbon nanostructures, including quisi-0D dots, 1D nanoribbons and 2D network with non-hexagonal carbon rings. The comparison of the electronic properties between benzenoid and nobenzenoid carbon nanostructures will also be presented.
3.7.1
Classification of graphene isomers
In fact, even before the discovery of graphene in 2004, dozens of graphene isomers were proposed by theorists when they attempted to construct new carbon allotropes similar to graphite. These isomers of graphene were initially called the planar 3-connected carbon nets [371], which also consists of all sp2 carbon atoms similar to graphene, however connecting in a (or partially) nonhexagonal manner. Since then, many of these single-layered 2D carbon nets [372-385], i.e., isomers of graphene, have been theoretically studied and predicted with exotic electronic properties [386-390]. Compared to the semimetal graphene, these isomers could be metal [391] or even superconductor [381]. Nevertheless, these predictions have seldomly been demonstrated experimentally so far due to the difficulty of their synthesis.
Compared to graphene with all six-membered rings, isomers of graphene can be generated by tiling of exclusively nonhexagonal rings or a mixture of hexagonal and nonhexagonal rings. According to the carbocycles contained, isomers of graphene can be classified into different types. Graphene isomers with even and odd-membered rings are divided into alternant and nonalternant structures, respectively. They could also be grouped by containing hexagonal rings or not, the former corresponds to benzenoid structure (only graphene is benzenoid), the latter is therefore nonbenzenoid category. As illustrated in Fig. 31, the first row shows three typical nonbenzenoid isomers of graphene containing soley nonhexagonal rings, including the T-graphene [381] with 4,8-membered rings, carbon pentaheptite [391] with 5,7-embered rings, and Kagome graphene [392] with 3,12-membered rings. While T-graphene contains only even-numbered rings and therefore belongs to an alternant structure. The carbon pentaheptite consisting of solely odd-numbered rings, which is nonalternant. Despite such type of graphene isomers with pure even or odd-numbered rings, most graphene isomers containing a mixture of them, e.g., the Kagome graphene.
Figure 31
Figure 31.
Skeletons of isomers of graphene classified by non-/benzenoid and non-/alternant characters. Apart from the above-mentioned nonbenzenoid graphene isomers with pure nonhexagonal rings, most of the predicted isomers contain a mixture of hexagonal and nonhexagonal rings, e.g., biphenylene sheet with 4,6,8-membered rings and Phagraphene with 5,6,7-membered rings. Regarding the graphene isomer containing the same type of carbocycles, tuning their tiling manners and the ratio of different carbocycles could also generate different types of graphene isomers. For instance, the biphenylene sheet contains alternative six and four-membered rings along the vertical direction [393]. Adding one more six-membered ring between two four-membered ring results in a new isomer of graphene [394] with also 4,6,8-membered ring with different properties.
These listed isomers of graphene have been predicted with contrasting properties compared to graphene. Graphene with all the hexagonal rings showing a higher lattice symmetry compared to its isomers, generating a band structure with Dirac cone [395]. Nevertheless, some of the graphene isomers turns into metals with overlapped valence and conduction bands, e.g., the Biphenylene sheet [393] and carbon pentaheptite [391] were predicted with considerable density of states located at Fermi level, in principle exhibiting conductance better than metallic carbon nanotubes. Phagraphene retains the Dirac cone feature in its band structure, however, asymmetry is induced along different orientations, showing promising transportation anisotropy [386]. More intriguing graphene isomers are T-graphene and Kagome graphene, which are predicted to be correlated systems, exhibiting superconductivity and flat band magnetism, respectively. Up to date, only bipheneylene sheet has been successfully synthesized on a gold surface and has been confirmed with a metallic nature. The synthesis of other graphene isomer is still of great challenge and requires continuous effort from chemists and material scientists to make them. In the following, synthesis of nonbenzenoid quasi-0D nanocarbon, 1D polymers and 2D network on surfaces will be presented aiming at the development of potential synthetic protocol towards various 2D graphene isomers.
3.7.2
On-surface synthesis of nonbenzenoid polycyclic hydrocarbons
In recent years, the successful synthesis of 2D (conjugated) polymers [396] or nanoporous graphene [397] has shed light on the possible realization of 2D isomers of graphene. Particularly, the emerging on-surface synthetic techniques can be applied to guarantee their single-layered growths, which also have a high tolerance for the solubility of the reactants and products. These features perfectly meet the needs of the synthesis of single-layered insoluble isomers of graphene. Its huge potential has been demonstrated by the successful synthesis of segments of phagraphene (5–6–7, an isomer of graphene with 5-, 6-, and 7-membered rings), TPH-graphene (4–5–7) from a nonbenzenoid 2,6-dibromoazulene molecule and the synthesis of biphenylene sheet (4–6–8) on gold surface. Although the domain size is currently limited as a starting point, the future developments of this technique may finally help to increase their domain to considerable size that is sufficient for electronic device applications. In this part, only nonbenzenoid 0D nanocarbons, 1D chain and 2D network with fully annulated nonhexagonal rings are reviewed since the most challenge part for the synthesis of graphene isomers is how to annulate all the carbocycles not just connecting them through a single C—C bond.
The synthesis of nonbenzenoid nanocarbons, as the nonbenzenoid counterparts of nanographene is the starting point of on-surface synthesis of nonbenzenoid structures. A representative sample is the synthesis of dibenzo[a,m]dicyclohepta[bcde, nopq]rubicene (Fig. 32a), a nonbenzenoid nonalternant structural isomer of peri-tetracene with two embedded azulene units [398]. The key goal of this starting work is to compare their electronic structures which help to elucidate the influence of the altered bond topologies on their properties. Both the peri-tetracene and its nonalternant counterpart were achieved through on-surface cyclodehydrogenation reaction, which is a unique reaction particularly works well on metal surfaces discovered during the on-surface synthesis of the armchair edged graphene nanoribbon with a width of seven carbon atoms (7-AGNR) [330]. Later on, more nonbenzenoid nanocarbons were achieved by the simple cyclodehydrogenation of a nonplanar precursor [399].
Figure 32
Figure 32.
On-surface synthesis of nonbenzenoid nanocarbons including peri-tetracene and its nonbezenoid isomer with two azulene moieties (a), nanocarbons with 4 to 8-membered rings (b), nanographenes with six azulene units (c) and biphenylene dimmer (d). The corresponding nc-AFM or bond-resolved STM images were displayed at the right sides. Reproduced with permission [398,400-402]. Copyright 2018, 2020–2022, American Chemical Society.
To achieve odd-numbered rings in nonbenzenoid nanocarbons, precursors based on benzenoid/mainly benzenoid carbon skeletons were typically functionalized with methyl groups such as in the synthesis of dibenzo[a,m]dicyclohepta[bcde, nopq]rubicene. Another interesting case is the synthesis of nonbenzenoid nanocarbon containing four- to eight-membered rings from a precursor with already existing five and six-membered rings as illustrated in Fig. 32b [401]. The examples show that cyclodehydrogenation reaction succeeds to generate quasi 0D nonbenzenoid nanocarbons. However, extending such nonbenzenoids unit into 1D or 2D periodic structures requires additional reaction steps. One promising reaction for the on-surface polymerization is the surface Ullmann coupling [403], which has been applied to many benzenoid precursors for the synthesis of graphene nanoribbons and 2D polymers. G. Lohr et al. employed a singly brominated nonplanar precursor that is designed for two step hierarchical reaction (Fig. 32c) [400]. The first step Ullmann coupling of the nonplanar monomer leads to the formation of a covalently coupled dimer, which is still nonplanar. At the second step, cyclodehydrogenation reaction planarizes the dimer into an elongated nanocarbon with 5, 6, and 7-membered rings. The planarization is expected to form four pairs of 5, 7-membered rings and two singly sited 7-membered rings where an unpaired electron on each is expected. Unexpectedly, the carbon skeleton rearranged into a structure with six pairs of 5, 7 rings by forming additional five-membered rings that is fused to the singly sited 7-membered rings. The breaking of hexagonal rings to a five-membered ring that combines a seven-membered ring forming an azulene units is owing to the lower down of the molecular energy by eliminating the unpaired electrons.
Construction of even-numbered rings other than hexagon, e.g., four and eight membered rings can be achieved directly by the coupling between precursor monomers with all benzenoid skeletons. Biphenylene is such a case, which can be formed by 2 + 2 cycloaddition reaction of 1,2-dibromobenzene. The analogue of this reaction on surface was demonstrated by Fasel et al. on Ag(111) surface with a dibromotetracene precursor [344], a considerable percentage of dimerized tetracene molecules with a cyclobutadiene joint ring (4-membered ring) has been observed. Similar work has also been done on gold surface by Grill et al. A 1,2-dibromoanthracene monomer has been employed first on an Au(111) surface yielding majorly starphene by 2 + 2 + 2 cycloaddition reaction [346]. Nevertheless, on an Au(100) reconstructed corrugated surface, the 1D confinement effect steers the reaction into a 2 + 2 cycloaddition type, resulting in the nearly exclusive formation of biphenylene bridged acene. Coupling of halogenated biphenyls is another way to the biphenylene containing structures. Kawai et al. reported the synthesis of dibenzo[b,h]biphenylene from a 3,3′-dibromo-2,2′-binaphthalene precursor and further demonstrated that the four-membered ring is a radialene structure rather than the annulene structure to reduce the antiaromaticity [404]. Zeng et al. employed a similar strategy however with four brominated sites that are ortho to the C—C bond bridge of the biphenyl skeleton (Fig. 32d). In such a case, the first two brominated sites at one side of biphenyl were activated and C—C coupled forming a biphenylene with additional two radicals bonded to the surface lattice. At the second step, two chemisorbed biphenylene connect into a biphenylene dimer representing a limited case of extension of biphenylene units [402].
To extend the nonbenzenoid nanocarbon units into 1D can be achieved similarly using surface Ullmann coupling and cyclodehydrogenation reactions, however with doubled reactive sites. Taking the formation of biphenylene bridged acene from 1,2-dibromotetracene as example, a variant tetrabrominated tetracene precursor could undergo 2 + 2 cycloaddition reactions at its two terminals leading to 1D extended chain of tetracene periodically bridged with four-membered rings as illustrated by Fig. 33a [348]. The 2 + 2 cycloaddition reactions also have several variant types including simultaneous C-X(halogen) and C—H activation and bond rearrangement induced generation of ortho-arynes. Zhong et al. reported the formation of 1D nanoribbon consisting of periodically arrayed PTCDA moieties bridged with four and eight-membered rings from a tetrachlorinated PTCDA molecules on Au(111) surface (Fig. 33b) [405]. The 2 + 2 cycloaddition reaction here is achieved by the activation of both C—Cl and C—H bonds owing to a proximity catalytic effect. Ecija et al. reported a bond rearrangement induced generation of ortho-aryne, which then subsequently undergo the 2 + 2 cycloaddition reaction leading to the formation of four-membered rings and subsequent formation of zigzag-shaped polymer chain bridge with biphenylene units (Fig. 33c) [406]. The precursor is dibromoethylene functionalized at the two ends of a terphenyl moiety, the anchoring of dibromoethylene units on a five-membered ring rearrange into a six-membered ring with two otho-positioned radicals bonded to gold atom, which is ready for their 2 + 2 cycloaddition reaction.
Figure 33
Figure 33.
On-surface synthesis of nonbenzenoid alternant 1D structures by 2 + 2 cycloaddition reactions including (a) tetracene polymers periodically bridge with four-membered rings, (b) graphene like nanoribbons periodically embedded with four- and eight-membered rings, and (c) dibenz[a,h]anthracene polymers bridge with biphenylene units. The corresponding nc-AFM and bond-resolved STM images were displayed accordingly. Reproduced with permission [348,405,406]. Copyright 2019, John Wiley and sons; Copyright 2017, CC-BY 4.0; Copyright 2023, Springer Nature.
The using of 2 + 2 cyloaddition reaction generates only four-membered rings and the derived eight-membered rings. The synthesis of nonbenzenoid 1D nanoribbons with odd-numbered rings, such as five and seven-membered rings requires the design of building block containing directly odd-numbered rings or motifs that could generate them at a second reaction step. For instance, Ullmann coupling of the dibromo-para-terphenyl with three methyl groups leads to the formation of poly(paraphenylene) chain functionalized with methyl groups [407]. At an elevated temperature, cyclodehydrogenation involving the methyl and phenyl groups results in the formation of five-membered rings bridging all the phenyl rings yielding indenfluorene polymers with alternating five and six-membered rings. Generation of five-membered rings is also possible from the intrachain C—C coupling by activation of cumulenic C—C double bonds. As illustrated in Fig. 34b, the precursor consisting of a bianthracene skeleton functionalized with two dibromoethylene terminals in a first step undergoes Ullmann coupling and flattening of bianthracene moieties on Au(111) at 500 K into bisanthene chain bridged with cumulene structure [408]. Further annealing of the polymer chain to 650 K triggers the activation of the C—H bonds that is adjacent to the bridging C—C bond on the bisanthene units, resulting in the formation of five membered rings and subsequently the pentalene-bridged bisanthene polymers. These two examples both employs additional single carbon atoms either from the methyl or the ethylene groups that is ready to couple with the benzenoid carbon skeletons forming the five-membered rings. Alternatively, odd-numbered rings including five- and seven-membered rings could also be introduced directly from the precursor monomers such as the azulene containing molecules. Sun et al. reported the on-surface synthesis of C—C covalently linked azulene polymer chain from a dibromo-biazulene precursor connected at the 1, 3 positions [409]. Interestingly, further annealing of the polymer chain to 523 K triggers not only the cyclodehydrogenation reaction but also the carbon rearrangement into short curved chains with alternatively fused 5- and 7-membered rings, representing a promising surface reaction for the generation of 1D or 2D structures with rich 5- and 7-membered rings.
Figure 34
Figure 34.
On-surface synthesis of nonbenzenoid 1D oligomer or polymer chains including (a) polyindenfluorene with alternating 5- and 6-membered rings, (b) anthracene polymers bridged with pentalene units, and (c) short chain consisting of alternatively fused 5, 7-membered rings. The corresponding nc-AFM images were displayed accordingly. Reproduced with permission [407-409]. Copyright 2020, American Chemical Society; Copyright 2020, CC-BY 4.0.
The above discussed nonbenzenoid 1D structures are more close to the definition of polymers with limited widths. To make 1D nonbenzenoid nanoribbons with enlarged widths is even more challenging owing to the annulation of more ring along the direction perpendicular to the chain axes. One possible approach is to lateral fuse single polymer with nonbezenoid rings to a wider nanoribbon. Azulene as a molecule containing only odd-membered rings is an appropriate candidate for the building of nonbenzenoid polymer chains. As illustrated in Fig. 35a, Ullmann coupling of 2,6-dibromoazulene on an Au(111) surface at 473 K leads to the formation of polyazulene chains that are well aligned together [410]. Further annealing to 673 K leads to their lateral fusion into wider nanoribbons. According to the different relative positions of the polyazulene chains that are fused together, phagraphene nanoribbon with 5, 6, 7-membered rings and TPH-graphene nanoribbon with 4, 5, 7-membered rings were obtained. Owing to the richness of 5, 7-membered rings in polyazulene chains, the formed nanoribbons are therefore also rich of these odd-membered rings. Particularly, this leads to the formation of TPH-graphene without any six-membered rings. The synthesis of wide carbon nanoribbon with nonalternant rings could also be achieved by direct design of precursor. Fig. 35b shows the synthesis of carbon nanoribbons with 5, 6, 8-membered rings with also an hierarchical surface reaction on Ag(111) surface [411]. The first step is the Ullmann coupling of a tetrabrominated precursor with cove-edges into a porous graphene nanoribbon with periodically arrayed divacancy defects. Further annealing to elevated temperature leads to the dehydrogenation of the C—H bonds inside the divacancies leading to the C—C coupling of cove-edges into five-membered rings and thus the transformation of divacancy defect into 5–8–5 ring defect. However, owing to the nonplanar nature of 5–8–5 defects, only one of every two divacancy defects can be converted, which could release the structural strain for the stability consideration. The remaining divacancies with removed inner hydrogen atoms therefore was inserted by an Ag adatom forming ultimately graphene nanoribbons with alternatively arrayed 5–8–5 defects and organometallic boned Ag centers.
Figure 35
Figure 35.
On-surface synthesis of nonbenzenoid 1D nanoribbons including (a) phagraphene and TPH-graphene nanoribbons from Ullmann coupling of 2,6-dibromoazulene and subsequent lateral fusion of polyazulene chains, (b) graphene nanoribbons embedded with periodically arrayed 5–8–5 ring defects and Ag centers. The corresponding nc-AFM images were displayed at the right sides. Reproduced with permission [410,411]. Copyright 2019, 2023, American Chemical Society.
3.7.4
Extending nonbenzenoid structure into two-dimensional networks
Further extension of the nonbenzenoid structures along the second dimension is rather challenging. For instance, in the study of fusion of polyazulene into phagraphene and TPH-graphene nanoribbons, it is very difficult to laterally fuse more than two chains together since the azulene units tends to undergo carbon rearrangement into the naphthalene moieties. This caused the bending of the polyazulene chains and the alignment between two chains is absent which is necessary for the lateral fusion reaction. Moreover, the polyazulene chain involved in the lateral fusion reaction has two possible relative orientations, which also leads to different structures. Fan et al. developed an intrachain dehydrofluorination reaction for the lateral fusion of polyparaphenylene (PPP) chain skeletons forming four- and eight-membered rings in between [340]. As shown by the reaction scheme in Fig. 36a, the dibromo-para-terphenyl monomer is functionalized with additional six C-F groups. The first step employs the surface Ullmann coupling leading to the formation of fluorinated PPP chains. The suitable fluorination leads to the lateral fusion of the PPP chain in a ladder type arrangement that guarantees the formation of four- and eight-membered rather than the six-membered rings that typically occurs for the fusion unsubstituted PPP chains [412]. Furthermore, owing to the high stability of the phenylene moieties (Clar's Sextet), carbon skeleton rearrangements of PPP chains is absent favoring their continuous lateral fusion. As shown by Fig. 36b, a section of 2D domains of biphenylene network were successfully achieve by the lateral fusion of at least seven fluorinated PPP chains. With the first realization of this biphenylene network, its predicted metallicity has been confirmed by the STS. However, up to date, apart from the success in the on-surface synthesis of biphenylene network, attempts to prepare other 2D nonbenzenoid isomers of graphene are still elusive.
Figure 36
Figure 36.
On-surface synthesis of nonbenzenoid graphene isomer, biphenylene network, through the reaction scheme (a) lateral fusion of fluorinated poly(para-phenylene) chains. (b) The corresponding STM and nc-AFM images were displayed underneath the structures in panel (a). (c) Differential conductance spectra of the biphenylene ribbon with different widths. The red frame marked out the metallicity of biphenylene ribbon with 21 carbon atoms wide. Reproduced with permission [340]. Copyright 2021, American Association for the Advancement of Science.
The on-surface synthesis of 2D nonbenzenoid isomers of graphene is still at a starting point. To realize more of them, the development of more nonbenzenoid polycyclic aromatic molecules in solution as building block for the on-surface polymerization is of great importance. Currently, the nonbenzenoid building blocks is limited to azulene based molecules, other nonbenzenoid molecules such as pentalene with two five-membered rings (5–5), heptalene (7–7), and cyclooctatetraene functionalized with halogens or other reactive groups is still challenging. This blocks the initial starting of couple them in a 2D manner, which may finally result in the formation of various isomers of graphene. Additional limitations for the on-surface synthesis of graphene isomers lie in the limited domain sizes, which is the general problem faced by the 2D polymers formed on solid/vacuum interfaces. Performing the same strategy of polymerization of nonbenzenoid building blocks into 2D at a solid/liquid interface that allows a reversible C—C bond formation may help to substantially improve the quality of the formed 2D isomers of graphene.
3.8
Substrate effect on-surface synthesis
The on-surface reactions are initiated by the adsorption of specific precursor molecules on surfaces. Therefore, it is important to investigate the influence of the detailed adsorption configurations of precursor molecules to the reaction pathways. In 2018, Zhong et al. reported the lattice mismatch induced dissymmetric adsorption of 4,4′'-diamino-p-terphenyl (DATP) on Cu(111) [413]. As shown in Fig. 37a, different image performance is observed on 2 equiv. amino terminals. Careful investigations reveal that the phenomenon arises from that two amino groups of DATP are located at top site and hollow site, respectively. Subsequently, the influence of the detailed adsorption configuration on the reaction pathway was reported by Wang et al. [414]. As depicted in Fig. 37b, desulfonylation homocoupling reactions were studied on Au(111) and Ag(111), respectively. The activation temperature on Ag(111) is 50 ℃ higher than on the Au(111) surface, which is in contradiction to the fact that silver is generally more chemically active than gold. Combining the SRPES observations with DFT calculations, they elucidated that the phenomenon arises from the different adsorption configurations of the dechlorinated monomer on Au(111) and Ag(111) surfaces. On Au(111), Au-S interactions efficiently weaken the bonds between phenyl and S, facilitating the desulfonylation reactions. On the other hand, the precursors anchor on Ag(111) via Ag-O interactions, which lifts the S atoms and results in a high activation barrier of desulfonylation reactions.
Figure 37
Figure 37.
Adsorption configuration and its influence. (a) Abnormal hopping behavior of DATP molecules on Cu(111) induced by the asymmetry adsorption [413]. (b) The influence of different adsorption configurations of precursor molecules on Ag(111) and Au(111) to the reaction barrier [414]. Reproduced with permission [413,414]. Copyright 2022, American Chemical Society; Copyright 2018, CC-BY 4.0.
Metal adatoms, especially external ones, exhibit promising perspectives on promoting selective synthesis of certain products in high yields [357,415,416]. During the on-surface reactions, the radical intermediate states tend to interact with metal adatoms (e.g., Cu or Ag), forming metal-organic structures on surfaces. When the precursor molecules possess abundant functional groups, various organometallic structures may be constructed through external excitations on surfaces. Moreover, various metal adatoms can trigger distinct reaction pathways and induce remarkable different final products [417]. Comprehension on how metal adatoms influence selective synthesis on surfaces can help for exploit of other precise surface reactions for fabrication of more advanced robust nanoarchitectures, and achieving controlled hierarchical formation of metal-organic bonds thus acquired much attention. As shown in Fig. 38a, Li et al. successfully fabricated multiple metal-organic bonds (N—Cu bonds and C—Cu bonds) through one-by-one scission of X-H bonds (X = N and C) in aromatic amines on the Cu(111) surface [418]. Each step of dehydrogenation reaction lead to the formation of corresponding metal-organic coordination products, as monitored by high resolution STM and AFM. The multiple metal-organic bonds (aryl-Au and aryl-Ag bonds) formation can also be observed during the synthesis of 5-AGNRs on Au(111) and Ag(111) surfaces [322,419]. Afterwards, Zhong and colleagues demonstrated the synthesis of multiple aryl-metal bonds via the stepwise thermal dehalogenation reactions [420]. Dehalogenated monomers interact with each other through multiple aryl-metal bonds, forming 1D and 2D metal-organic hybrids on Au(111) and Ag(111), respectively (Fig. 38b).
Figure 38
Figure 38.
Formation of multiple metal-organic bonds. (a) The hierarchical dehydrogenation reaction of aniline on the Cu(111) surface [418]. (b) The substrate-modulated synthesis of 1D and 2D metal-organic hybrids with the organic building blocks (perylene) on Au(111) and Ag(111), respectively [420]. Reproduced with permission [418,420]. Copyright 2018, 2022, American Chemical Society.
Xu's group has recently presented a series of studies on unraveling the role of metal atoms on the tautomerization reaction of nucleobases on the inert Au(111) surface [126,421]. It is reported that, Ni atoms are able to inhibit the tautomerization of G molecules from the canonical G/9H form to non-canonical G/7H one (left panel in Fig. 39a) [126]. The underlying mechanism is found to be the preferentially coordination of Ni atoms with the nitrogen atoms at N7 sites and thus the site of N7 is automatically screened for proton transfer to inhibit G tautomerization from G/9H to G/7H. Inspired from such coordination priorities, nickel atoms are applied as a catalyst to trigger keto-enol tautomeric dehydrogenation reaction of thymine molecule on Au(111) (right panel in Fig. 39a), which is induced by Ni atoms preferentially coordinating with nitrogen atoms instead of oxygen atoms [421]. Among a large number of surface-assisted reaction, the surface-assisted Ullmann coupling has been most frequently used for construction of distinct robust covalent bonded nanostructures. It is reported that metal atoms can also assist the Ullmann reaction. For example, Liu et al. reported that for 3-bromonaphthalen-2-ol (3,2BNOL) and 2-bromonaphthalen-1-ol (2,1-BNOL) molecules with multiple active site, no uniform reaction products can be formed on Ag(111) (Fig. 39b) [422]. After introducing Fe atoms, selective Ullmann couplings are triggered and complex exo- and endo-bent organic ligands are selectively synthesized on Ag(111) in high yield. More interestingly, the exo- and endo-bent ligands in turn promote the buildup of mono- and bi-iron chelated complexes respectively, which show distinctly different electronic properties.
Figure 39
Figure 39.
(a) Illustration on metal-modulated tautomerization. Left panel: Ni atom inhibit the G tautomerization from G/9H to G/7H form on Au(111) by preferential coordination at N7 site. Right panel: Ni atom induced tautomeric dehydrogenation of thymine molecules on Au(111) by the preferential coordination of Ni atoms with nitrogen atoms [126,421]. (b) Illustration on Fe mediated selective synthesis of stereoselective organic ligands and mono-/bi-iron chelated complexes on Ag(111) [422]. Reproduced with permission [126,421,422]. Copyright 2014, 2018, American Chemical Society; Copyright 2021, John Wiley and sons.
More than half a century ago, the surface science of heterogeneous catalysis started with using metal single crystals as the model systems to study elementary processes in catalysis on metal surfaces [423,424], accompanying which an arsenal of surface techniques that are capable of molecular level characterization have been developed and reach into maturation. Along this line, surface science studies over metal single crystals have achieved tremendous progress in opening up the "black box" of catalysis by revealing the elementary processes of heterogeneous catalysis. Yet, the problems of "materials gap" and "pressure gap" have gradually surfaced that confronted whether single-crystal model catalysts and ultra-high vacuum (UHV) conditions used in surface science studies could demonstrate physical or chemical properties of practical catalysts, and as such to facilitate their rational design. To bridge these gaps, tremendous progress have been made over the past decades in the construction of nanoscale model catalytic systems with the atomic precision and the development of ambient-pressure (AP) surface science techniques that could study the catalytic surfaces/interfaces under realistic working conditions.
The employment of AP techniques is highly desired for current research focus on the catalytic hydrogenation of COx (CO and CO2) [425], a process center to carbon capture and utilization (CCU). Studies of catalytic hydrogenation reactions typically necessitate high H2 pressures as evidenced in powder catalysis, because active hydrogen species typically adsorb weakly and tend to desorb at above room temperature under vacuum conditions. While oxide catalysts for hydrogenation reactions [426-428] have increasingly caught attention due to their high selectivity, mechanistic studies of hydrogenation catalysis on oxides are almost exclusively performed on powder catalysts [429], which limited the accurate identification of key intermediates/reaction path due to their complex surface composition and morphology. In particular, ZnO-based catalysts have achieved significant attention in the past years owing to their promising catalytic performances for syngas conversion [427,428,430], methanol synthesis [431] and the reverse water gas shift reaction (RWGS) [432,433]. The role of ZnO in hydrogenation catalysis has remained largely unknown, making it a central focus while there has been a lack of mechanistic studies on well-defined metal oxide surfaces regarding hydrogenation reactions of small molecules owing to the difficulties [434] in detecting hydrogenated intermediates. AP-STM provides an apt tool for atomic-scale studies of hydrogenation catalysis, but the challenge of maintaining a stable tunneling junction on bulk oxide surfaces in a reactive environment has hampered its application.
By improving the stability of AP-STM, we can now visualize the atomic-scale dynamics of oxide surfaces in response to ambient reactant gasses (Fig. 40) [435,436]. Despite the weak interaction between CO and ZnO, physisorption of an ordered (2 × 1)-CO structure on ZnO(1010) could be observed in ambient CO gas. Meanwhile, the adsorption of CO also led to simultaneous reaction with ZnO at 300 K to form CO2. The etching of step edges of ZnO(1010) demonstrated that CO reacted with step oxygen atoms. In comparison, chemisorption of CO2 on ZnO(1010) occurs readily in UHV and forms tridentate carbonate species. Yet, the co-presence of physisorbed CO2 under ambient conditions could alter the locations of chemisorbed CO2, enhance its adsorption and lead to the formation of a new adsorbate structure, (3 × 1)-CO2, on ZnO(1010). The heterolytic dissociation of H2 on ZnO(1010) under ambient pressure was also directly visualized and the dissociation reaction did not require the assistance of surface defects. In H2/CO or H2/CO2 mixture gas, the presence of CO or CO2 on ZnO at 300 K does not impede the availability of surface sites for H2 dissociation; instead, CO can even enhance the stability of coadsorbed hydride species, thereby facilitating their dissociative adsorption.
Figure 40
Figure 40.
(a) Atomically-resolved and (b) simulated STM images of hydride species on ZnO(1010) in 0.4 mbar H2 at 300 K. The corresponding structural model of hydride chains is displayed in (b). Gray, red, and white spheres represent Zn, lattice O, and H atoms, respectively. (c) STM images of hydride species on ZnO(1010) in 2 mbar H2/CO (3:1) mixture gas at 300 K. (d–f) STM images and STM simulation of ZnO(1010) in 1 mbar CO at room temperature. The formation of (2 × 1)-CO structure is calculated by DFT with the optimized structural model and simulated STM image depicted in (e), showing 1/2 ML CO adsorbed on ZnO(1010). Gray, red, green and brown spheres represent Zn, lattice O, O in adsorbents and C atoms, respectively. (f) STM image showing the presence of bright spots within the (2 × 1) unit cell. The bright spots are diffusing during the imaging process and could be assigned as CO2 species. (g) AP-XPS C 1s of ZnO(1010) in 0.5 mbar CO or after the evacuation of CO gas. (h) A (3 × 1) STM image observed on ZnO(1010) in 3 mbar CO2 at room temperature. (i) Simulated STM image and (j) the optimized (3 × 1)−1 ML CO2 structure by DFT, including 2/3 ML chemisorbed CO2 plus 1/3 ML physisorbed CO2. Gray, red, green and brown spheres represent Zn, lattice O, O in adsorbents and C atoms, respectively. Reproduced with permission [435,436]. Copyright 2023, American Chemical Society; Copyright 2022, Elsevier.
Hydrides are highly effective at hydrogenating CO or CO2, evidenced by formation of formate species in H2/CO and H2/CO2 at elevated temperatures (Fig. 41). Meanwhile, the transformation from hydrides to hydroxyls is facile on ZnO at elevated temperatures, leading to a dominance of hydroxyl groups on the surface of ZnO, which would lead to weakened adsorption of CO and CO2 and hindered H2 adsorption. As a result, hydrogenation of COx is unlikely to occur on a fully hydroxylated ZnO surface, and must be carried out at above 450 K to facilitate desorption of hydroxyls and enable hydrogenation to take place. Both AP studies and DFT calculations showed that CO2 hydrogenation on ZnO is thermodynamically and kinetically more favorable compared to CO hydrogenation. These results point towards a two-step mechanism for CO hydrogenation, involving initial oxidation to CO2 at step sites on ZnO followed by reaction with hydride to form formate. This proposition is supported by the substantial energy barriers for CO binding with hydride and the absence of oxygen vacancies detected on the ZnO terrace following hydrogenation reaction. By shedding light on the COx hydrogenation reaction on ZnO, this study offers valuable molecular insights that deepen our understanding of syngas conversion and oxide catalysis in a broader context.
Figure 41
Figure 41.
(a) STM image of ZnO(1010) during the exposure to 1 mbar H2/CO2 (3:1) mixture gas at 300 K. (b) STM image of surface adsorbates on ZnO(1010) after being annealed in 1 mbar H2/CO2 (3:1) mixture gas at 400 K. (c) AP-XPS C 1s and O 1s of ZnO(1010) in 0.6 mbar H2/CO2 (3:1) mixture gas at different temperatures. (d) STM images of ZnO(1010) during the exposure to H2/CO mixture gas at 300 K. (e) STM image of surface adsorbates on ZnO(1010) after being annealed in 1 mbar H2/CO (3:1) mixture gas at 450 K. The formate species is indicated by white arrows. (f) AP-XPS C 1s and O 1s of ZnO(1010) in the 0.6 mbar H2/CO (3:1) mixture gas at different temperatures. STM images of hydroxylated ZnO(1010) surface during the exposure to (g) 2.1 mbar CO at 300 K, (h) 1.1 mbar CO at 450 K. The black arrows indicate linear adsorbed CO2. (i) AP-XPS C 1s and O 1s spectra of clean ZnO(1010), hydroxylated ZnO(1010) prepared by being annealed in 1 mbar H2 at 400 K and the hydroxylated ZnO(1010) exposed to CO at elevated temperatures. Reproduced with permission [436]. Copyright 2023, American Chemical Society.
The development of model catalysts that could capture the complexity of technological catalysts [437] has been another major thrust of surface science studies in catalysis. Since this century, together with the advancement of nanocatalysis, and the development of synchrotron-based techniques and advanced electron microscopies, great progress has been made in our understanding on the structure-function relationship in heterogenous catalysis. The design of catalytic materials could now be more focused on the design of active centers, and these heterogeneous catalysts by their forms could be divided into three categories: nanostructured catalysts (usually with size below 10 nm), cluster catalysts, and single site/atom catalysts. These catalysts are all associated with the prominent interfacial effects [438-440] or size effects [441], i.e., two main phenomena or long-lasting puzzles in catalysis research. The challenges now have come to solve these puzzles, to understand the nature of these catalytically active sites and to reveal reaction mechanism and dynamics at the molecular level [442], such that the rational design of new and highly efficient catalysts could be achieved. In a larger scope, meeting these challenges could lead to the unification of catalysis theory and bridging the gaps among heterogeneous catalysis, homogeneous catalysis and enzyme catalysis [443].
In the FeO-Pt interface, which is active for low-temperature CO oxidation, it was found that interface confinement effect controls the growth of supported nanostructures [444], which could be utilized to synthesize well-defined nanocrystals with identical shape and structures, a direction long-desired in model catalysis for the elucidation of size effects (Fig. 42). FeO nanostructures (NSs) on Pt(111) with diameter below 10 nm show typically the shapes of triangles or truncated triangles. The size distribution and structure of FeO NSs demonstrate the growth of FeO NSs is dictated by the strong interaction between FeO and Pt, i.e., an interface-confined growth. Due to lattice mismatch, Fe atoms occupy successively fcc, hcp and top sites on Pt(111), rendering the formation of fcc-, hcp- and top-FeO domains and a moiré pattern [445-447], which could be distinguished by their different apparent heights in STM. The periodicity of the moiré pattern is ~25 Å, corresponding to the coincidence lattice between FeO and Pt(111). The edges of FeO NSs typically expose two-coordinated Fe atoms, or the so-call CUF sites after UHV annealing. Interestingly, all corners and edges of FeO NSs consist of fcc-FeO domains, while top-FeO domains are always at the center. FeO triangles with the side length below 4 nm consist of only fcc-FeO and hcp-FeO domains (Fig. 42). DFT calculations have shown that fcc-FeO domains are thermodynamically most stable, while top-FeO domains are the least stable. Thus, with all edges located in the fcc-FeO domains could help stabilize the edges of FeO NSs and lower the total energy of FeO NSs.
Figure 42
Figure 42.
STM images and structural models of FeO triangles with different sizes and exposing CUF edge sites. n is the number of Fe atoms at each edge. Reproduced with permission [444]. Copyright 2017, American Chemical Society.
With these different-sized FeO NSs, a dynamic size effect that could control the chemical properties of supported clusters using their oxidation kinetics is shown as an example (Fig. 43). As we looked into the oxidation of different-sized FeO NSs, it could be found that while most FeO NSs were oxidized into FeO2, small FeO NSs remained the FeO phase under the same UHV oxidation conditions, although all could be oxidized into FeO2 under ambient O2. In UHV, as the area of FeO2 domains increases with the increasing NS size (d), the oxidation ratio went down drastically for FeO NSs with d < 5 nm and no FeO2 domains were observed for FeO NSs with d < 3.2 nm. In-situ STM images have shown that FeO2 domains were developed by oxygen penetration from the step edges of FeO NSs. By examining the interaction of FeO NSs with O2, a dynamic size governing the oxidation kinetics of FeO NSs was observed.
Figure 43
Figure 43.
Dynamic size effect in the catalytic oxidation of FeO NSs supported on Pt(111). Reproduced with permission [448]. Copyright 2017, CC-BY 4.0.
The interaction of O2 with different-sized FeO triangles is compared to expose exclusively CUF steps. Despite their same shape and structure, O2 dissociation over Fe78O66 NS led to a spontaneous and complete reconstruction of NS, which was also the case for smaller FeO NSs. A collective shift of surface oxygen atoms was observed upon O2 exposure, which causes edge CUF sites to be saturated by CUO atoms. In contrast, only a partial reconstruction was observed for larger NSs, i.e., starting from the edges, only a portion of O atoms were shifted to the adjacent three-fold hollow sites. Oxygen dislocation lines were formed at the boundary between the reconstructed and unreconstructed oxygen domains. DFT calculations showed that upon reconstruction, the diffusion of CUO atoms was hindered significantly with a barrier of 2.37 eV/O atom to enter into the FeO-Pt interface. Meanwhile, the diffusion barriers for dislocation O atoms entering the FeO-Pt interface is ~0.50 eV/O atom and for the insertion of dangling O into the FeO-Pt interface at 1.41 eV/O atom. Thus, the spontaneous reconstruction prevented the oxidation transition to FeO2 and FeO NSs with d < 3.2 nm are likely passivated from oxidation by stabilizing all O adatoms via the complete reconstruction in O2. The partial reconstruction of larger FeO NS led to unreconstructed steps and oxygen dislocation lines, both of which are vulnerable for oxygen insertion. Therefore, rather than the structure of active sites as often perceived, the dynamic size effect governs the oxidation kinetics by tuning the stability of edge O atoms. The same dynamic size effect were also found for the oxidation of CoO NSs on Pt(111) and Au(111), where CoO NSs with d < 3 nm remained the CoO phase while larger CoO NSs were susceptible for further oxidation. Dynamic size effect in catalytic CO oxidation was further studied at the Pt-FeO interface, which will be reported later.
To demonstrate the role of interfacial interaction in tuning the catalytic properties of supported oxide nanostructures, well-defined Cu2O nanostructures have been synthesized on Pt(111), Pd(111), Au(111) and Ag(111) surfaces [449,450]. Despite the same Cu2O(111)-like structure, these supported Cu2O NSs exhibited variable thermal stability depending on the metal substrate (Fig. 44). Cu2O NSs on Pt exhibit notably lower thermal stability compared to those on Au and Ag, which could be attributed to the difference in the oxide-metal interaction (OMI). The catalytic activities for CO oxidation at these interfaces are found to follow the order Cu2O/Pt > Cu2O/Au > Cu2O/Ag. A direct correlation was found between these activities and the OMI — particularly the formation energy of interfacial oxygen vacancies and electronic interactions between Cu+ and the metal substrates. In-situ AP-STM allowed further analysis of the reaction mechanism, where CO2 formation was found rate-limiting for CO oxidation at the Cu2O–metal interface. The understandings from model catalysts could be further extended to practical powder catalysts, which exhibited the same reaction trend, corroborated model findings and thus established the design principles acquired from model catalysts for low-temperature CO oxidation. The role of OMI in determining both activity and stability of supported oxide NSs offered new avenues for the rational design of highly efficient catalytic systems.
Figure 44
Figure 44.
Role of oxide-metal interaction (OMI) in determining the activity and stability of supported Cu2O NSs for catalytic CO oxidation. Reproduced with permission [449]. Copyright 2020, CC-BY-4.0.
Overall, this section demonstrated how advanced model catalyst studies can establish atomic-scale structure-activity relationships and descriptors to understand and optimize complex practical catalysts, where the identification of active sites has long been challenging. This study marks a leap toward model-guided catalyst design, where model structures could inform quantitative optimization of complex catalysts.
4.2
Single-atom catalyst
Single-atom catalyst [451-457] refers to active metals of a catalyst in the form of individual atoms. They offer superior atomic efficiency and clearly defined active centers compared to conventional catalysts, resulting in enhanced catalytic performance, including activity and selectivity. The stable creation, structural characterization, and exploration of their catalytic processes are vital to understanding and utilizing single-atom catalysis. Kai Wu and his team [458-461] have devised a technique to prepare single atoms stably by managing the surface free energy of oxides. While metals tend to have higher surface free energy than oxides, leading them to aggregate on oxide surfaces, a thin oxide layer on a metal's surface can change this dynamic. The metal's electronic properties raise the oxide's surface free energy, promoting the dispersion of the deposited metal. For instance, in their study, a CuO thin film layer on a Cu(110) single crystal surface acted as the support, and it successfully stabilized individual Au atoms at a coverage of 0.05 monolayers (Fig. 45a). Remarkably, these atoms remained stable even at 400 K. This approach to crafting single atoms is straightforward and doesn't necessitate stabilization via adsorbed molecules or lattice inclusion, paving a fresh path for single-atom catalyst preparation.
Figure 45
Figure 45.
Preparation of single atoms and the mechanism of CO oxidation. (a) STM imaging of Au single atoms on a CuO monolayer film, accompanied by a schematic representation of the catalytic mechanism for CO oxidation [459]. (b) STM imaging of Ni single cations on a CuO monolayer film, accompanied by a schematic representation of the catalytic mechanism for CO oxidation [461]. Reproduced with permission [459,461]. Copyright 2018, 2022, American Chemical Society.
Kai Wu et al. utilized Au single atoms as a model catalyst, delving into their CO oxidation reaction mechanism [459]. Initially, the Au atoms were negatively charged, with nearby CuO oxygen ions donating electrons. This negative charge facilitated the D-π* feedback between the metal and CO molecules, activating the adsorbed CO molecules. Consequently, CO molecules reacted with nearby O ions at room temperature to produce CO2 molecules, generating O vacancies. After the reaction, the Au atoms became neutral, deactivating the reaction. Introducing O2 at 400 K repaired these defects, recharging the Au atoms and restoring their activity. This process followed the typical Mars-van Krevelen (MvK) mechanism. The study enabled atomic-scale observation of these charge state evolutions, providing essential evidence for explaining the high activity of highly dispersed Au.
Additionally, Kai Wu et al. employed the CuO thin film on Cu(110) to support a Ni single cation model catalyst (Fig. 45b), by depositing Ni atoms at a temperature of 370 K [461]. Notably, the behavior of Ni single atoms varied during CO oxidation based on their chemical states. Ni atoms in a metallic state had a CO desorption peak at 400 K, while cationic Ni barely adsorbed CO. Yet, cationic Ni displayed a strong tendency to break down O2. At room temperature, O2 split on Ni single cations, forming reactive "Ni-O" species that combined with CO gas to yield CO2. This behavior indicates that Ni single cations drive the CO oxidation via the Eley-Rideal mechanism, contrasting with the MvK method seen in metallic Ni. This study provides a detailed characterization and analysis of the catalytic properties of cationic and metallic Ni single atoms in the CO oxidation reaction, contributing to a deeper understanding of the single-atom catalytic mechanisms and the chemical properties of single metal cations on surfaces.
Although single-atom catalysts offer advantages, they have limitations, especially in intricate reactions. Single cluster catalysts [463-465], with atomic precision, emerge as another potential promising catalysts due to their adjustable electronic structures and multifunctional active sites. In-depth research on cluster catalysis helps us gain a deeper understanding of the inherent principles behind catalytic reactions, enabling the targeted design and development of efficient catalysts. Therefore, Kai Wu et al. innovatively prepared uniform single Ni2 clusters on a CuO monolayer (Fig. 46) [462]. Their method involved depositing Ni atoms on a CuO monolayer at a low temperature of 100 K. This resulted in two Ni species: Ni cations (Nic) and mobile Ni atom precursors (Nip). Nip combined with Nic during migration, forming transient NipNic clusters. Subsequent heating gave rise to stable Ni2 clusters linked by oxygen atoms.
Figure 46
Figure 46.
STM imaging of single Ni2 clusters on CuO monolayer and their catalytic low-temperature CO2 dissociation [462]. Reproduced with permission [462]. Copyright 2023, American Chemical Society.
Distinctly, only Ni2 clusters could split CO2 at low temperatures. While metallic Ni did not hold onto CO2, cationic Ni did, but could not break it down. Conversely, Ni2 clusters adsorbed CO2 laterally and activated it with one oxygen atom of CO2 bonded with the Nic atom and the carbon attaching to Cu substrate. Upon further heating, CO2 decomposed into CO and oxygen. Theoretical calculations revealed that variances in CO2 adsorption configurations between the Ni2 clusters and Nic atoms stemmed from orbital symmetry deviations. The formation of Ni2 clusters introduced horizontal components to the previously vertical orbitals of Nic atoms, facilitating CO2's lateral absorption and activation. This study deepened the understanding of Ni2 clusters' catalytic behavior in heterogeneous catalysis, especially regarding CO2 dissociation at lower temperatures.
Apart from single clusters, single sites [468,469], composed of multiple atoms, can have distinct catalytic attributes. Polyethylene, the main component of many plastics with a global production of 100 million metric tons annually, is deeply integrated into human society. Its creation, ethylene polymerization, splits into two primary processes: free radical and coordination polymerization. The latter follows the Cossee-Arlman mechanism, where ethylene inserts itself between the bond of the growing polyethylene chain and the catalyst's metal center. A longstanding challenge in polymer science has been initiating chain formation on Phillips catalysts for ethylene polymerization without using alkyl aluminum.
To address this challenge, Kai Wu, Xiong Zhou, and their team utilized a substrate made of an atomically flat iron carbide thin film, derived from the carbon aggregation of an Fe(110) single crystal [466]. The resulting surface featured parallel domain strips, closely resembling the θ-Fe3C(102) surface. Continuous in situ STM imaging of this surface, under an ethylene atmosphere at room temperature, revealed the growth of polyethylene chains at the boundary sites. These chains were anchored at one end and expanding in a singular direction. Notably, once the polymerization is initialized, its subsequent chain growth becomes very fast, reflecting the characteristics of the chain-reaction polymerization mechanism. All experimental and computational results well fit the scenario that a C2H4 molecule is inserted into the initial CHCH3 intermediate at the triangular Fe3 site to form a chain (Fig. 47b). This sheds light on the formation of polyethylene through ethylene self-initiation over the Phillips catalyst without requiring an activator. These findings validate the molecular insertion mechanism for ethylene polymerization and unveil the self-initiation via ethylene isomerization without an initiator, settling the debate about the chain initiation process on the Phillips catalyst. Importantly, this study suggests a method to harness the polyethylene-catalyst interaction to intentionally end the polyethylene chain at specific lengths, refining product selectivity and introducing a novel method to control chain length distribution.
Figure 47
Figure 47.
Visualization of on surface ethylene polymerization at boundary Fe3 sites. (a) STM snapshots of the carburized iron surface when exposed to C2H4 at room temperature [466]. (b) Schematic illustration of the ethylene polymerization mechanism at boundary Fe3 sites on the iron carbide surface [467]. Reproduced with permission [466,467]. Copyright 2022, The American Association for the Advancement of Science.
5.
Microscopic inspirations of surface electrochemical processes by ECSTM
The investigations of the solid/liquid interfaces under electrochemical condition are of unique significance for revealing the important interface processes in catalysis, electronic science, material science and many other fields [470-472]. Due to the complexity of the electric double layer (EDL), the study of the electrochemical interfaces in electrolytes has become one of the challenges in surface science [473,474]. The EDL is localized in a thin region (several nanometers) at the electrode/electrolyte interface, and the distributions of species, charge and potential in the EDL are microscopic and dynamic, which are closely related to the electrolyte components and the electrode structures. Various techniques have been developed for investigating the interface electrochemical processes in the EDL [475-477], and the electrochemical STM (ECSTM) has become one of the state-of-the-art characterization methods due to the high spatial resolution, in-situ characterization and flexible operating condition [478-480]. Binnig and Rohrer [481] developed the first STM in 1982, which makes it possible to observe the surface structure directly at the atomic scale. After a few years, Bard and coworkers [482] operated the STM in water for the first time. Soon after, Itaya and Tomita [483] invented the first ECSTM by combining the STM technique and the electrochemical measurement system for controlling the potential of the sample in the electrolyte during STM imaging. As with the STM in ambient environment and ultra-high vacuum, the ECSTM possesses the high spatial resolution to the atomic and molecular scale. In addition, the ECSTM can be operated in a broad range of electrolytes, which enables the in-situ observations of the interface processes under the applied potentials. Therefore, since it was invented, the ECSTM has attracted wide attention in the surface science and electrochemistry community and has been used to investigate the critical electrochemical processes at the electrode/electrolyte interface, which provides microscopic insights for understanding the mechanisms of these processes [484-488]. In this section, the progresses in the applications of the ECSTM in investigating the electrochemical interfaces are systematically discussed. First, the basic principles and the features of the ECSTM are introduced. Then, we focus on the investigations of the interface electrochemical processes by the ECSTM, including the adsorption, diffusion, reconstruction and catalytic reaction.
5.1
Basic principles of ECSTM
The STM is operated on the basis of the tunneling effect. When the atomically sharp metallic probe (STM tip) approaches the sample surface (less than a few nanometers), the electron transfer could take place between the tip and the sample under the external electric field. By detecting and processing this tunneling current, the surface morphology of the sample can be imaged. According to the quantum mechanics, the tunneling current is very sensitive to the distance between the tip and the sample surface, so the vertical resolution of the STM could achieve 0.01 nm. For the ECSTM operated in aqueous solutions, the tunneling current was found to be influenced by the water layer. The arrangements of the water molecules and clusters around the tip and the sample lead to a distinct tunneling barrier compared to vacuum, and the bias and the adsorbates are also important [489]. Various models for electron tunneling through the water layer have been proposed to reveal the imaging mechanism of the ECSTM. For instance, Lindsay et al. [490,491] investigated the asymmetric and nonexponential electron tunneling in water, and the corresponding mechanisms were illustrated.
In the ECSTM experiments, an electrochemical cell with reference and counter electrodes is used for imaging. As shown in Fig. 48, the cell is a four-electrode configuration including the sample and tip as two working electrodes. The potentials of the sample and the tip can be applied independently with the control of bipotentiostat. In addition, the inlet and outlet of the gas and liquid are provided for controlling the electrolyte component and atmosphere in the experiments.
Figure 48
Figure 48.
Schematic illustration of the ECSTM setup. WE, RE and CE indicate working electrode, reference electrode and counter electrode respectively.
The sample for ECSTM characterization should be electric conductive for measuring the tunneling current, and the insulator cannot be imaged. Meanwhile, the surface of the sample should be clean and flat. Excessive impurities on surfaces or in electrolytes would affect the tip and lead to experimental artifacts, which make it difficult to process and analyze the STM images. The well-defined surfaces, such as single-crystal metals and freshly cleaved two-dimensional materials, are widely used as the substrates in the ECSTM experiments. The substrates should have a wide potential window to avoid influencing the samples at the experimental potentials. By deposition, self-assembly and surface reaction, various samples with different structures and properties can be constructed on substrate surfaces. It is worth mentioning that the ECSTM is a microscopic characterization technique, so the samples with uniform and well-defined structures are more favored for the ECSTM investigation.
The tips commonly used in the ECSTM experiments are prepared from W or Pt/Ir alloy wires by electrochemical etching or mechanical cutting. The mechanical cutting is suitable for tips with lower hardness, such as the Pt/Ir alloy wires, and the electrochemical etching is applicable to tips of various materials. The alkaline solutions are often used as the etchants in electrochemical etching, and the concentrations and applied potentials can be adjusted for different materials. It is worth mentioning that the faradaic currents and charging/discharging currents of the EDL on tips in the ECSTM experiments could disturb the desired tunneling currents for imaging. Therefore, nail polish or Apiezon wax could be used for coating the tip with an insulating layer to reduce the area exposed to electrolytes, thus reducing the current disturbances. Ideally, only a few atoms at the end of the tip are exposed, and the disturbances can be minimized.
5.2
Surface adsorption
The adsorption is one of the critical initial steps of the interfacial electrochemical processes, which plays a key role in corrosion, catalysis and electroplating fields [492-495]. The species in electrolytes adsorb on the electrode surface by various interactions, and the adsorption strength, configuration and kinetic have an important influence on the sequential reactions. The ECSTM can be used to study the surface (interface) adsorption in electrolytes at the atomic and molecular scale, and help reveal the correlations between the adsorption behaviors and the interfacial processes. For instance, Agnoli et al. [496] investigated the adsorption of hydrogen atoms on the Pt(111) surface in hydrogen evolution reaction (HER). The monolayer graphene was prepared on Pt(111) to construct the model system. The hydrogen adsorption/desorption peaks (hydrogen underpotential deposition, HUPD) can be observed in the cyclic voltammetry (CV) curve, and ECSTM experiments were performed around the HUPD potential to observe the hydrogen adsorption/desorption processes. As shown in Fig. 49a and b, when the sample potential was 0 mV (vs. reversible hydrogen electrode, RHE), hydrogen could adsorb on Pt(111) under the graphene monolayer and the (1 × 1) unit cell can be observed in the STM image. When the sample potential was set as 500 mV (vs. RHE), the hydrogen desorbed from the surface and the (3 × 3) pattern corresponds to the graphene monolayer appeared. The STM images of the graphene/Pt(111) and graphene/H/Pt(111) were simulated by density functional theory (DFT) calculations (Figs. 49c and d), which are consistent with the experimental results. The authors believed that the adsorption of hydrogen on Pt(111) is beneficial for improving the catalytic activity.
Figure 49
Figure 49.
(a, b) STM images of the graphene/Pt(111) surface at different potentials in 0.1 mol/L HClO4. (c, d) Constant-height simulated STM images and corresponding models at 78 meV and 2 Å for (c) graphene/Pt(111) and (d) graphene/H (1 monolayer)/Pt(111), where graphene is depicted by black sticks, hydrogen and platinum by blue and gray spheres, respectively. STM images of the self-assembled CoPc monolayer on the Au(111) substrate in the CO2 environment in the 0.1 mol/L NaClO4 electrolyte (e) and 0.05 mol/L Mg(ClO4)2 electrolyte (f). STM images of a CoTPP adlayer on Au(111) in 0.1 mol/L HClO4 (g) and 0.1 mol/L KOH (h). Reproduced with permission [496,499,500]. Copyright 2019, 2022, American Chemical Society; Copyright 2021, Springer Nature.
The adsorption of molecules and ions on the metal surface can also be investigated by ECSTM. Baricuatro et al. [497] observed the adsorption of CO molecules on Cu(100) surface in alkaline solutions. The CO adsorption was found to be a potential-dependent process which took place only at potentials lower than −0.80 V (vs standard hydrogen electrode, SHE). After adsorption, the surface structure was determined as Cu(100)-c(2 × 2)-CO, or Cu(100)-()R45°-CO, and the coverage of CO was 0.5. The authors attributed the stoichiometry of Cu2CO coordination structure to the fully-filled 3d orbital of Cu. Maurice et al. [498] performed in-situ ECSTM to investigate the adsorption of the hydroxyl group on Cu(111) in 0.1 mol/L NaOH. It can be observed that the Cu(111) surface became rough and Cu islands were formed in the adsorption stage. The hydroxyl adsorption was considered as the initial stage of Cu oxidation, which occurred preferentially at the step edges of the surface. Further in-situ experiments indicated that the desorption process could take place reversibly before the formation of Cu2O. Combined with the charge transfer measurements and STM data, the hydroxyl was determined to be the dominant adsorbed species rather than O2 molecule.
For revealing the species adsorption behavior on functional molecular systems, the ECSTM experiments were performed to investigate the self-assembled monolayer constructed on the substrate surface. For instance, Wan et al. [500] investigated the adsorption of CO2 on cobalt phthalocyanine (CoPc) in the CO2 reduction reaction. The well-defined self-assembled monolayer of CoPc was constructed on the Au(111) surface. In the CO2 environment, two adsorbed species with different contrasts can be observed in the STM image (Fig. 49e), and the proportion of the high contrast species decreased significantly as the environment changed to argon. Combining with the theoretical simulations, the high contrast species was attributed to the CoPc-CO2 complex, which was formed by the adsorption of CO2 on Co-N4 site in the initial stage of the reaction. In addition, when the Mg2+ ions were added to the electrolytes, the adsorbed species with a higher contrast than the CoPc-CO2 complex were observed, which was due to the further adsorption of the Mg2+ on CO2 (Fig. 49f). The formation of the CoPc-CO2-Mg2+ complex may have an influence of the catalytic mechanism and activity of the reaction. The adsorption of ions on functional molecules can also be observed by the ECSTM. As shown in Fig. 49g [499], when the meso-tetraphenylporphyrin cobalt (CoTPP) was imaged in the acidic electrolyte, each molecule appeared as a round spot; while in the alkaline environment (Fig. 49h), the adsorbed species exhibited a 2-fold symmetry and consisted of two bright spots with a low contrast in the middle, which was attributed to the adsorption of OH− on CoTPP. Combined with the electrochemical measurements, it was found that the formation of CoTPP-OH− species correlated with the catalytic activity of CoTPP towards the oxygen evolution reaction (OER).
5.3
Surface diffusion
The surface diffusion is one of the important processes in many surface reactions, which is highly related to the substrate-adsorbate interactions and the adsorbate-adsorbate interactions. Different from the UHV condition, the diffusion at the electrode/electrolyte interface can be influenced by the electrochemical environment. For revealing the process, in-situ characterization techniques with high temporal resolution should be used. Magnussen et al. developed the high-speed STM (video-STM) which can be operated in solutions to investigate the diffusion process at the solid/liquid interface [501]. The temporal resolution of the video-STM can achieve 0.1 ms and has been widely used in investigating the diffusion of various surface species. For instance, the diffusion behavior of the adsorbed sulfide (Sad) on Cu(100) in 0.01 mol/L HCl solution was revealed by the in-situ video-STM [502]. The stable c(2 × 2) lattice of the Sad can be observed in the STM image. On this basis, the hopping of the isolated Sad adsorbates between the c(2 × 2) lattice sites was illustrated in the video sequence, and the hopping rates can be calculated. By analyzing the results, a potential-dependent behavior of the diffusion barrier of Sad on Cu(100) was determined, which may be due to the variation in the Sad dipole moment during the hopping process. In addition, the surface dynamics of the Sad on Cu(100) and Ag(100) in the presence of the coadsorbates were investigated [503]. The chloride and bromide-covered Cu(100) and Ag(100) surfaces with the c(2 × 2) structures were constructed. It was found that the hopping rate of Sad decreased with increasing potential on the Cl-covered Cu(100) surface, Cl-covered Ag(100) surface, and Br-covered Ag(100) surface, while it increased on the Br-covered Cu(100) surface. The difference in the potential dependence behavior was due to the different diffusion mechanisms of Sad on surfaces. The exchange diffusion was adopted for the Sad on the Br-covered Cu(100) surface, and on the other surface, the rotation diffusion dominates. The results indicate that the coadsorbates have an important influence on the diffusion mechanism of the surface species. Magnussen et al. [504] investigated the structure and dynamic of the Cl adlayers on Au(100) by video-STM. It was found that the adsorption behavior of Cl on Au(100) was close to that on Ag(100) and Cu(100), and the adsorbed monolayer exhibited a c(2 × 2) surface structure. The hopping motion of Cl atoms at domain boundaries can be observed, and the surface mobility on the Au substrate was much lower than that on the other substrates. A higher defect density of the adlayer on Au(100) was considered to be one of the reasons for the difference. The difference in the ionicity of the M-Cl bond, which is caused by the different surface charges on different metals, can also make a difference in the surface mobility.
The ionic liquids are widely used as the electrolytes for batteries, and the dynamic behaviors of the ionic liquids on electrode surfaces can also be investigated by video-STM. For instance, the 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl) imide ([BMP][TFSA]) was widely used as one of the air and water stable ionic liquids [505]. It was found that with the decreasing potential, the surface structure of the [BMP]+ adlayer on Au(111) changed from disordered adsorption at −1.0 V (vs. the Pt quasi-reference electrode), to () at −1.4 V (vs. the Pt quasi-reference electrode) and finally to ( × 2) at −1.6 V (vs. the Pt quasi-reference electrode). At −1.4 V (vs. the Pt quasi-reference electrode), rapid dynamic fluctuations could be observed for [BMP]+, while at −1.6 V (vs. the Pt quasi-reference electrode), the surface mobility was decreased and no similar fluctuations could be observed. The improved charge screening effect at a negative potential, the polar-nonpolar separation of [BMP]+ and the counter-ions near the electrode surface are considered as the possible reasons for the changes.
The surface dynamic of the adsorbed CO molecules on Pt(111) was also investigated by video-STM [506]. The ordered (2 × 2)-CO structure was observed in the STM image. As the potential became more positive in the pre-oxidation stage of CO, the local mobility of CO increased simultaneously. Combined with the DFT calculations, a small number of point defects with high mobility on Pt(111) play a key role in this dynamic effect.
5.4
Surface reconstruction
The surface reconstruction, which can be induced by the potential, adsorption and stress, can be observed in a variety of interfacial electrochemical processes. The reconstruction of materials in electrolytes is different from that in UHV. For instance, the well-defined (7 × 7) structure of Si(111) was revealed by the UHV-STM after annealing treatment [507], while in 0.05 mol/L H2SO4, only the (1 × 1) structure can be formed [508]. Therefore, it is important to understand the surface reconstruction behavior in the electrochemical environment, and the ECSTM has been widely used in investigating the reconstruction of electrode surfaces. For instance, Soriaga et al. [509] investigated the reconstruction of the polycrystalline Cu electrode at −0.9 V (vs. SHE) in 0.1 mol/L KOH. Cu is one of the widely used electrocatalysts for the CO2 reduction reaction, and the surface structure has a decisive influence on the electrocatalytic activity and product selectivity. The STM results indicate that the polycrystalline Cu transformed to Cu(111) in 30 min, then to Cu(100) after another 30 min. According to the electrochemical measurements, the dominant product of polycrystalline Cu and Cu(100) is ethylene; while for Cu(111), methane is more favored. The structure transformation revealed by ECSTM provides direct evidence for understanding the selectivity changes in the reaction. Magnussen et al. [510] performed in-situ ECSTM to investigate the surface reconstruction of Cu(100) electrodes in CO2-saturated 0.1 mol/L KHCO3. As shown in Figs. 50a-d, when the potential decreased to −0.22 V (vs. RHE), the nanoclusters appeared in the STM image. The cluster density increased as the potential further decreased to −0.27 V (vs. RHE). When the potential was changed back to −0.12 V (vs. RHE), the clusters on Cu(100) disappeared, which indicates that the reconstruction process is reversible at different potentials. The crystal truncation rod (CTR) analysis in the in-situ surface X-ray diffraction (SXRD) characterizations further evidenced the surface roughening in the reconstruction process (Fig. 50e). By analyzing the full sequence of CTR data, the surface density of lattice defects and the vertical expansion of the topmost atomic Cu layer were obtained (Fig. 50f), and on this basis, the structure model of the reconstructed surface was proposed (Fig. 50g). In addition, the X-ray intensities at selected reciprocal space positions were plotted against the potential (Fig. 50h), and it was found that the surface reconstruction took place rather quickly. The authors proposed that the reconstruction of Cu(100) is a CO-promoted process, which occurred at the pre-reaction stage. To modulate the reconstruction process, Lingenfelder et al. [511] constructed a monolayer graphene-covered Cu electrode (g-Cu), and found that the g-Cu underwent more moderate structure changes compared to the pristine Cu, which makes the g-Cu an ideal model system for investigating the reaction dynamics and mechanisms. Meanwhile, the graphene layer remained stable in the reaction, which is beneficial for improving the stability of Cu catalysts in the CO2 reduction reaction.
Figure 50
Figure 50.
(a-d) In-situ STM images the Cu(100) electrode in CO2-saturated 0.1 mol/L KHCO3 at different potentials. Gray arrows indicate scan direction; regions of constant potential are separated by dashed lines. (e) In-situ SXRD measurements of the CTRs (11l) and (20l) at 0, −0.3 and −0.7 V (vs. RHE) and the corresponding best fits (solid colored lines). (f) Coverage of surface defects and vertical expansion of the d12 spacing between the top two Cu layers as a function of the applied potential, obtained from the CTR fits. (g) The proposed Cu(100) surface model used for the CTR fitting. (h) Potential-dependent scattered intensity at the reciprocal space positions during CV measurement (shown as black line) at a scan rate of 5 mV/s. (i-l) In-situ STM images of the Pt(111) electrode after different numbers of ORCs: (i) initial; (j) 8; (k) 31; (l) 170. Scan size is 230 nm × 230 nm. (m) The surface width (red) and the hydrogen desorption charge (blue) plotted against the cycle numbers. Both sets of data are plotted logarithmically. The two black dotted lines indicate the two stages as discussed. Reproduced with permission [510,512]. Copyright 2018, Springer Nature; Copyright 2023, CC-BY 4.0.
The surface reconstruction of other materials can also be investigated by the ECSTM. For instance, Rost et al. [512] performed in-situ ECSTM to reveal the roughening process of the Pt(111) electrode in the oxidation–reduction cycles (ORCs). As shown in Figs. 50i-l, with the increasing number of the ORCs, the formation of Pt nanoislands can be observed in the STM images. Combined with the analysis of the cross-section profiles, the entire roughening process can be divided into the ''nucleation and early growth'' stage and the ''late growth'' stage. The surface width of the nanoislands and the hydrogen desorption (Hdes) charge measured by CV were correlated with the number of ORCs (Fig. 50m). It was found that the Hdes charge was mainly contributed by the step sites generated in the ''late growth'' stage. Watanabe et al. [513] investigated the structural evolution of the Pt(111) step in the CO oxidation reaction. The disordered Pt(111) steps became straight during potential cycles, and the electrocatalytic activity decreased. The low activity of the straight Pt(111) steps was attributed to the formation of the tight, protective CO adlayer. Brummel et al. [514] constructed the cobalt oxide nanoislands on Au(111) and studied the surface structural transformation by the ECSTM. The experimental results indicate that the bilayer cobalt oxide nanoislands became mobile at 0.5 V (vs. RHE) and dissolved at lower potentials in the cathodic scan, which is different from the stable double-bilayer nanoislands. In the anodic scan, both the bilayer and double-bilayer nanoislands retained their morphology up to 1.5 V (vs. RHE), which provides microscopic evidence for the good electrochemical stability of cobalt oxides in the OER.
5.5
Electrocatalytic reactions
An in-depth understanding of the electrocatalytic mechanisms is of great significance for the rational design and synthesis of high-performance electrocatalysts [515-519]. Due to the high spatial resolution and in-situ characterization, the ECSTM has been widely used in investigating the electrocatalytic processes at the microscopic scale, which provides solid foundations for revealing the catalytic mechanisms of the reactions and establishing the structure-activity relationship of the catalysts [485,487,520].
Understanding the spatial distribution of the active sites on catalyst surface is important for constructing highly active materials. Based on the in-situ ECSTM technique, Bandarenka et al. [521] developed the current noise analysis for identifying the catalytic activity of different sites on the electrode surface. As shown in Fig. 51a, when the electrocatalytic reaction is triggered, the current noise between the tip and the highly reactive region should be higher than the non-reactive region. Therefore, by analyzing the level of the current noise in STM images, the reactivity of different surface sites can be revealed. For instance, when the HER was triggered on Pt(111), the current noises at the step sites were much higher than that on the terrace (Fig. 51b), which indicates the higher catalytic activity of the steps. The current noise analysis technique has been proven to be effective for a variety of model catalysts such as metal oxide, alloy and carbon-based materials [522-524], which provides experimental references for the optimization of the catalysts.
Figure 51
Figure 51.
(a) Schematic illustration of the current noise analysis technique. (b) STM line scans over a Pt(111) surface in 0.1 mol/L HClO4 when HER is on (red) and off (blue). (c) Time dependence of the surface coverage of CoPc-O2 and CoOEP-O2 in potential step experiments. (d-h) In-situ STM images of the CoOEP monolayer in O2-saturated 0.1 mol/L HClO4 in potential step experiments. Reproduced with permission [521,530]. Copyright 2017, Springer Nature; Copyright 2023, American Chemical Society.
Metal catalyst is one of the practical catalytic materials with good activity and high stability. Single-crystal electrodes are extensively constructed and investigated as the model systems for metal catalysts to reveal the catalytic mechanism. For instance, Kunze-Liebhäuser et al. [525] performed in-situ ECSTM to investigate the catalytic process of the Cu(111) electrode in the CO oxidation reaction. The electrochemical measurements indicate that the Cu(111) exhibited good activity towards the CO electrooxidation in the alkaline electrolyte. The self-activation process of the Cu catalyst in the reaction was revealed by the ECSTM experiments, which involves the ejection of Cu atoms by OH onto the surface and the stabilization of the undercoordinated Cu adatoms and adclusters by CO binding. The authors proposed that the formation of the high-energy undercoordinated Cu structures on Cu(111) surfaces is important for the catalytic activity. The catalytic process of the Cu(111)-catalyzed HER was also investigated by the in-situ ECSTM [526]. The formation of the Cu adislands on electrode surface in the reaction was observed upon the deposition of Ni(OH)2, which could generate a highly disordered water adlayer and promote the electron transfer in the reaction.
Single-atom catalysts (SACs) are widely considered as one of the emerging catalytic materials with enormous application potentials. Metal porphyrin and metal phthalocyanine, which possess the well-defined M-N4 coordination structure and good catalytic performance, have been widely investigated as the molecular model catalysts for SACs by the ECSTM [527,528]. For instance, Wang et al. [529] studied the formation and conversion processes of the OOH−-adsorbed cobalt porphyrin (CoTMPP) in the oxygen reduction reaction (ORR) in 0.1 mol/L NaClO4 by in-situ ECSTM. The CoTMPP-OOH− species with a higher contrast can be observed in the STM image. The two stages of the CoTMPP-catalyzed ORR, including the reduction of O2 to H2O2 and the further reduction of H2O2 to H2O, were revealed by the rotating disk electrode experiments. In the in-situ experiments, it was found that the CoTMPP-OOH− species formed in the first stage of the reaction, and then transformed to pristine CoTMPP in the following stage. The similar processes could not be observed in the acidic electrolyte, which indicates that the reaction pathway of the ORR may vary in electrolytes with different pH values. Wan et al. [530] investigated the reaction processes of the CoPc and cobalt octaethylporphyrin (CoOEP)-catalyzed ORR. The experimental results indicate that the electrocatalytic activity of CoOEP is better than CoPc. In the reaction, the valence states of Co sites were preferentially reduced from +3 to +2 to bind with O2 before the catalytic conversion. The ECSTM potential step experiments were performed to investigate the dynamics of the O2 adsorption and desorption processes in the reaction. As shown in Figs. 51c-h, the coverage of the high contrast O2-adsorbed catalysts increased after the potential step. By fitting the curve, the rate constants of O2 adsorption (kad) and dissociation (kdi) can be determined. It was found that the kad and kdi of CoOEP are higher than that of CoPc, which may be one of the reasons for its better activity.
The organic electrocatalytic conversion can also be revealed by the ECSTM. For instance, the dynamic processes of the adsorption and electrooxidation of methanol on rhodium porphyrin (RhOEP) were demonstrated by ECSTM characterizations [531]. It was found that the methanol adsorption process was correlated with the oxidation of Rh sites in the reaction. In addition, the rate constants of different stages of the reaction were calculated by analyzing the coverage of the high contrast RhOEP-CH3OH complex in the sequential in-situ ECSTM images, and the desorption/conversion of CO was determined as the rate-limiting step in the reaction.
6.
Summary
Surface chemistry is an interdisciplinary approach and has significantly blurred the traditional borders between chemistry, physics and materials science. Despite a tremendous progress has been made in surface chemistry, several exciting directions are intriguing to be explored. For example, surface chemistry, bearing the ability to the fabrication and characterization of atomically precise nanostructures, may be used to construct surface structures with specifically designed properties and functionalities. Owing to weak spin orbit coupling, magnetic graphene nanostructures provide a highly engineerable platform to study quantum magnetism. It would be interesting to synthesize extended two-dimensional covalent structures with periodical spin lattice. Such 2D quantum spin lattice would host intriguing low-lying spin excitations, allowing real-space study of spinons and quantum spin liquids. Considering the synthesis of well-defined 2D covalent structures remains challenge, it would be of great importance to explore quantum magnetism in metal-coordinated and hydrogen-bonded magnetic nanostructures, which can be effectively fabricated on different surfaces.
Another challenge is the synthesis of molecular nanostructures directly on insulating substrates for exploring their intrinsic properties as well as for device applications. Albeit intensive theoretical studies, topological flat bands and strong electron correlation effects have been seldom realized in organic materials. When it comes to quantum applications, the direct measurements of quantum coherence and ultra-fast dynamics of different organic materials remain to be explored, which should provide a highly tunable platform for understanding quantum behaviors and shed light on quantum applications, including quantum sensors, spin qubits, single-photo emitter, to name a few. For technological applications, the ability to synthesize mesoscale materials and fabricate actual devices is greatly demanded. With increasing number of scientists working in surface chemistry, we are convinced more and more important and original advances will come up in near future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Xin Li: Writing – original draft. Zhen Xu: Writing – original draft. Donglei Bu: Writing – original draft. Jinming Cai: Writing – original draft. Huamei Chen: Writing – original draft. Qi Chen: Writing – original draft. Ting Chen: Writing – original draft. Fang Cheng: Writing – original draft. Lifeng Chi: Writing – original draft. Wenjie Dong: Writing – original draft. Zhenchao Dong: Writing – original draft. Shixuan Du: Writing – original draft. Qitang Fan: Writing – original draft. Xing Fan: Writing – original draft. Qiang Fu: Writing – original draft. Song Gao: Writing – original draft. Jing Guo: Writing – original draft. Weijun Guo: Writing – original draft. Yang He: Writing – original draft. Shimin Hou: Writing – original draft. Ying Jiang: Writing – original draft. Huihui Kong: Writing – original draft. Baojun Li: Writing – original draft. Dengyuan Li: Writing – original draft. Jie Li: Writing – original draft. Qing Li: Writing – original draft. Ruoning Li: Writing – original draft. Shuying Li: Writing – original draft. Yuxuan Lin: Writing – original draft. Mengxi Liu: Writing – original draft. Peinian Liu: Writing – original draft. Yanyan Liu: Writing – original draft. Jingtao Lü: Writing – original draft. Chuanxu Ma: Writing – original draft. Haoyang Pan: Writing – original draft. JinLiang Pan: Writing – original draft. Minghu Pan: Writing – original draft. Xiaohui Qiu: Writing – original draft. Ziyong Shen: Writing – original draft. Shijing Tan: Writing – original draft. Bing Wang: Writing – original draft. Dong Wang: Writing – original draft. Li Wang: Writing – original draft. Lili Wang: Writing – original draft. Tao Wang: Writing – original draft. Xiang Wang: Writing – original draft. Xingyue Wang: Writing – original draft. Xueyan Wang: Writing – original draft. Yansong Wang: Writing – original draft. Yu Wang: Writing – original draft. Kai Wu: Writing – original draft. Wei Xu: Writing – original draft. Na Xue: Writing – original draft. Linghao Yan: Writing – original draft. Fan Yang: Writing – original draft. Zhiyong Yang: Writing – original draft. Chi Zhang: Writing – original draft. Xue Zhang: Writing – original draft. Yang Zhang: Writing – original draft. Yao Zhang: Writing – original draft. Xiong Zhou: Writing – original draft. Junfa Zhu: Writing – original draft. Yajie Zhang: Writing – review & editing, Writing – original draft, Supervision, Project administration, Funding acquisition. Feixue Gao: Writing – review & editing, Writing – original draft, Supervision, Project administration. Yongfeng Wang: Writing – review & editing, Writing – original draft, Supervision, Project administration, Funding acquisition.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (Nos. 22225202, 92356309, 22132007, 21991132, 22172002). Experiments are supported by Peking Nanofab.
Z.F. Wang, Z. Liu, F. Liu, Nat. Commun. 4 (2013) 1471. doi: 10.1002/sec.415
[248]
W. Jiang, F. Liu, Organic Topological Insulators, in: C. Boehme (Ed.) World Scientific Reference On Spin in Organics, Word Scientific 2018, pp. 201-224.
[249]
W. Huang, Y. Zhang, M. Song, et al., Chin. Chem. Lett. 33 (2022) 2281–2290. doi: 10.1016/j.cclet.2021.08.086
Y.C. Feng, X. Wang, Z.Y. Yi, et al., Sci. China Chem. 66 (2023) 273–278. doi: 10.1007/s11426-022-1465-8
[530]
Y.Q. Wang, X.H. Dan, Z.Y. Yi, et al., J. Phys. Chem. C 127 (2023) 2929–2935. doi: 10.1021/acs.jpcc.2c07891
[531]
X. Wang, Z.Y. Yi, Y.Q. Wang, D. Wang, L.J. Wan, J. Phys. Chem. Lett. 14 (2023) 9448–9455. doi: 10.1021/acs.jpclett.3c02514
Figure 1
(a) Schematic of the qPlus-AFM setup. The inset shows the calculated charge distribution of the CO at the tip apex. (b) High-resolution AFM images and the atomic models of water dimer (left panel), tetramer (middle panel) and sodium ion hydrates (right panel) on NaCl(001)/Au(111) surface. (c) Left panel: Force curves of individual hydroxyls on the In2O3(111) surface with OH-terminated tip. Right panel: Schematic of the experiment setup. (d) The correlation between H bond length and the force minima. (e) Schematic of the experimental setup and d2I/dV2 spectra of a water monomer on NaCl(001)/Au(111) surface with a Cl-tip (R for rotational, B for bending, and S for stretching). Reproduced with permission [58-62]. Copyright 2018, 2021, Springer Nature; Copyright 2016, American Chemical Society; Copyright 2016, The American Association for the Advancement of Science; Copyright 2018, CC-BY 4.0.
Figure 2
(a) 3D STM image of water hexamer on Au(111) surface. (b) High-resolution STM image and corresponding atomic model of water hexamer on Cu(111) surface. (c) STM, height-dependent AFM, and proposed atomic model of water clusters on Pt(111) and Cu(111) surface. (d) Height-dependent AFM, and proposed atomic model of () R19° monolayer ice on Ni(111) surface. (e) Height-dependent AFM and atomic model of buckled BHI on corrugated Au(110)-1 × 2 surface. (f) Constant-height AFM images and corresponding atomic model of bilayer hexagonal ice on Au(111) surface. (g) Growing process for zigzag (left) and armchair (right) edges. Reproduced with permission [40,56,70,71,73,89]. Copyright 2018, American Chemical Society; Copyright 2007, 2020, Springer Nature; Copyright 2019, 2022, American Physical Society; Copyright 2023, CC-BY 4.0.
Figure 3
(a) Top: STM image of the water-adsorbed anatase-TiO2(001)-(1 × 4) surface (setpoint: 1.2 V, 10 pA). Bottom: The UPS spectra obtained after the water adsorption. (b) Top: Simulated water splitting pathways, considering 1H2O, 2H2O, and 3H2O configurations. Bottom: Calculated potential energy surface along the water splitting pathways. (c) Dissociated water forming a "dynamic ion pair" on the SrO-terminated surface of Sr3Ru2O7. Top: Adsorption geometry of the lowest-energy configuration (left) and consecutive STM images of water monomer motion at 78 K (right). Bottom: The hopping model with DFT, showing the initial, transition, and the final state. (d) Arrhenius plot for the "dynamic ion pair" motion. (e) Top: Height-dependent AFM images of different water coverage (left: 2.5 H2O/unit cell, right: 8 H2O/unit cell). Bottom: O 1s XPS data show that water molecules are partially dissociated. (f) Top view of the minimum-energy structures calculated by DFT. Reproduced with permission [47,49,101]. Copyright 2022, American Chemical Society; Copyright 2015, Springer Nature; Copyright 2018, National Academy of Sciences.
Figure 4
(a) Left panel: Symmetric double-well potential. Middle panel: Schematic of the harmonic (red) and an-harmonic (blue) zero-point motion of the proton. Right: Model of symmetric H bond. (b) Tunneling current on the chirality interconversion of tetramer on NaCl(001) surface. (c) NQEs on the H-bonding strength. (d) STM image and atomic model of water-hydroxyl cluster on Cu(110) surface with a symmetric H-bond. (e) AFM image (left panel) of Zundel cation and the snapshot (middle panel) from PIMD calculation. Right panel: The probability distribution of proton from PIMD and MD simulations of Zundel-water structures. Reproduced with permission [62,102,104,107,108]. Copyright 2010, American Physical Society; Copyright 2015, Springer Nature; Copyright 2016, 2022, The American Association for the Advancement of Science; Copyright 2017, Elsevier.
Figure 5
High-resolution AFM images and the schematic models of (a) Eigen-water and (b) Zundel-water overlayers on Au(111) surface. (c) High-resolution AFM images and the schematic models of the Na+·nD2O, n = 2,3,5 cluster on NaCl(001)/Au(111) surface. (d) Tip-induced diffusion dynamics of Na+ hydrates. Top: Schematic diagram of the Au-mediated inelastic electron excitation of the Na+ hydrates with the STM tip on NaCl(001). Inset of Top: Current-bias relationship of Na+·3D2O with a Cl− tip. Bottom: Comparison of Veff for Na+·nD2O (n = 1–5) at d × the lattice constant. Reproduced with permission [59,107]. Copyright 2022, The American Association for the Advancement of Science; Copyright 2018, Springer Nature.
Figure 6
STM images showing the structural transformation of cytosines from (a, b) disordered networks with T-junctions to (c, d) one-dimensional cytosine chains on Au(111) after water exposure. Nc-AFM images of the typical (e), T-junctions (f) and one-dimensional chains superimposed with the corresponding structural models (g, h). (i-k) Representative zoomed-in images of molecular dynamics simulations of the transformation of cytosine chains, where red, blue, and green dots represent weak, water-involved, and strong hydrogen bonds, respectively. Reproduced with permission [109]. Copyright 2020, American Chemical Society.
Figure 7
(a) Schematic illustration showing the local dehydration of water-involved T structure by removing the target water molecules via in-situ STM manipulation, leading to the local chiral inversion of certain T dimers. (b-e, i, j) Sequential STM manipulations on the water molecules (highlighted in blue solid circles) recorded in the same regions. (f) STM images showing the naturally existing water vacancies (depicted by white dotted circles) in the water-involved T structure. (g, h) Magnified STM and nc-AFM images confirming the formation of a blue T dimer in the absence of water molecules on both sides (white dotted circles). Reproduced with permission [135]. Copyright 2022, American Chemical Society.
Figure 8
(a) Model depicting a coordination node formed via coordination interaction between three BPyB and one Fe atom. (b) STM image of a fifth-order STs on Au(111). (c) A series of BPyB-Fe-ST-n structures, ranging from 0 to 5, along with their respective models. (d-g) STM images and accompanying models of 2D crystals composed of ST-(n + n). Reproduced with permission [167]. Copyright 2020, American Chemical Society.
Figure 9
(a) Approach towards creating biomolecular STs with global organizational chirality. STs formation is possible when 120° V-shaped organic molecules interact with metal atoms on surfaces. If chiral biomolecules imitate metal atoms in forming nodes with these V-shaped ligands, biomolecular ST formation would occur. It is expected that all resulting STs would have consistent handedness. (b) L-Trp-BPyB-ST-n prepared via a co-assembly method with n ranging from 0 to 3. Reproduced with permission [120]. Copyright 2021, American Chemical Society.
Figure 10
(a) C3PC molecules and Fe atoms formed STs on Au(111) through three-fold coordination interactions. (b) Upon dosing CO molecule on the STs/Au(111) with the pressure of 5.0 × 10−6 torr for 120 s at room temperature, stable coordination motifs changed from Fe(C3PC)3 to Fe(C3PC)4 with an axially bonded CO molecule, transforming STs into one-dimensional chains where CO coassembled with Fe(C3PC)3 units. (c) Subsequently, dosing CO2 of 650 L on the sample induced structural transformation from STs to chains via a molecular catassembly process. Reproduced with permission [124]. Copyright 2023, CC-BY 4.0.
Figure 11
(a) Illustration for the heterochiral and homochiral recognition. (b) 2D crystallization of racemic mixtures on surface. (c) Br···Br contacts mediated chiral recognition in the self-assembled structure of achiral molecules [191]. (d) Water-induced chiral separation via hydrogen bonding interactions on an Au(111) Surface [110]. Reproduced with permission [110,191]. Copyright 2021, American Chemical Society; Copyright 2022, CC-BY 4.0.
Figure 12
(a) Schematic diagram of chiral control on 2D surface. (b-d) Co-adsorber induced homochirality at the liquid/solid interface [192]. (e-g) Biasing enantiomorph formation through geometric confinement [193]. Reproduced with permission [192,193]. Copyright 2013, CC-BY-NC—ND; Copyright 2018, American Chemical Society.
Figure 13
(a) Schematic diagram of the sergeants and soldiers principle. (b) Manifesting the chiral amplification process controlled by sergeants-and-soldiers principle in the co-adsorber induced homochiral assemblies at the liquid/solid interface [200]. (c) Schematic diagram of the majority rules. (d) Concentration–modulated supramolecular chirality following the majority rules [212]. Reproduced with permission [200,212]. Copyright 2016, Royal Society of Chemistry. Copyright 2023, CC-BY-NC 4.0.
Figure 14
(a) The transmission of chirality from the self-assembly of achiral TiOPc to the TiOPc molecule situated on the top of chiral void [214]. (b) STM image and the suggested model for the multilayer growth, with the first and second layers exhibiting opposite interlocking patterns [215]. (c) Characterization of the structures, computational models, and proposed reaction mechanism for the transformation products P1, P2, and P3 resulting from the annealing of (P)-DBH on Ag(111) in the temperature range of 520–670 K [217]. (d) Transferring two-dimensional chirality from self-assembled structures to covalently bonded products [220]. Reproduced with permission [214,215,217,220]. Copyright 2016, 2019, 2020, American Chemical Society; Copyright 2016, Springer Nature.
Figure 15
(a-d) Transferring of the chiral information from the tetramer intermediate states to the dimer reaction products on Ag(100) [219]. Reproduced with permission [219]. Copyright 2019, American Chemical Society.
Figure 16
(a) Schematic illustration showing the orientation selectivity of a Na-TPyP molecule in a "Klotski Puzzle", which was realized by STM manipulations. (b) Two possible pathways for the orientation change of a Na-TPyP molecule, i.e., molecular rotation and conformational change. (c-n) Sequential STM images showing the orientation selectivity of the target sliding-block molecules after STM manipulations (as indicated by red arrows) in "Klotski Puzzles" containing (c-f) a single-cavity, (g-j) a 2 × 2 cavity, and (k-n) a 3 × 3 cavity, respectively. The apparent long axes of the molecules are indicated by blue dashed lines. Reproduced with permission [223]. Copyright 2023, American Chemical Society.
Figure 17
(a) Schematic illustration of the effects of anchoring groups, spacer groups, and terminal groups on the properties of self-assembled molecule monolayer (SAM) [226]. (b) The optimization of interfacial band bending of perovskite solar cells with a fullerene derivative SAM. [232]. Reproduced with permission [226,232]. Copyright 2019, CC-BY 4.0; Copyright 2020, John Wiley and sons.
Figure 18
(a) Dehydrogenative polymerization of linear alkane on Au(110) [260]. (b) Direct transformation of n-alkane into all-trans conjugated polyene via cascade dehydrogenation on Cu(110) [263]. (c) Transformation of poly(o-naphthylene vinylidene) into its conjugated derivative poly(o-naphthylene vinylene) via dehydrogenation reaction [266]. Reproduced with permission [260,263,266]. Copyright 2011, American Association for the Advancement of Science; Copyright 2021, 2022, CC-BY 4.0.
Figure 19
The selectivity of reaction pathways. (a) Mono- and di-selective ortho C—H bond activation of phenol derivatives on Au(111) and Ag(111), respectively [268]. (b) Homocoupling reaction of polarized terminal alkynes (TAs) on Au(111) [270]. Reproduced with permission [268,270]. Copyright 2016, 2023, American Chemical Society.
Figure 20
(a) Schematic illustration showing the selection between C–C coupling reaction and C–H activation of terminal alkynes on Ag(111) under different reaction conditions. (b) STM-TERS measurement on the two newly generated sp-sp2-carbon skeletons (i.e., enyne and cumulene). (c) Charge-density-distribution of enyne and cumulene on Ag(111) (left), and the corresponding CC stretching modes of sp-carbon involved (right). (d) STM-TERS spectra collected at the positions marked in the inset [271]. (e) STM image showing the self-assembled structure of DEBP on Ag(111). (f) Formation of long C–C coupled chains upon annealing at ~370 K. (g–i) Gradual formation of organometallic dimers and chains after O2 dosage. (j) Energy diagram of DFT-calculated reaction pathways of C–H activation on Ag(111) without and with the assistance of oxygen species. (k) Charge-density-difference map of IS2. (l) Structural models involved in the C–H activation pathway in the presence of atomic oxygen [278]. Reproduced with permission [271,278]. Copyright 2021, 2022, American Chemical Society.
Figure 21
(a) Scheme of on-surface synthesis of cis-, trans-polyacetylenes, and organometallic Cu-carbynes on Cu(110) from C2H2. (b, c) Nc-AFM and STM images of cis- and trans-polyacetylenes on Cu(110). (d) STS characterization of an individual trans-polyacetylene on Cu (black), at the Cu/oxide interface (blue), and on the oxide (red), respectively. (e) Nc-AFM and STM images, DFT model, and STM simulation of an individual Cu-carbyne on Cu(110). (f) On-surface synthesis and STM/nc-AFM characterization of close-packed Cu-carbyne ribbons from CBr4via an elimination reaction. (g-i) STM/nc-AFM/STS characterization of diacetylenic Au-carbynes on Au(111) synthesized from C4Br4. (j, k) Characterization of triacetylenic Ag-carbynes on Ag(111) synthesized from C6Br6. Reproduced with permission [280-284]. Copyright 2019, Springer Nature; Copyright 2016, 2020, 2022, 2023, American Chemical Society.
Figure 22
Surface synthesis strategies and chemical structure characterization of C10 and C14. (a) The upper panel shows the reaction scheme for the formation of C10. The lower panels show the AFM image, Laplace-filtered AFM image, and atomic structure overlapped on the image of C10, respectively. (b) The upper panel shows the reaction scheme for the formation of C14. The lower panels show the AFM image, Laplace-filtered AFM image, and atomic structure overlapped on the image of C14, respectively. Reproduced with permission [286]. Copyright 2023, Springer Nature.
Figure 23
Formation and breaking of non-carbon-carbon chemical bonds. (a) Formation of amide bonds between carboxyl and amine species on Au(111) [289]. (b) The formation of phenols through the catalytic dealkylation of ethers on metal surfaces [292]. Reproduced with permission [289,292]. Copyright 2016, 2021, John Wiley and sons.
Figure 24
(a) Illustration of the first example of Glaser coupling on Ag(111) [295]. (b) Illustration of the synthesis of anthracene oligomers linkered by diacetylene moieties by desilylation followed by C—C homocoupling on Cu(111) [298]. (c) Left panel: illustration of the synthesis of graphyne nanowires by sonogashira coupling anthracene oligomers linked by diacetylene moieties by desilylation followed by C—C homocoupling. Right panel: Illustration of the synthesis of bitadiyne sigments by stepwise silylene tethering and elimination of the ME3Si group on Ag(111) [302,303]. (d) Illustration of the synthesis of poly(diynes) by decarboxylative coupling after deprotonation on Ag(111) [299]. Reproduced with permission [295,298,299,302,303]. Copyright 2012, Springer Nature; Copyright 2018, American Chemical Society; Copyright 2021, 2023, John Wiley and sons.
Figure 25
(a) Illustration of competition between dehalogenation coupling and dehydrogenation/dehydration reaction of DN molecules on Au(111) and Ag(111) [321]. (b) illustration of synthesis of carbon nanoribbon consisting of period 4–5–6–8 membered rings on Au(111) formed by dehalogenative homocoupling followed by face-to-face dehydrogenation coupling of DIF molecules [324]. (c) Illustration of stepwise dehalogenative homocoupling reaction of precursor molecules where function group of C(sp3)–Br are activated firstly followed by the cleavage of C(sp2)–Br bond [308]. Reproduced with permission [308,321,324]. Copyright 2017, 2023, American Chemical Society; Copyright 2020, Royal Society of Chemistry.
Figure 27
Formation of benzenoid conjugated rings on surfaces. (a) On-surface hexadehydro-Diels–Alder reaction [332]. (b) On-surface Bergman cyclization [312]. (c) On-surface cyclotrimerization of terminal alkynes [333]. (d) On-surface dehydration cyclotrimerization of acetyls [334]. (e) On-surface formation of pyrazine via dehydration cyclocondensation of o-diamine and o-diketone [335]. (f) On-surface formation of triazine via a special dehydration reaction [336]. Reproduced with permission [312,332-336]. Copyright 2021, CC-BY-NC—ND; Copyright 2013, 2015, 2016 American Chemical Society; Copyright 2014, Royal Society of Chemistry; Copyright 2022, CC-BY 4.0.
Figure 29
On-surface generation of polycyclic aromatic hydrocarbons A1, A2, A3, A4, and A5 via a phenyl group migration reaction. Reproduced with permission [352]. Copyright 2023, CC-BY 4.0.
Figure 30
(a) The reaction pathway of M-DPP (M = 2H, Cu, Fe) monomers on metal surfaces. (b) High regioselectivity of H2-DPP in cyclodehydrogenation reaction. (c) No selectivity of Fe-DPP in cyclodehydrogenation reaction. Reproduced with permission [359]. Copyright 2023, American Chemical Society.
Figure 31
Skeletons of isomers of graphene classified by non-/benzenoid and non-/alternant characters. Apart from the above-mentioned nonbenzenoid graphene isomers with pure nonhexagonal rings, most of the predicted isomers contain a mixture of hexagonal and nonhexagonal rings, e.g., biphenylene sheet with 4,6,8-membered rings and Phagraphene with 5,6,7-membered rings. Regarding the graphene isomer containing the same type of carbocycles, tuning their tiling manners and the ratio of different carbocycles could also generate different types of graphene isomers. For instance, the biphenylene sheet contains alternative six and four-membered rings along the vertical direction [393]. Adding one more six-membered ring between two four-membered ring results in a new isomer of graphene [394] with also 4,6,8-membered ring with different properties.
Figure 32
On-surface synthesis of nonbenzenoid nanocarbons including peri-tetracene and its nonbezenoid isomer with two azulene moieties (a), nanocarbons with 4 to 8-membered rings (b), nanographenes with six azulene units (c) and biphenylene dimmer (d). The corresponding nc-AFM or bond-resolved STM images were displayed at the right sides. Reproduced with permission [398,400-402]. Copyright 2018, 2020–2022, American Chemical Society.
Figure 33
On-surface synthesis of nonbenzenoid alternant 1D structures by 2 + 2 cycloaddition reactions including (a) tetracene polymers periodically bridge with four-membered rings, (b) graphene like nanoribbons periodically embedded with four- and eight-membered rings, and (c) dibenz[a,h]anthracene polymers bridge with biphenylene units. The corresponding nc-AFM and bond-resolved STM images were displayed accordingly. Reproduced with permission [348,405,406]. Copyright 2019, John Wiley and sons; Copyright 2017, CC-BY 4.0; Copyright 2023, Springer Nature.
Figure 34
On-surface synthesis of nonbenzenoid 1D oligomer or polymer chains including (a) polyindenfluorene with alternating 5- and 6-membered rings, (b) anthracene polymers bridged with pentalene units, and (c) short chain consisting of alternatively fused 5, 7-membered rings. The corresponding nc-AFM images were displayed accordingly. Reproduced with permission [407-409]. Copyright 2020, American Chemical Society; Copyright 2020, CC-BY 4.0.
Figure 35
On-surface synthesis of nonbenzenoid 1D nanoribbons including (a) phagraphene and TPH-graphene nanoribbons from Ullmann coupling of 2,6-dibromoazulene and subsequent lateral fusion of polyazulene chains, (b) graphene nanoribbons embedded with periodically arrayed 5–8–5 ring defects and Ag centers. The corresponding nc-AFM images were displayed at the right sides. Reproduced with permission [410,411]. Copyright 2019, 2023, American Chemical Society.
Figure 36
On-surface synthesis of nonbenzenoid graphene isomer, biphenylene network, through the reaction scheme (a) lateral fusion of fluorinated poly(para-phenylene) chains. (b) The corresponding STM and nc-AFM images were displayed underneath the structures in panel (a). (c) Differential conductance spectra of the biphenylene ribbon with different widths. The red frame marked out the metallicity of biphenylene ribbon with 21 carbon atoms wide. Reproduced with permission [340]. Copyright 2021, American Association for the Advancement of Science.
Figure 37
Adsorption configuration and its influence. (a) Abnormal hopping behavior of DATP molecules on Cu(111) induced by the asymmetry adsorption [413]. (b) The influence of different adsorption configurations of precursor molecules on Ag(111) and Au(111) to the reaction barrier [414]. Reproduced with permission [413,414]. Copyright 2022, American Chemical Society; Copyright 2018, CC-BY 4.0.
Figure 38
Formation of multiple metal-organic bonds. (a) The hierarchical dehydrogenation reaction of aniline on the Cu(111) surface [418]. (b) The substrate-modulated synthesis of 1D and 2D metal-organic hybrids with the organic building blocks (perylene) on Au(111) and Ag(111), respectively [420]. Reproduced with permission [418,420]. Copyright 2018, 2022, American Chemical Society.
Figure 39
(a) Illustration on metal-modulated tautomerization. Left panel: Ni atom inhibit the G tautomerization from G/9H to G/7H form on Au(111) by preferential coordination at N7 site. Right panel: Ni atom induced tautomeric dehydrogenation of thymine molecules on Au(111) by the preferential coordination of Ni atoms with nitrogen atoms [126,421]. (b) Illustration on Fe mediated selective synthesis of stereoselective organic ligands and mono-/bi-iron chelated complexes on Ag(111) [422]. Reproduced with permission [126,421,422]. Copyright 2014, 2018, American Chemical Society; Copyright 2021, John Wiley and sons.
Figure 40
(a) Atomically-resolved and (b) simulated STM images of hydride species on ZnO(1010) in 0.4 mbar H2 at 300 K. The corresponding structural model of hydride chains is displayed in (b). Gray, red, and white spheres represent Zn, lattice O, and H atoms, respectively. (c) STM images of hydride species on ZnO(1010) in 2 mbar H2/CO (3:1) mixture gas at 300 K. (d–f) STM images and STM simulation of ZnO(1010) in 1 mbar CO at room temperature. The formation of (2 × 1)-CO structure is calculated by DFT with the optimized structural model and simulated STM image depicted in (e), showing 1/2 ML CO adsorbed on ZnO(1010). Gray, red, green and brown spheres represent Zn, lattice O, O in adsorbents and C atoms, respectively. (f) STM image showing the presence of bright spots within the (2 × 1) unit cell. The bright spots are diffusing during the imaging process and could be assigned as CO2 species. (g) AP-XPS C 1s of ZnO(1010) in 0.5 mbar CO or after the evacuation of CO gas. (h) A (3 × 1) STM image observed on ZnO(1010) in 3 mbar CO2 at room temperature. (i) Simulated STM image and (j) the optimized (3 × 1)−1 ML CO2 structure by DFT, including 2/3 ML chemisorbed CO2 plus 1/3 ML physisorbed CO2. Gray, red, green and brown spheres represent Zn, lattice O, O in adsorbents and C atoms, respectively. Reproduced with permission [435,436]. Copyright 2023, American Chemical Society; Copyright 2022, Elsevier.
Figure 41
(a) STM image of ZnO(1010) during the exposure to 1 mbar H2/CO2 (3:1) mixture gas at 300 K. (b) STM image of surface adsorbates on ZnO(1010) after being annealed in 1 mbar H2/CO2 (3:1) mixture gas at 400 K. (c) AP-XPS C 1s and O 1s of ZnO(1010) in 0.6 mbar H2/CO2 (3:1) mixture gas at different temperatures. (d) STM images of ZnO(1010) during the exposure to H2/CO mixture gas at 300 K. (e) STM image of surface adsorbates on ZnO(1010) after being annealed in 1 mbar H2/CO (3:1) mixture gas at 450 K. The formate species is indicated by white arrows. (f) AP-XPS C 1s and O 1s of ZnO(1010) in the 0.6 mbar H2/CO (3:1) mixture gas at different temperatures. STM images of hydroxylated ZnO(1010) surface during the exposure to (g) 2.1 mbar CO at 300 K, (h) 1.1 mbar CO at 450 K. The black arrows indicate linear adsorbed CO2. (i) AP-XPS C 1s and O 1s spectra of clean ZnO(1010), hydroxylated ZnO(1010) prepared by being annealed in 1 mbar H2 at 400 K and the hydroxylated ZnO(1010) exposed to CO at elevated temperatures. Reproduced with permission [436]. Copyright 2023, American Chemical Society.
Figure 42
STM images and structural models of FeO triangles with different sizes and exposing CUF edge sites. n is the number of Fe atoms at each edge. Reproduced with permission [444]. Copyright 2017, American Chemical Society.
Figure 44
Role of oxide-metal interaction (OMI) in determining the activity and stability of supported Cu2O NSs for catalytic CO oxidation. Reproduced with permission [449]. Copyright 2020, CC-BY-4.0.
Figure 45
Preparation of single atoms and the mechanism of CO oxidation. (a) STM imaging of Au single atoms on a CuO monolayer film, accompanied by a schematic representation of the catalytic mechanism for CO oxidation [459]. (b) STM imaging of Ni single cations on a CuO monolayer film, accompanied by a schematic representation of the catalytic mechanism for CO oxidation [461]. Reproduced with permission [459,461]. Copyright 2018, 2022, American Chemical Society.
Figure 46
STM imaging of single Ni2 clusters on CuO monolayer and their catalytic low-temperature CO2 dissociation [462]. Reproduced with permission [462]. Copyright 2023, American Chemical Society.
Figure 47
Visualization of on surface ethylene polymerization at boundary Fe3 sites. (a) STM snapshots of the carburized iron surface when exposed to C2H4 at room temperature [466]. (b) Schematic illustration of the ethylene polymerization mechanism at boundary Fe3 sites on the iron carbide surface [467]. Reproduced with permission [466,467]. Copyright 2022, The American Association for the Advancement of Science.
Figure 49
(a, b) STM images of the graphene/Pt(111) surface at different potentials in 0.1 mol/L HClO4. (c, d) Constant-height simulated STM images and corresponding models at 78 meV and 2 Å for (c) graphene/Pt(111) and (d) graphene/H (1 monolayer)/Pt(111), where graphene is depicted by black sticks, hydrogen and platinum by blue and gray spheres, respectively. STM images of the self-assembled CoPc monolayer on the Au(111) substrate in the CO2 environment in the 0.1 mol/L NaClO4 electrolyte (e) and 0.05 mol/L Mg(ClO4)2 electrolyte (f). STM images of a CoTPP adlayer on Au(111) in 0.1 mol/L HClO4 (g) and 0.1 mol/L KOH (h). Reproduced with permission [496,499,500]. Copyright 2019, 2022, American Chemical Society; Copyright 2021, Springer Nature.
Figure 50
(a-d) In-situ STM images the Cu(100) electrode in CO2-saturated 0.1 mol/L KHCO3 at different potentials. Gray arrows indicate scan direction; regions of constant potential are separated by dashed lines. (e) In-situ SXRD measurements of the CTRs (11l) and (20l) at 0, −0.3 and −0.7 V (vs. RHE) and the corresponding best fits (solid colored lines). (f) Coverage of surface defects and vertical expansion of the d12 spacing between the top two Cu layers as a function of the applied potential, obtained from the CTR fits. (g) The proposed Cu(100) surface model used for the CTR fitting. (h) Potential-dependent scattered intensity at the reciprocal space positions during CV measurement (shown as black line) at a scan rate of 5 mV/s. (i-l) In-situ STM images of the Pt(111) electrode after different numbers of ORCs: (i) initial; (j) 8; (k) 31; (l) 170. Scan size is 230 nm × 230 nm. (m) The surface width (red) and the hydrogen desorption charge (blue) plotted against the cycle numbers. Both sets of data are plotted logarithmically. The two black dotted lines indicate the two stages as discussed. Reproduced with permission [510,512]. Copyright 2018, Springer Nature; Copyright 2023, CC-BY 4.0.
Figure 51
(a) Schematic illustration of the current noise analysis technique. (b) STM line scans over a Pt(111) surface in 0.1 mol/L HClO4 when HER is on (red) and off (blue). (c) Time dependence of the surface coverage of CoPc-O2 and CoOEP-O2 in potential step experiments. (d-h) In-situ STM images of the CoOEP monolayer in O2-saturated 0.1 mol/L HClO4 in potential step experiments. Reproduced with permission [521,530]. Copyright 2017, Springer Nature; Copyright 2023, American Chemical Society.

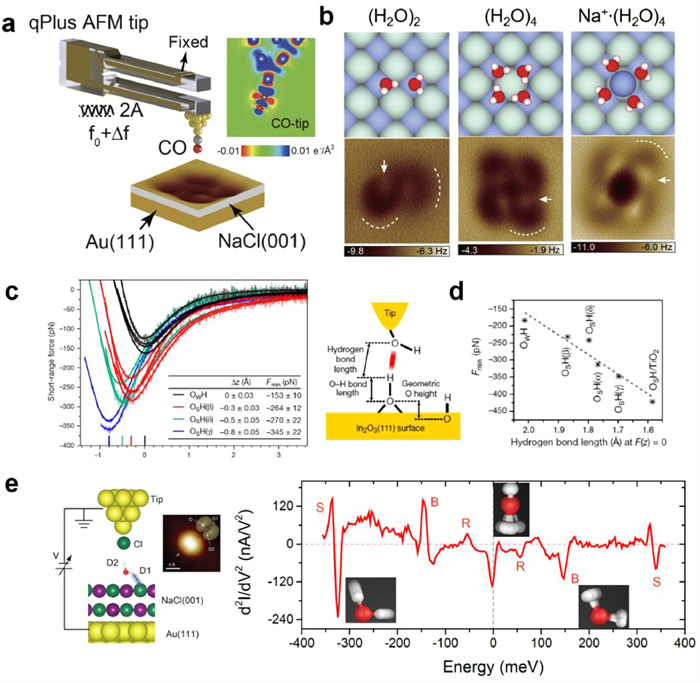
 DownLoad:
DownLoad:



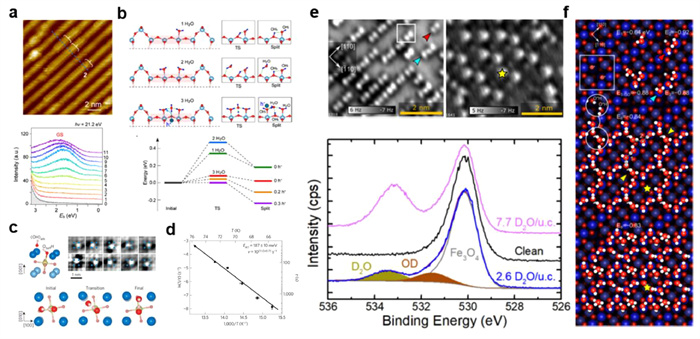

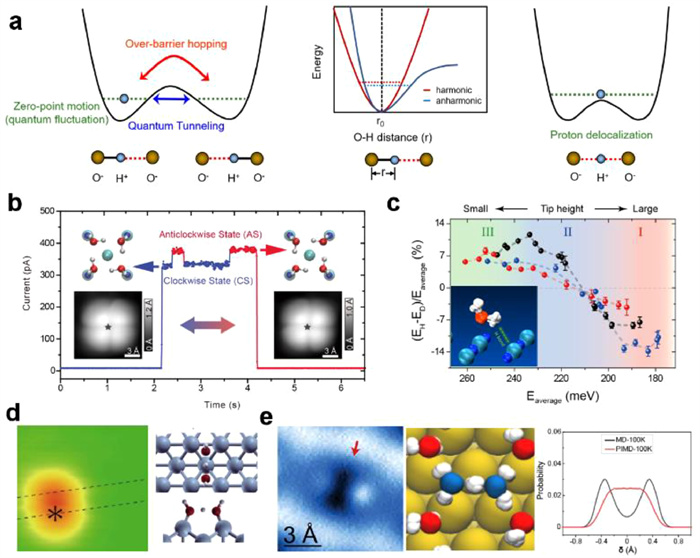
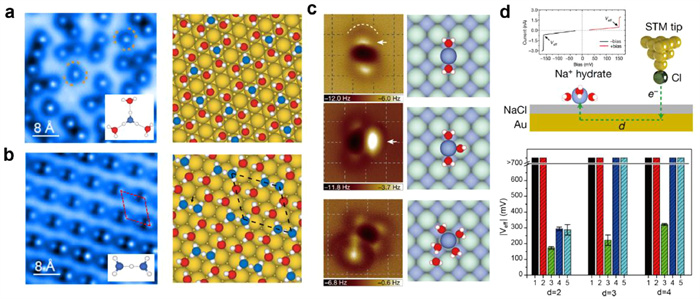
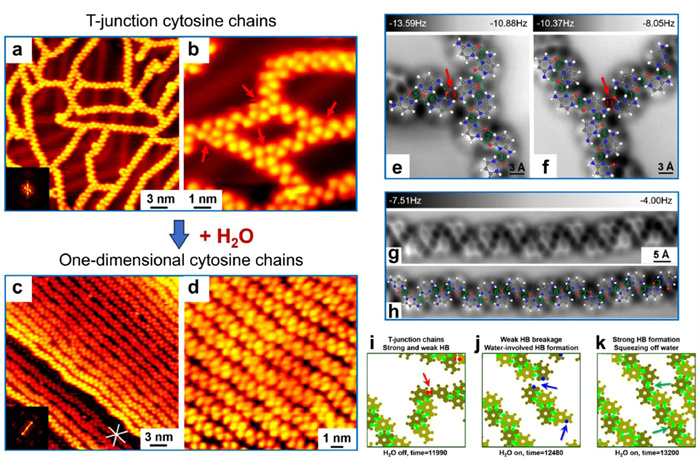
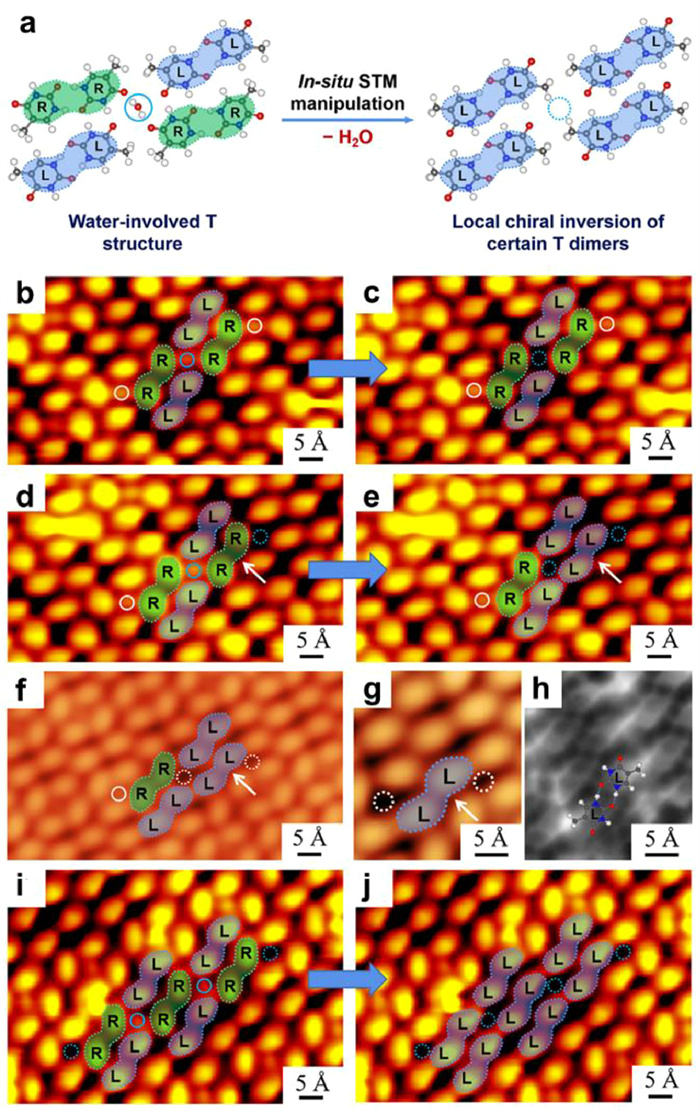


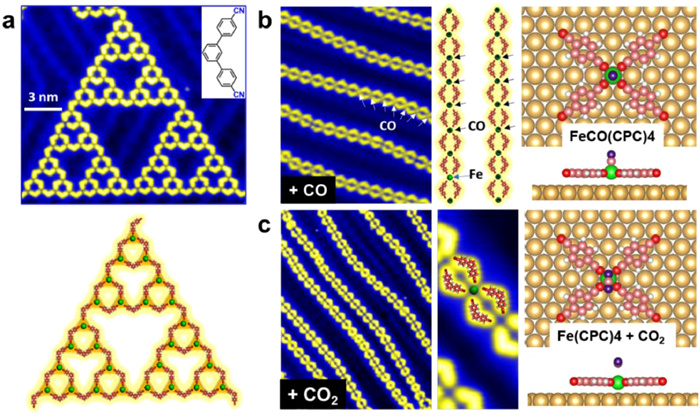
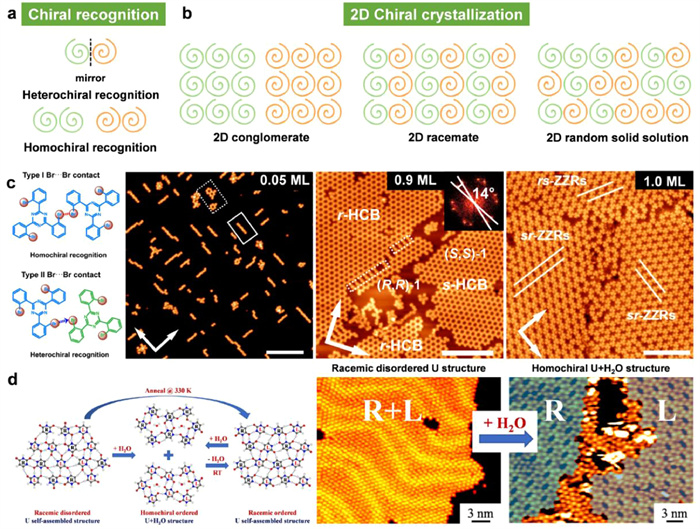
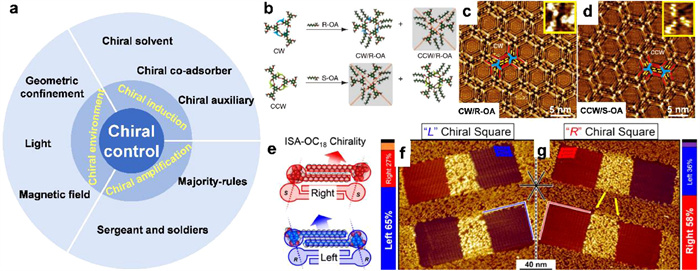
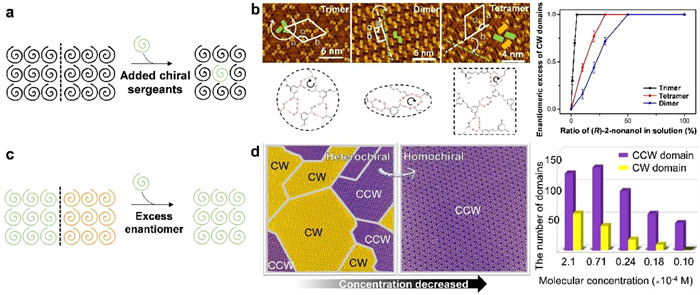




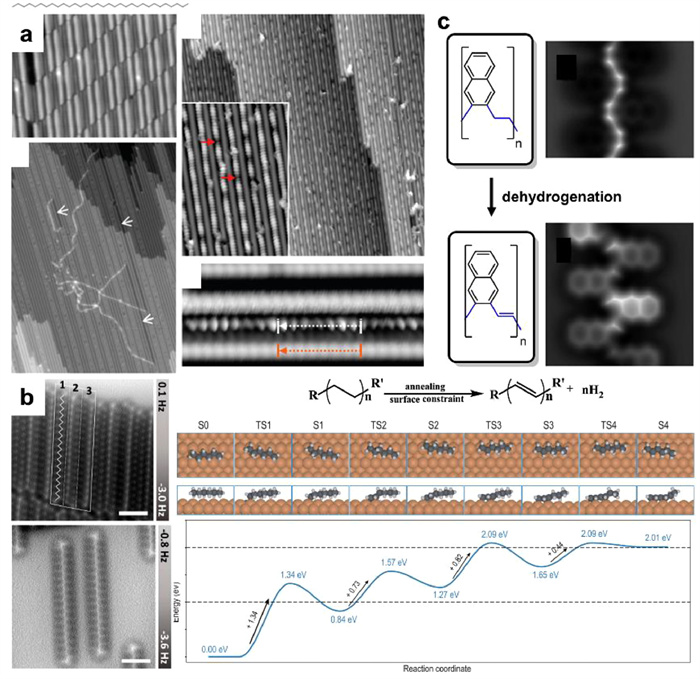
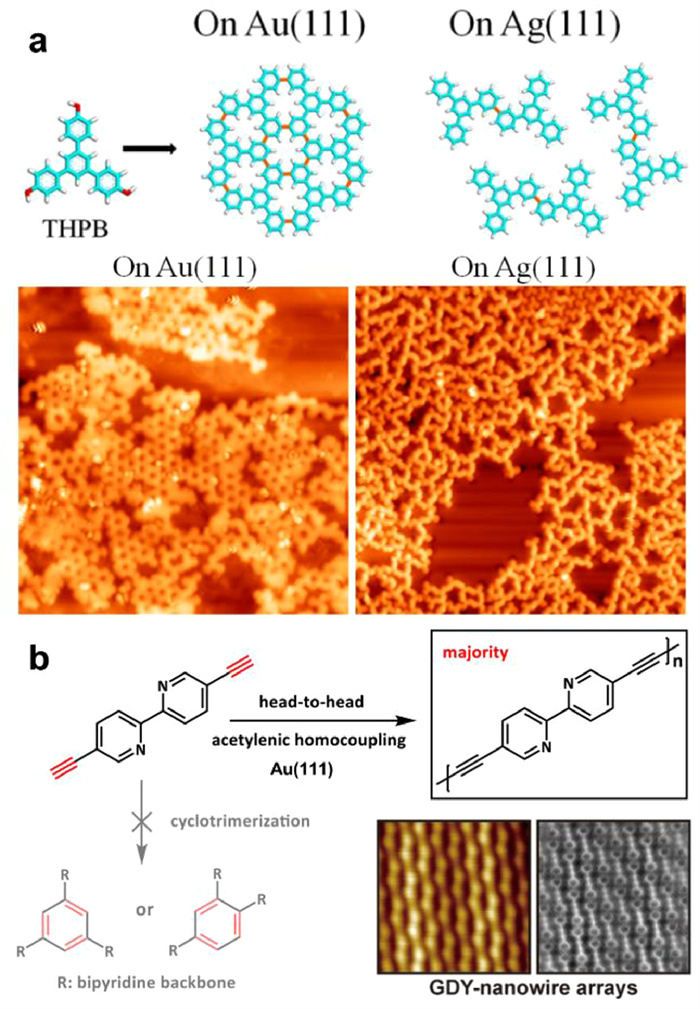
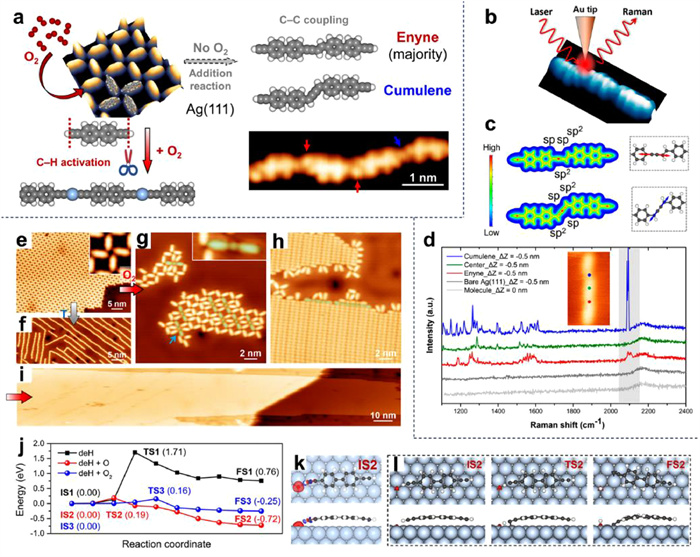


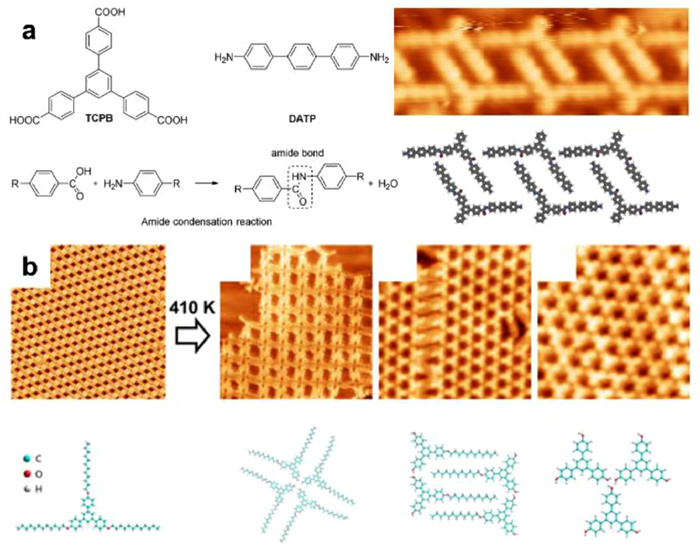
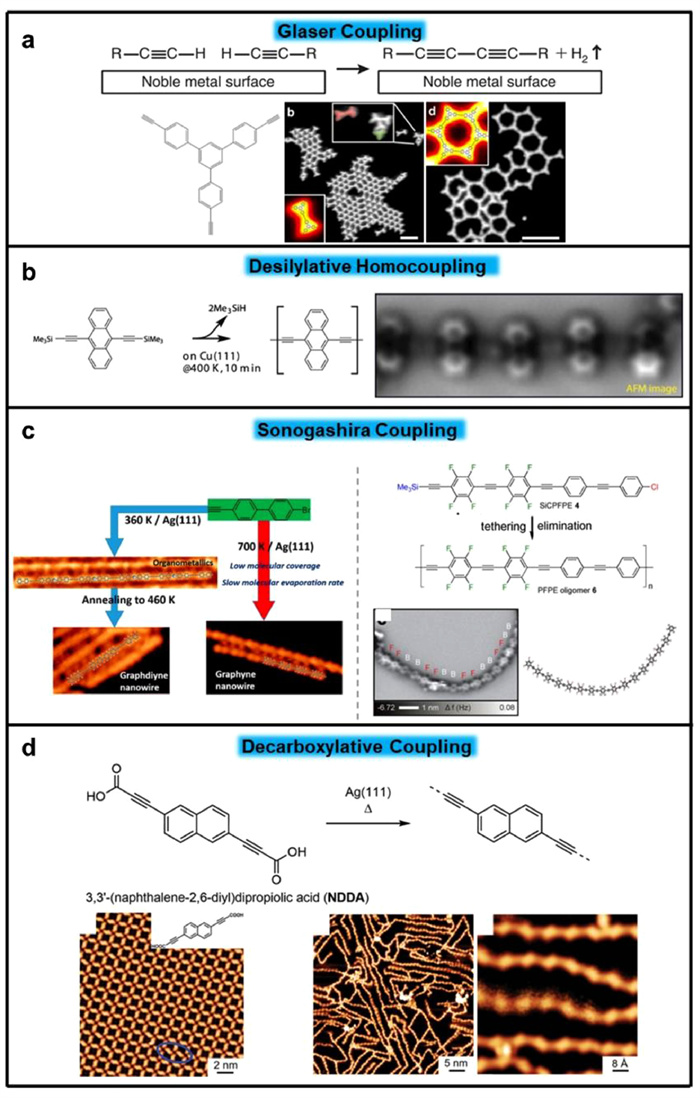
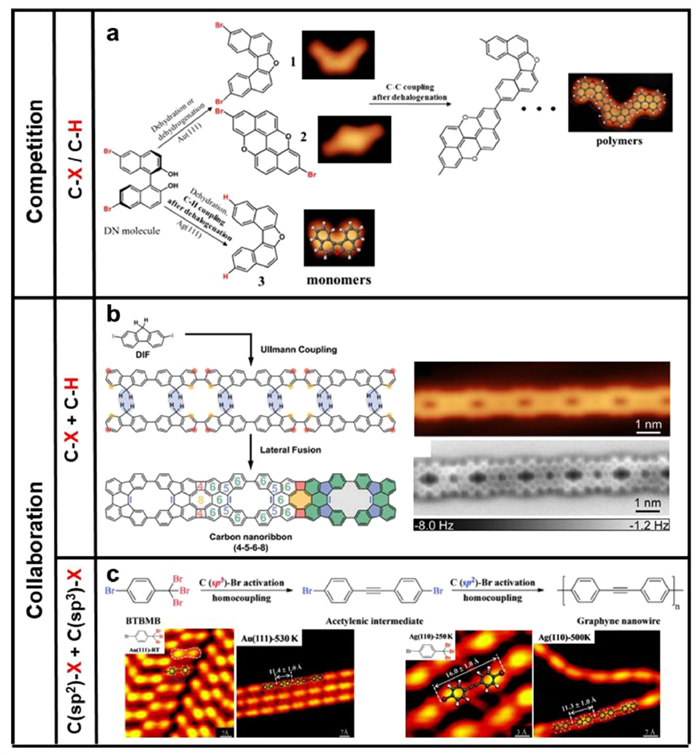
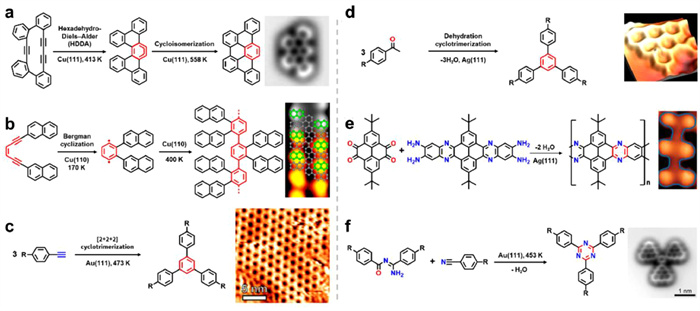
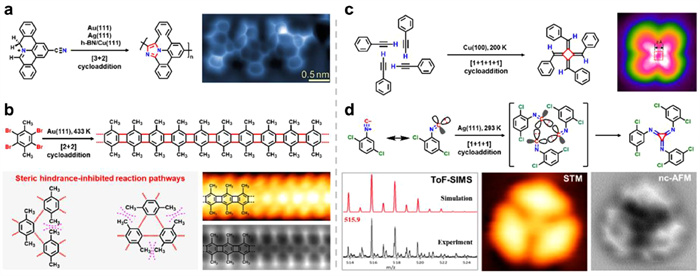

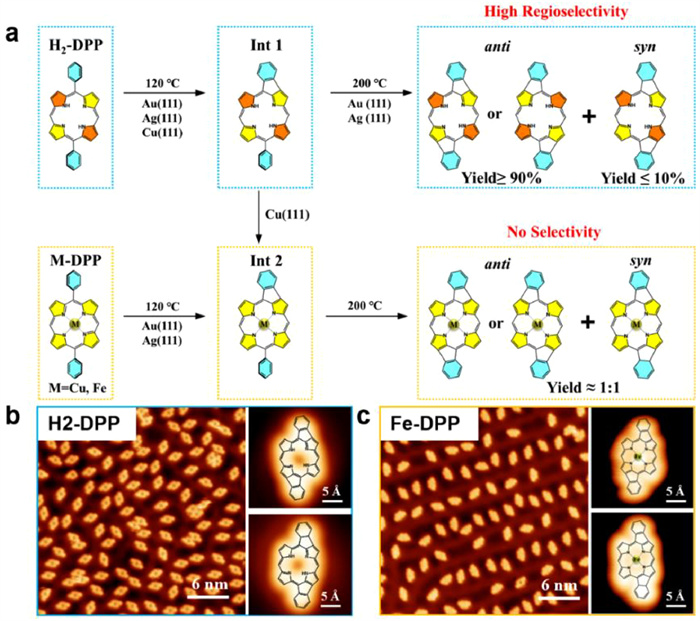
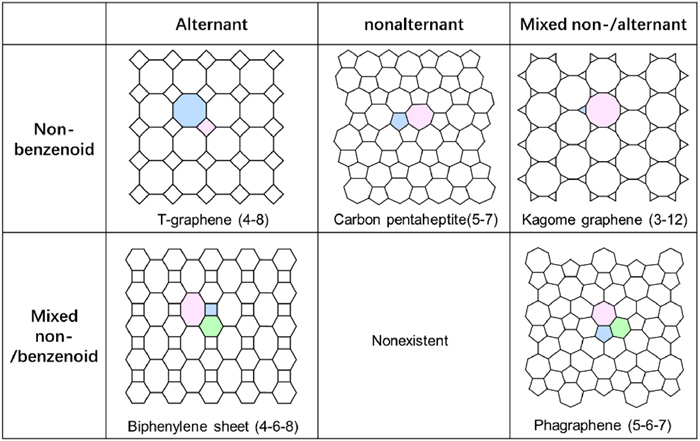
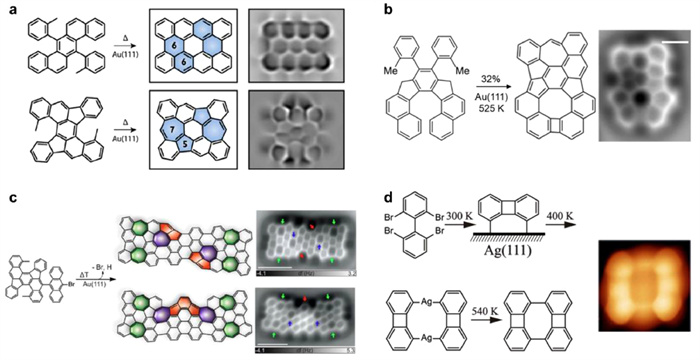
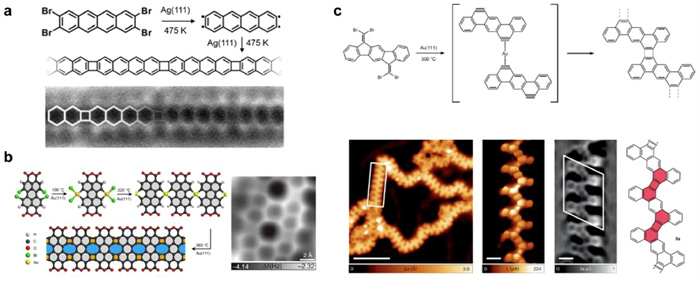
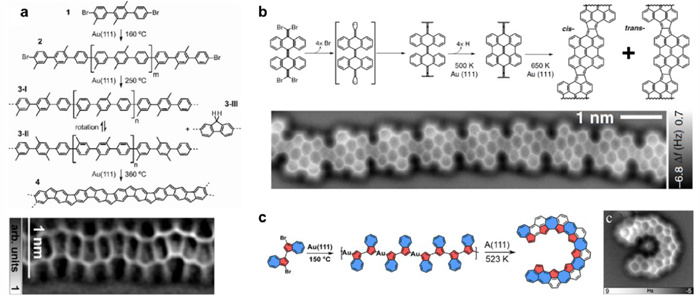





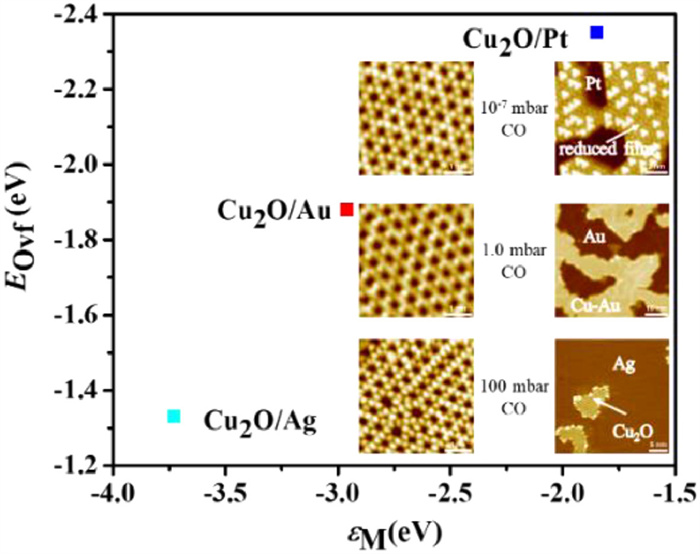

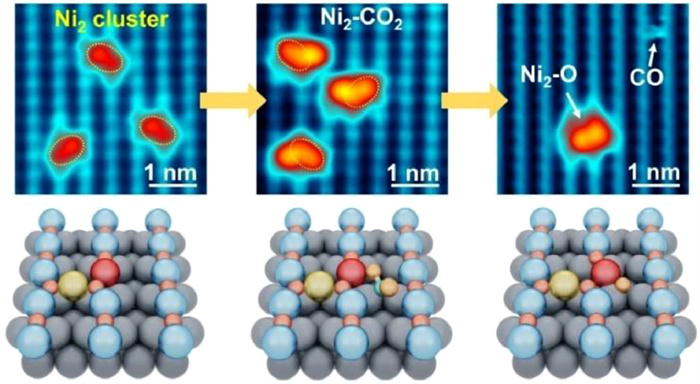
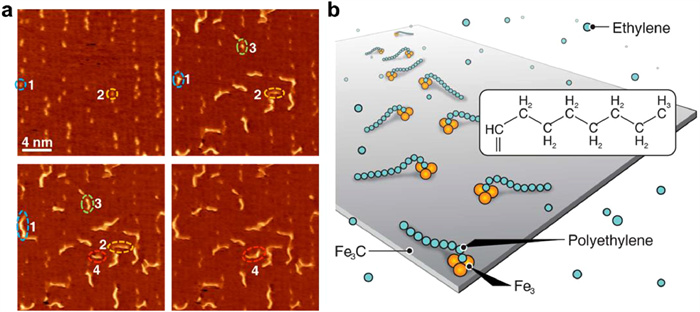
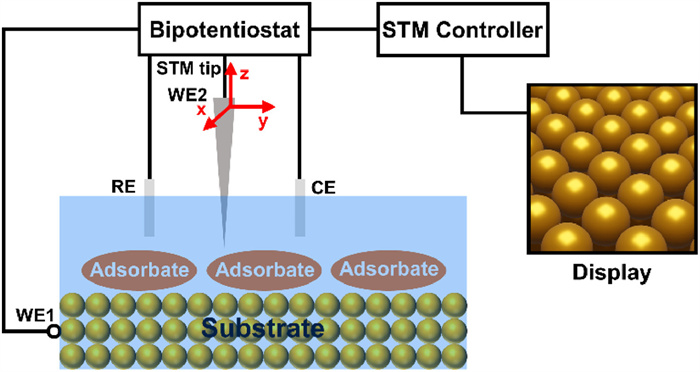



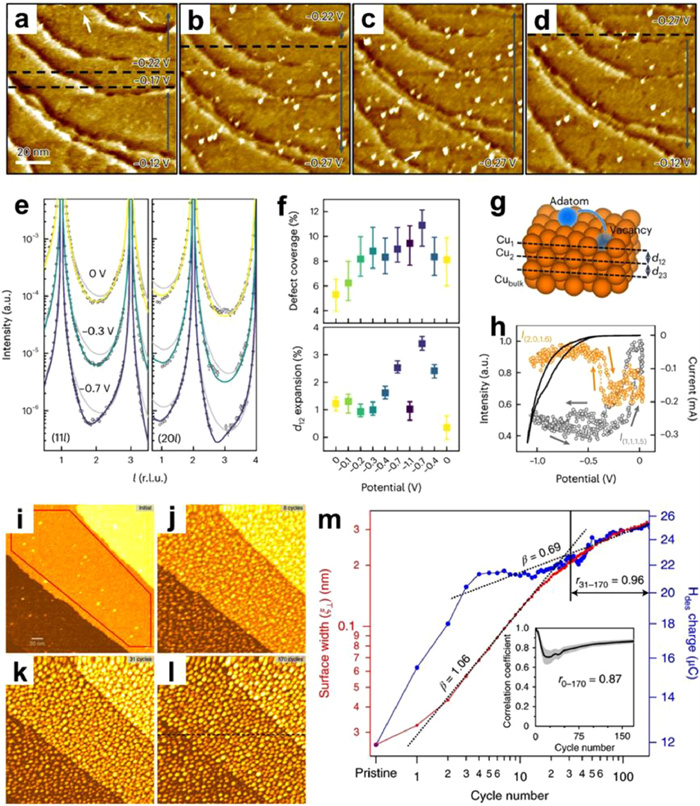

 下载:
下载:




















































 下载:
下载:
