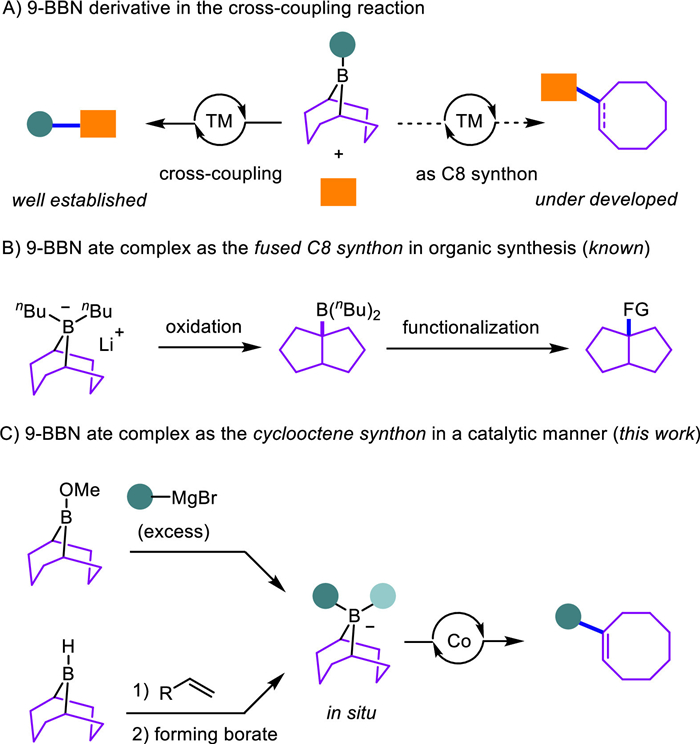
Scheme 1.
Contents of this study.
Cobalt-catalyzed migratory carbon-carbon cross-coupling of borabicyclo[3.3.1]nonane (9-BBN) borates
Peng Guo , Shicheng Dong , Xiang-Gui Zhang , Bing-Bin Yang , Jun Zhu , Ke-Yin Ye

Transition-metal-catalyzed cross-coupling of organoborons [1] and organic (pseudo)halides, namely Suzuki–Miyaura coupling [2], constitutes one of the most intensively investigated and widely applied carbon-carbon (C—C) bond-forming reactions. Owing to their ready preparation [3], the 9-BBN scaffold [4] serves as a very reliable and versatile auxiliary in diverse C—C cross-coupling reactions (Scheme 1A, left) [5,6]. Surprisingly, there are rare investigations of using 9-BBN derivatives as the potentially diverse C8 building blocks, which are typically neglected and discarded as the by-products in most Suzuki–Miyaura cross-coupling reactions (Scheme 1A, right).
In this context, early studies by Brown [7,8] have revealed that lithium dibutyl-9-borabicyclo[3.3.1]nonane ate complexes underwent an oxidant-mediated migration to form cis-bicyclo[3.3.0]oct-1-yldibutylborane, which could be transformed into functionalized fused hydrocarbons via the follow-up functional-group-interconversions (Scheme 1B) [9,10]. Despite the intriguing mechanistic merits, this reaction tolerated only very limited substrates and cannot be used for the preparation of other types of C8 skeletons. Therefore, catalytic approaches that allow diverse 9-BBN derivatives to function as novel C8 synthons are still elusive [11].
In continuance with our research interests in redox cobalt catalysis [12-17], we found the in situ formed 9-BBN ate complexes proceed via a cobalt-catalyzed migratory C—C cross-coupling reaction to afford diverse aryl- and alkyl-functionalized cyclooctenes (Scheme 1C). Preliminary mechanistic studies, including control experiments, cyclic voltammetry studies, and density functional theory (DFT) calculations suggest the oxidation-induced cis-bicyclo[3.3.0]oct-1-ylborane [18,19] being the key intermediate in this migratory cross-coupling reaction [20-23].
We initiated our investigations by identifying an optimal synthetic procedure and cobalt catalyst for this migratory C—C cross-coupling reaction of 9-BBN ate complexes (Scheme 2). Treatment of the commercially available 9-BBN-OMe (1) with 3.0 equiv. of para-tolylmagnesium bromide in THF readily generated the diarylated borate complex (2), which was evident by the chemical shift of 11B NMR from trisubstituted borane (δ 56.4) to tetrasubstituted borate (δ −15.0). Without further purification, the in situ formed borate [24] was then directly subjected to the Co(salen)/N-fluorobenzenesulfonimide (NFSI) catalytic system. Interestingly, a conjugated tolylcyclooctene (3) was obtained in 34% yield with Co(salen) A as the catalyst. Notably, this product represents a rare example of the migratory C—C cross-coupling of Grignard agent and 9-BBN derivative as the cyclooctene synthon.
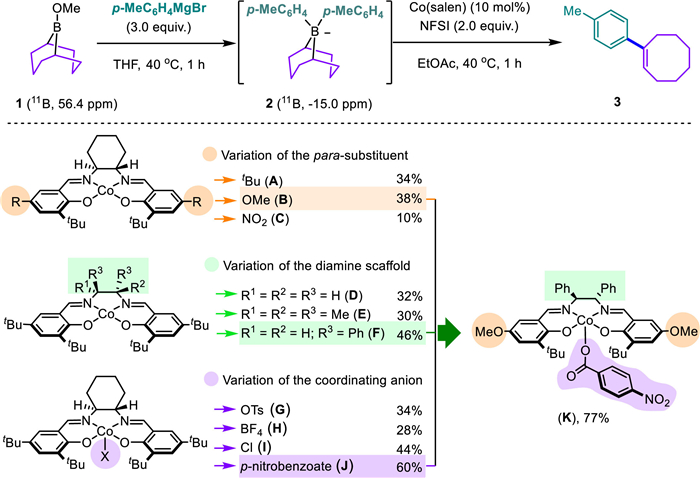
Extensive optimization suggested the CoⅡ(salen) catalyst featuring a para-methoxy substituent (B) or a 1,2-diphenylethylenediamine backbone (F) was effective in this catalytic transformation. While both the di- and trivalent Co(salen) were competent, the coordinating anion in CoⅢ(salen)–X catalyst provided an extra handle for tuning reactivity. Indeed, the incorporation of the para-benzoate coordinating anion (J) dramatically improved the catalytic efficiency. Assembly of these privileged motifs from the above investigations led to an optimal catalyst (K), which efficiently promoted the formation of the anticipated tolylcyclooctene (3) in 77% yield. Besides phenylmagnesium bromide, the corresponding lithium agent also afforded compatible results. However, a survey of various oxidants other than NFSI only resulted in no reactivity or much lower yields (for details, see Supporting information).
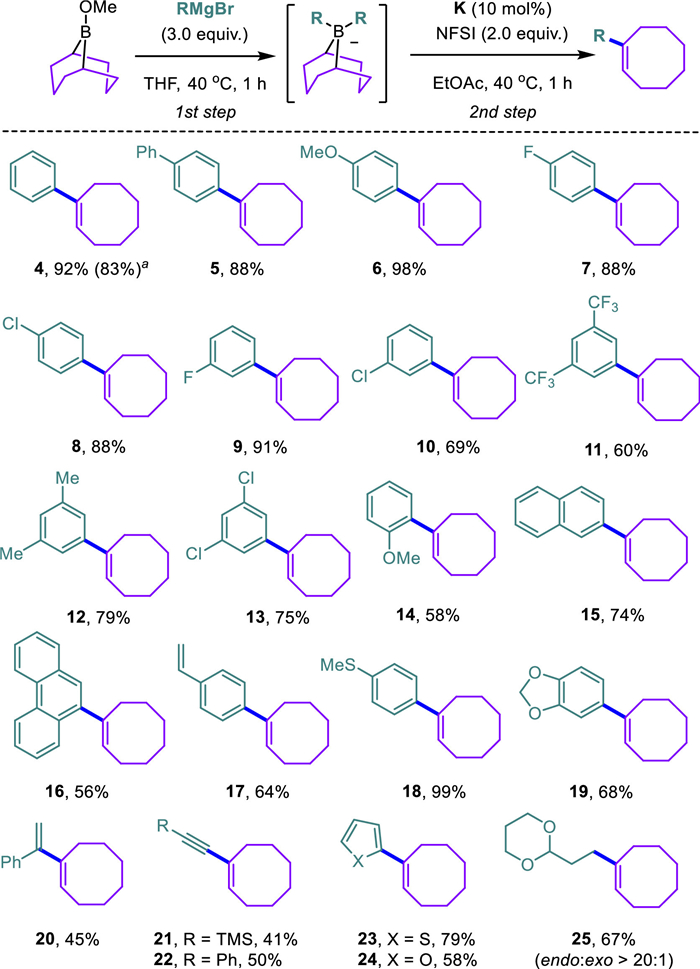
The substrate scope of this cobalt-catalyzed migratory C—C bond-forming reaction was delineated as Grignard reagents are either commercially available or readily accessible from organohalides (Scheme 3). This reaction tolerated diverse para-(4-8), meta-(9-13), and ortho-substituted (14) phenylmagnesium bromides. Polycyclic aromatic hydrocarbons, including naphthalene (15) and phenanthrene (16), can be readily hinged onto the cyclooctene framework. This protocol also tolerated many functional groups, such as alkene (17), thioether (18), and acetal (19), saving plenty of opportunities for further derivatization of the cyclooctenes. Besides arylmagnesium bromides, diverse Grignard reagents were amenable in the functionalization of cyclooctene to afford the corresponding conjugated diene (20), enynes (21, 22), and heteroaryl cyclooctenes (23, 24). Though (1,3-dioxan-2-ylethyl)magnesiumbromide can be used in this transformation to afford the anticipated product (25) with high endo/exo regioselectivity (vide infra), attempts to incorporate other aliphatic chains into the cyclooctene framework were not successful yet.
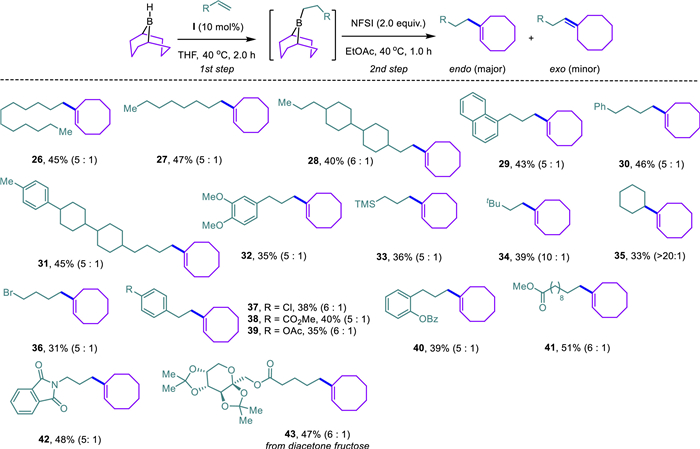
To access α-alkylated cyclooctenes, we found the in situ formed alkyl 9-BBN derivatives from hydroboration served as competent candidates in the analogously migratory cross-coupling reaction with Co(salen) Ⅰ as the catalyst (Scheme 4). Though hydroboration of 9-BBN and alkene generally proceeds without a catalyst, early incorporation of this cobalt complex throughout the current one-pot synthesis is beneficial to reaction yields. The anticipated α-alkylated cyclooctenes were obtained as the major products accompanied by minor amounts of exo-isomers. Diverse aliphatic terminal alkenes were well-tolerated to afford the corresponding α-alkylated cyclooctenes (26–33) in synthetically useful yields. Note that the endo/exo selectivity dramatically increased by using a bulky 3,3-dimethyl-1-butene (34) or a cyclic alkene (35). This approach displayed remarkable functionality tolerance, which could not be replicated by using organometallic reagents such as Grignard or organolithium agents. For instance, the mild reaction conditions rendered a wide array of aliphatic/aromatic halides (36, 37), esters (38, 41), phenol derivatives (39, 40), o-phthalimide (42), and the diacetonefructose-derived alkene (43) intact.
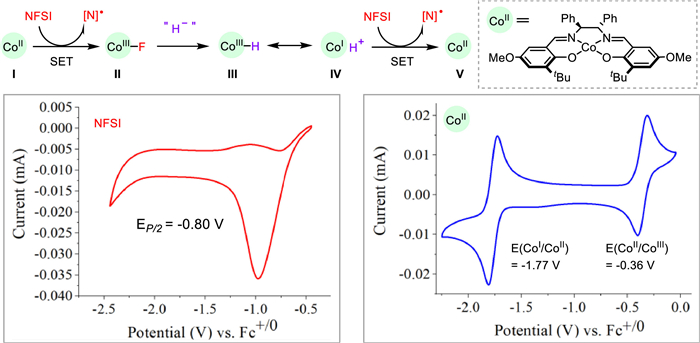
Cyclic voltammetry studies of Co(salen) and NFSI provided some mechanistic insights (Scheme 5). While NFSI only displayed one irreversible reduction peak (EP/2 = −0.80 V), two reversible redox couples of Co(salen) appeared wherein −0.36 V and −1.77 V were assigned to E(CoⅡ/CoⅢ) and E(CoI/CoⅡ), respectively [25]. Therefore, NFSI was competent to mediate both the single-electron transfer processes of CoI to CoⅡ (Ⅳ-to-Ⅴ) and CoⅡ to CoⅢ (Ⅰ-to-Ⅱ), the latter of which is likely via an inner-sphere manner [26]. Remarkably, the CoⅢ‒F (Ⅱ) underwent a facile fluoride-to-hydride exchange reaction [27,28] to afford the formal CoⅢ‒H (Ⅲ) [29,30], which is acidic (pKa 10−15) [31,32] and thus can be interpreted as CoI(H+) (Ⅳ) [33].
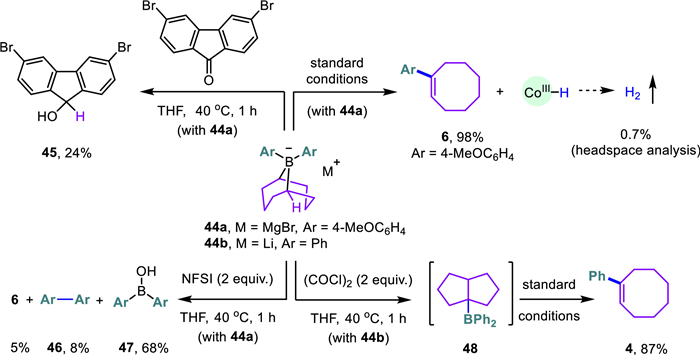
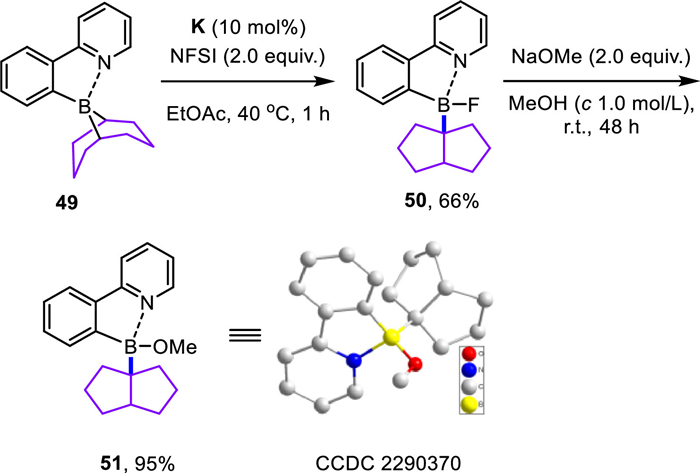
Early studies have shown that the bridge-head hydrogen atoms of dibutyl-9-borabicyclo[3.3.1]nonane ate complexes are competent organic hydrides in various reduction reactions [34-37]. Indeed, the diarylated 9-BBN borate complex (44a) in Scheme 6 exhibited a similar hydride feature but was much less effective in reducing the 9-fluorenone derivative (45). The headspace analysis detected a small amount of dihydrogen (0.7%), a minor hydrogen evolution reaction pathway from the CoⅢ‒H species [38-41]. Without the cobalt catalyst, the borate complex (44a) reacted with NFSI only to afford the diarylborinic acid (47, 68% yield) accompanied by trace amounts of cyclooctene (6) and biaryl (46). The extensive screening of various oxidants revealed the use of oxalyl chloride transformed the borate complex (44b) into a new boron species (48, 11B, δ 80.6). Unfortunately, attempts to isolate this boron species or its tetra-coordinated variant were not successful yet, probably owing to its ease of decomposition, which was tentatively proposed as the diphenyl analog of the known cis-bicyclo[3.3.0]oct-1-yldibutylborane (11B, δ 81.8) [10,18,19]. While organomagnesium and organolithium compounds were both effective coupling agents in the current transformation, the formation of intermediate (48) could be detected only by using lithium borate (44b). Exposure of this in situ generated boron species to the standard reaction conditions resulted in a high conversion forming the anticipated cyclooctene (4). Under the standard reaction conditions, ortho-borylated 2-phenylpyridine (49) [42] was readily converted into its fluoroborate derivative with a cis-bicyclo[3.3.0]oct-1-yl scaffold (50) in 66% yield (Scheme 7). Further treatment with 2.0 equiv. NaOMe in methanol resulted in a quantitative formation of its methoxide analog (51), which was unambiguously confirmed by X-ray diffraction analysis.
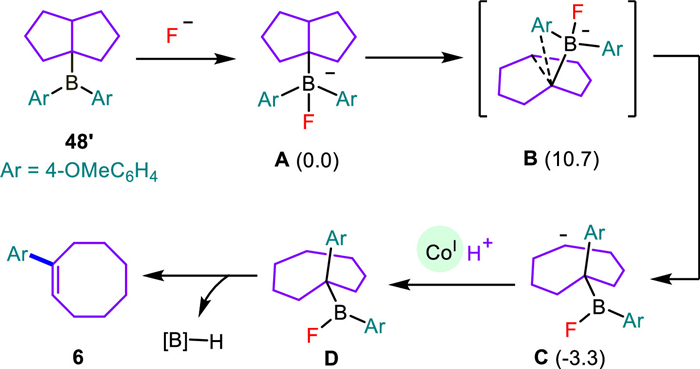
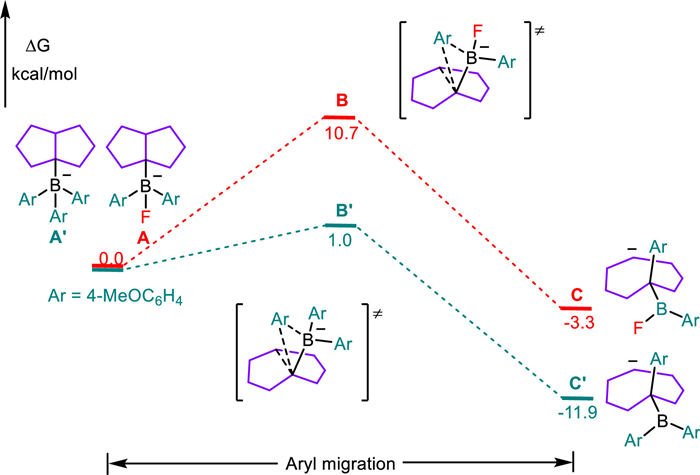
As illustrated in Scheme 8, DFT calculations were employed to elucidate the possible reaction pathway for the formation of α-aryl cyclooctene (6) from the cis-bicyclo[3.3.0]oct-1-yldiphenylborane (48'). We selected the fluorinated borate (A) as the model substrate, which might proceed with an ionic boron-to-carbon phenyl migration [43-45]. According to our calculations, the cis orientated, front migration of the phenyl group (B) required a moderate energy barrier (ΔG≠, 10.7 kcal/mol), giving rise to a thermodynamic more stable carbanion (C, −3.3 kcal/mol) through the cleavage of the bridged C—C bond. By contrast, the back migration of the phenyl group trans to the bridged C—C bond was disfavored both by an exceptionally high energy barrier (ΔG≠, 79.0 kcal/mol) and generation of the undesired 9-BBN-type borate as the only located product (Fig. S14 in Supporting information). The subsequent protonation, likely by the in situ formed acidic CoI(H+), delivered a cyclooctylborane (D), which then proceeded with a kinetically (ΔG≠, 28.5 kcal/mol) and thermodynamically (ΔG, 4.9 kcal/mol) accessible dehydroboration to give the anticipated cyclooctene (Fig. S16 in Supporting information). The resultant borane is likely involved in the fluoride-to-hydride exchange reaction of CoⅢ‒F toward CoⅢ‒H [46]. Meanwhile, another reaction pathway for the conversion of cyclooctylborane (D) to α-aryl cyclooctene (6) through the oxidation of alkyl borane to the corresponding alkyl radical [47-49] and subsequent cobalt-mediated hydrogen-atom transfer (HAT) [50,51] is also possible (Scheme 9).
In summary, we have disclosed a cobalt-catalyzed migratory C—C cross-coupling reaction of the in situ formed 9-BBN ate complexes, affording a wide variety of aryl- and alkyl-functionalized cyclooctenes. Preliminary mechanistic studies suggested a cis-bicyclo[3.3.0]oct-1-yl-borane as the key intermediate, which was achieved by the oxidation-induced rearrangement of the 9-BBN ate complex. Mechanistic insights gained from the current studies should promote the development of other diverse migratory cross-coupling of borate complexes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Peng Guo: Writing – review & editing, Methodology, Investigation, Formal analysis. Shicheng Dong: Writing – review & editing, Formal analysis, Data curation. Xiang-Gui Zhang: Investigation. Bing-Bin Yang: Investigation. Jun Zhu: Writing – review & editing, Supervision. Ke-Yin Ye: Writing – review & editing, Writing – original draft, Supervision, Project administration, Funding acquisition, Conceptualization.
This work was supported by the National Natural Science Foundation of China (No. 22171046), the Hundred-Talent Project of Fujian (No. 50021113), and Fuzhou University (No. 0480-00489503). The authors thank Prof. Zhi-Xiang Yu (Peking University) for helpful discussions.
Supplementary material associated with this article can be found, in the online version, at doi:
E. Fernández, A. Whiting, Synthesis and Application of Organoboron Compounds, Springer, 2015, doi:
N. Miyaura, A. Suzuki, Chem. Rev. 95 (1995) 2457–2483. doi: 10.1021/cr00039a007
R.S. Dhillon, Hydroboration and Organic Synthesis: 9-Borabicyclo[3.3.1]nonane (9-BBN), Springer, 2007, doi:
G. Seidel, A. Fürstner, Chem. Commun. 48 (2012) 2055–2070. doi: 10.1039/c2cc17070a
S.R. Chemler, D. Trauner, S.J. Danishefsky, Angew. Chem. Int. Ed. 40 (2001) 4544–4568. doi: 10.1002/1521-3773(20011217)40:24<4544::AID-ANIE4544>3.0.CO;2-N
A.J.J. Lennox, G.C. Lloyd-Jones, Chem. Soc. Rev. 43 (2014) 412–443. doi: 10.1039/C3CS60197H
Y. Yamamoto, H.C. Brown, J. Org. Chem. 39 (1974) 861–862. doi: 10.1021/jo00920a034
G.W. Kramer, H.C. Brown, J. Organomet. Chem. 90 (1975) C1–C5. doi: 10.1016/S0022-328X(00)91618-8
G.W. Kramer, H.C. Brown, J. Am. Chem. Soc. 98 (1976) 1964–1965. doi: 10.1021/ja00423a054
G.W. Kramer, H.C. Brown, J. Org. Chem. 42 (1977) 2832–2836. doi: 10.1021/jo00437a010
V.K. Aggarwal, G.Y. Fang, X. Ginesta, D.M. Howells, M. Zaja, Pure Appl. Chem. 78 (2006) 215–229. doi: 10.1351/pac200678020215
S.W.M. Crossley, C. Obradors, R.M. Martinez, R.A. Shenvi, Chem. Rev. 116 (2016) 8912–9000. doi: 10.1021/acs.chemrev.6b00334
H. Shigehisa, Chem. Pharm. Bull. 66 (2018) 339–346. doi: 10.1248/cpb.c17-01006
M. Kojima, S. Matsunaga, Trends Chem. 2 (2020) 410–426. doi: 10.1016/j.trechm.2020.01.004
Y. Yin, D. Ouyang, J. Wang, R. Zhu, Sci. Sin. Chim. 50 (2020) 1217–1232. doi: 10.1360/SSC-2020-0106
J. Zhong, Y. Yu, D. Zhang, K. Ye, Chin. Chem. Lett. 32 (2021) 963–972. doi: 10.1016/j.cclet.2020.08.011
L. Wan, Y. Tong, X. Lu, Y. Fu, Chin. Chem. Lett. 35 (2024) 109283. doi: 10.1016/j.cclet.2023.109283
S. Pal, P.Y. Zavalij, A.N. Vedernikov, Chem. Commun. 50 (2014) 5376–5378. doi: 10.1039/C3CC47445C
S. Pal, P.Y. Zavalij, A.N. Vedernikov, Organometallics 34 (2015) 5183–5190. doi: 10.1021/acs.organomet.5b00737
D. Leonori, V.K. Aggarwal, Acc. Chem. Res. 47 (2014) 3174–3183. doi: 10.1021/ar5002473
S. Namirembe, J.P. Morken, Chem. Soc. Rev. 48 (2019) 3464–3474. doi: 10.1039/c9cs00180h
M. Kischkewitz, F.W. Friese, A. Studer, Adv. Synth. Catal. 362 (2020) 2077–2087. doi: 10.1002/adsc.201901503
H. Wang, C. Jing, A. Noble, V.K. Aggarwal, Angew. Chem. Int. Ed. 59 (2020) 16859–16872. doi: 10.1002/anie.202008096
K. Yang, Q. Song, Acc. Chem. Res. 54 (2021) 2298–2312. doi: 10.1021/acs.accounts.1c00132
X. Wu, C.N. Gannett, J. Liu, R. Zeng, et al., J. Am. Chem. Soc. 144 (2022) 17783–17791. doi: 10.1021/jacs.2c08278
P. Guo, Y. Li, X.-G. Zhang, et al., Org. Lett. 22 (2020) 3601–3606. doi: 10.1021/acs.orglett.0c01072
K. Ding, T.R. Dugan, W.W. Brennessel, E. Bill, P.L. Holland, Organometallics 28 (2009) 6650–6656. doi: 10.1021/om900394t
T.R. Dugan, X. Sun, E.V. Rybak-Akimova, et al., J. Am. Chem. Soc. 133 (2011) 12418–12421. doi: 10.1021/ja2052914
D.C. Lacy, G.M. Roberts, J.C. Peters, J. Am. Chem. Soc. 137 (2015) 4860–4864. doi: 10.1021/jacs.5b01838
M.J. Chalkley, P.H. Oyala, J.C. Peters, J. Am. Chem. Soc. 141 (2019) 4721–4729. doi: 10.1021/jacs.9b00193
G.N. Schrauzer, R.J. Windgassen, J. Kohnle, Chem. Ber. 98 (1965) 3324–3333. doi: 10.1002/cber.19650981032
D.P. Estes, D.C. Grills, J.R. Norton, J. Am. Chem. Soc. 136 (2014) 17362–17365. doi: 10.1021/ja508200g
K.-Y. Ye, T. McCallum, S. Lin, J. Am. Chem. Soc. 141 (2019) 9548–9554. doi: 10.1021/jacs.9b04993
Y. Yamamoto, H. Toi, S.-I. Murahashi, I. Moritani, J. Am. Chem. Soc. 97 (1975) 2558–2559. doi: 10.1021/ja00842a052
Y. Yamamoto, H. Toi, A. Sonoda, S.I. Murahashi, J. Am. Chem. Soc. 98 (1976) 1965–1967. doi: 10.1021/ja00423a055
Y. Yamamoto, H. Toi, A. Sonoda, S.I. Murahashi, J. Chem. Soc., Chem. Commun. (1976) 672–673.
H. Toi, Y. Yamamoto, A. Sonoda, S.I. Murahashi, Tetrahedron 37 (1981) 2261–2267. doi: 10.1016/S0040-4020(01)97982-7
V. Artero, M. Fontecave, Coord. Chem. Rev. 249 (2005) 1518–1535. doi: 10.1016/j.ccr.2005.01.014
C. Baffert, V. Artero, M. Fontecave, Inorg. Chem. 46 (2007) 1817–1824. doi: 10.1021/ic061625m
X. Hu, B.S. Brunschwig, J.C. Peters, J. Am. Chem. Soc. 129 (2007) 8988–8998. doi: 10.1021/ja067876b
J.L. Dempsey, B.S. Brunschwig, J.R. Winkler, H.B. Gray, Acc. Chem. Res. 42 (2009) 1995–2004. doi: 10.1021/ar900253e
Y. Kuninobu, T. Iwanaga, T. Omura, K. Takai, Angew. Chem. Int. Ed. 52 (2013) 4431–4434. doi: 10.1002/anie.201210328
G. Zweifel, H. Arzoumanian, C.C. Whitney, J. Am. Chem. Soc. 89 (1967) 3652–3653. doi: 10.1021/ja00990a061
H.D.A. Heck, J. Am. Chem. Soc. 93 (1971) 23–29. doi: 10.1021/ja00730a004
R.J. Armstrong, V.K. Aggarwal, Synthesis 49 (2017) 3323–3336. doi: 10.1055/s-0036-1589046
X.G. Zhang, Z.X. He, P. Guo, Z. Chen, K.Y. Ye, Org. Lett. 24 (2022) 22–26. doi: 10.1021/acs.orglett.1c03511
C. Ollivier, P. Renaud, Chem. Rev. 101 (2001) 3415–3434. doi: 10.1021/cr010001p
P. Renaud, A. Beauseigneur, A. Brecht-Forster, et al., Pure Appl. Chem. 79 (2007) 223–233. doi: 10.1351/pac200779020223
D.P. Curran, T.R. McFadden, J. Am. Chem. Soc. 138 (2016) 7741–7752. doi: 10.1021/jacs.6b04014
S.W.M. Crossley, F. Barabé, R.A. Shenvi, J. Am. Chem. Soc. 136 (2014) 16788–16791. doi: 10.1021/ja5105602
G. Li, J.L. Kuo, A. Han, et al., J. Am. Chem. Soc. 138 (2016) 7698–7704. doi: 10.1021/jacs.6b03509

Scheme 3 Substrate scope of α-arylated cyclooctenes. Reaction conditions: 1st step: 9-BBN-OMe (hexane solution, 1.0 mol/L, 0.2 mmol, 1.0 equiv.), Grignard reagent (0.6 mmol, 3.0 equiv.), 40 ℃, 1 h; 2nd step: Co(salen), K (0.02 mol, 10 mol%), NFSI (0.4 mmol, 2.0 equiv.), ethyl acetate (1.0 mL), 40 ℃, 1 h. a PhLi instead of PhMgBr.

Scheme 4 Substrate scope of α-alkylated cyclooctenes. Reaction conditions: 1st step: Co(salen) Ⅰ (0.02 mol, 10 mol%), 9-BBN (THF solution, 0.5 mol/L, 0.3 mmol, 1.5 equiv.), alkene (0.2 mmol, 1.0 equiv.), 40 ℃, 2 h; 2nd step: NFSI (0.4 mmol, 2.0 equiv.), ethyl acetate (1.0 mL), 40 ℃, 1 h.

Scheme 8 A plausible mechanism of the formation of α-aryl cyclooctene. The Gibbs free energies are given in kcal/mol (see Supporting information for calculation details).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载: