Citation:
Congyan Liu, Xueyao Zhou, Fei Ye, Bin Jiang, Bo Liu. Confined electric field in nano-sized channels of ionic porous framework towards unique adsorption selectivity[J]. Chinese Chemical Letters,
2025, 36(2): 109969.
doi:
10.1016/j.cclet.2024.109969
Confined electric field in nano-sized channels of ionic porous framework towards unique adsorption selectivity
English
Confined electric field in nano-sized channels of ionic porous framework towards unique adsorption selectivity
liuchem@ustc.edu.cn (B. Liu) 1 These authors contributed equally to this work.
Received Date:
08 January 2024 Accepted Date:
07 May 2024 Revised Date:
08 April 2024 Available Online:
15 February 2025
Abstract:
Efficient selective adsorption and separation using porous frameworks are critical in many industrial processes, where adsorption energy and dynamic diffusion rate are predominant factors governing selectivity. They are highly susceptible to framework charge, which plays a significant role in selective adsorption. Currently, ionic porous frameworks can be divided into two types. One of them is composed of a charged backbone and counter ions. The framework with zwitterionic channels is another type. It is composed of regular and alternating arrangements of cationic and anionic building units. Herein, we report a hydrogen-bonded ionic framework (HIF) of {(CN3H6)2[Ti(μ2O)(SO4)2]}n with 1D channel exhibits unique adsorption selectivity for Ar against N2 and CO2. Density functional theory (DFT) results suggest that CO2 cannot be adsorbed by HIF at the experimental temperature due to a positive adsorption free energy. In addition, due to a relatively large diffusion barrier at 77 K, N2 molecules hardly diffuse in HIF channels, while Ar has a negligible diffusion barrier. The unique net positively-charged space in the channel is the key to the unusual phenomena, based on DFT simulations and structural analysis. The findings in this work proposes the new adsorption mechanism and provides unique perspective for special separation applications, such as isotope and noble gasses separations.
Porous frameworks containing void spaces can accommodate guest molecules owing to their high specific surface area and surface energy [1]. Last decades have witnessed the fast development of porous frameworks, especially the booming growth of new types of porous structures including metal-organic frameworks (MOFs), covalent-organic frameworks (COFs) and hydrogen-bonded organic frameworks (HOFs) [2-5], with wide-range applications in gas adsorption and separation [6,7], energy storage [8,9], sensors [10,11] and catalysis [12,13], etc. Selective adsorption and separation using porous frameworks is critical in many industrial processes. This is predominated by the interaction between pores/channels/cavities and adsorbates in terms of adsorption energy and dynamic diffusion rate. Therefore, tuning the pore properties is essential towards the high selectivity of adsorption.
Various strategies have been designed to tailor pore properties including pore sizes and shapes, surface functionalities, pore polarity, etc. Pore size is the most intuitive factor influencing the process of adsorptive separation [14]. Reticular chemistry has demonstrated its strong power to create the MOFs and COFs with varied pore sizes [15,16]. In spite of massive reports on gas separation based on adjusting pore size [17,18], precise controlling size, especially for exact molecular sieving effect, is still challenging. Special pore structure featured with pockets connected to channels in a MOF of JNU-3 (Jinan University-3) exhibits efficient dynamic separation of propylene and propane [19]. Functional organic groups could be directly introduced into the framework during the synthetic process for improved adsorption selectivity [20]; however, it is restricted owing to their reactivity which may lead to coordinate with metal ions. Post-synthetic modification (PSM) provides an alternative way to incorporate functional sites into porous frameworks [21,22]. It may reduce the pore volume of materials with less adsorption capacity. For instance, the introduction of amino groups into MOFs has been widely investigated for achieving selective CO2 adsorption [23,24].
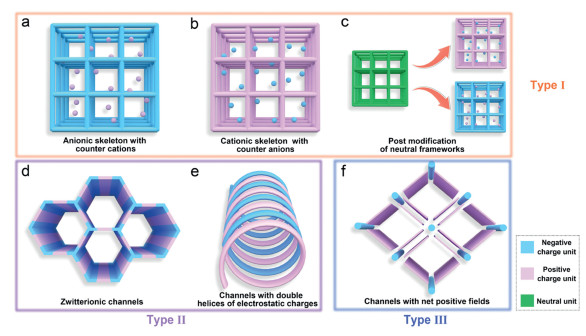
On the other hand, pore wall of high polarity could greatly affect the its adsorption and diffusion behaviors for adsorbates bearing different polarizability [25,26]. Typically, ionic porous frameworks containing negatively-charged or positively-charged skeleton possess high polarity on the pore wall, nevertheless, counter ions as compensated charges, will seriously change the polarity, depending on its distribution. As shown in Fig. 1, ionic porous frameworks are catalogued into different types according to the charge distribution, namely relative position of negative and positive motifs. The type Ⅰ is that the framework is charged and the counter ions are filled in the pores (Figs. 1a-c). Zeolite is a kind of typical charged porous framework materials. The presence of aluminum endows these frameworks with a net negative charge that is compensated by cations in the pores (Fig. 1a) [27]. The number, species and distribution of the cations surrounding the framework affect the electric field in the cavities of zeolites, and hence change the adsorption performance [28]. Reversely, cationic framework can be coupled with counter anions (Fig. 1b). MOFs comprising of positive-charged skeleton and counter anions such as ClO4−, BF4− and NO3−, registered higher C2H2 uptakes when competing with C2C1 gasses (C2H4, CO2 and CH4) [29]. For electroneutral frameworks bearing functional groups, it is possible to be ionized via a chemical reaction [30]. For example, amino-groups attached inside pores can be protonated, and be compensated with chloride ions in the form of an ionic porous framework, which is also catalogued into type Ⅰ (Fig. 1c) [31,32]. For type Ⅰ adsorbents, counterions have different effects on gas adsorption performance in various adsorbents. In case the adsorption site is on the charged skeleton, the counter ions will decrease the adsorption capacity as they occupy the pore volume [28]. On the contrary, if the adsorption site is the counter ion, the increase in the number of counter ions could lead to an increase in the adsorption amount [33]. However, more counter ions will also cause the free pore volume to decrease. The type Ⅱ ionic porous frameworks are charactered with zwitterionic channels assembled from regular and alternating arrangement of cationic and anionic building units or double helices with opposites charges (Figs. 1d and e). COFs with zwitterionic channels have been designed as attractive porous materials for SO2/CO2 separation (Fig. 1d) [34]. On the other hand, the framework with channels assembled from double helices with opposite charges can also be regarded as Type Ⅱ ionic porous frameworks owing to their regular and alternative arrangements and hence cancelation of electric fields (Fig. 1e). CPOS-5 (crystalline porous organosulfonate–amidinium salt-5) consisting of tetra-anionic and di-cationic motifs connected via hydrogen bonds, exhibits tailored sub-nanometer channels with double helices with opposite charges, and the structure governs the association and transport of CO2 molecules inside [35].
Figure 1
Figure 1.
Classification of ionic porous materials. Type Ⅰ, backbones are charged, and pores are filled with counterions. (a) Anionic skeleton with counter cations in the pores. (b) Cationic skeleton with counter anions and (c) electroneutral skeleton modified with organic salt. Type Ⅱ, Zwitterionic channels. Frameworks assembled from (d) regular and alternating arrangement of cationic and anionic building units and (e) double helices with opposites charges. Type Ⅲ, (f) the ionic porous frameworks with channels bearing net electric fields.
In spite of massive porous frameworks with charges on their channel surface as discussed above, ionic porous frameworks with net electric fields in the confined space have not been reported yet, to the best of our knowledge, as negative and positive charges usually appear in the form of ion pairs so that electric fields are canceled out. In order to build the net electric fields in confined pore/channel space, cationic and anionic components must follow the specific arrangement to make the empty space encircled by homoelectric charges as illustrated in Fig. 1f, a new type of ionic porous framework (Type Ⅲ).
The ionic building blocks can be assembled into highly ordered and porous frameworks by hydrogen bonds, such as HOFs [36,37]. We have previously proposed the concept of hydrogen-bonded ionic frameworks (HIFs) and reported the elasticity of the HIF sample [38]. HIF is assembled from organic and/or inorganic ions through hydrogen bonding and electrostatic interactions. It differs from ionic hydrogen-bonded organic frameworks (iHOFs) as it is limited to organic components [39]. In this work, we report an abnormal and unique adsorption phenomenon on the HIF of {(CN3H6)2[Ti(μ2O)(SO4)2]}n with one-dimensional (1D) channels assembled from 1D inorganic [Ti(μ2O)(SO4)2]n2n− chains and guanidium cations (Gua+) via hydrogen bonds and electrostatic interactions. The framework exhibited selective adsorption of argon (Ar) against CO2 and N2. The density functional theory (DFT) simulations revealed that HIF does not favor to adsorb CO2 because its adsorption free energy (ΔGad) is positive at the experimental temperature, while the large diffusion barrier at 77 K accounts for non-adsorption for N2. Combined with the detailed structure analyses, we find that the 1D channels are encircled by Gua+ ions with the unique positively-charged channels and thus net electric field in the confined space, which is responsible for the adsorption selectivity of HIF for Ar. The derived structure-property relationship could provide insights into the design of adsorbent materials for specific applications.
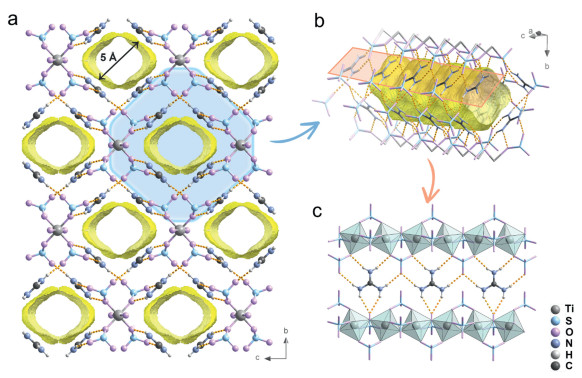
The HIF of {(CN3H6)2[Ti(μ2O)(SO4)2]}n was synthesized according to the reported procedures (see details in Supporting information) [38]. As shown in Fig. 2, the TiO6 octahedra are assembled into the 1D chain ([Ti(μ2O)(SO4)2]n2n−) along the crystallographic a-axis by sharing the oxygen sites. Each Gua+ cation forms six N—H···O hydrogen bonds with three SO42− groups from two 1D inorganic chains. And there are two bifurcated hydrogen bonds among these six hydrogen bonds. As a sequence, Gua+ cations and 1D anion chains are assembled into a framework via hydrogen bonds and electrostatic interaction with 1D channels (Fig. 2a). The Gua+ ions standing between 1D chains are arranged in sequence to form the ribbon structure, which constitutes the wall of the 1D channel (Figs. 2b and c). The cationic Gua+ ions and anionic 1D chains follows the specific arrangement and results in the channel being in a net positive charge field, which is different from the alternating or disordered distribution of cation and anion in ionic porous frameworks of Types Ⅰ and Ⅱ (Fig. 1). The channel diameter in HIF is measured to be ca. 5 Å and the porosity is calculated to be 21.7% (radius of the rolling sphere = 1.4 Å).
Figure 2
Figure 2.
The channel structure of {(CN3H6)2[Ti(μ2—O)(SO4)2]}n. (a) View of HIF along the a-axis. Space encircled by yellow represents empty channels (a rolling sphere of 1.4 Å was used). (b) The channel wall structure of HIF, Gua+ assembles into the channel wall (blue part in (a)). (c) The 1D chain structure of [Ti(μ2—O)(SO4)2]}n and arrangement of Gua+, which assembles into the channel wall (orange part in (b)). The dash line represents the hydrogen bonds.
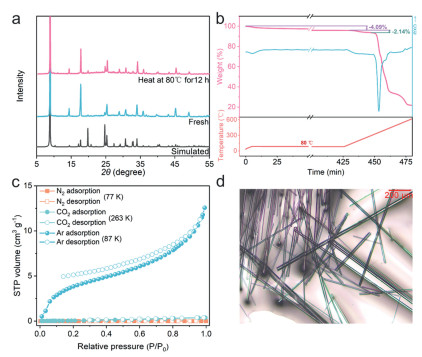
Powder X-ray diffraction (PXRD) analysis demonstrated a high degree of crystallinity and phase purity of the HIF, even after heating at 80 ℃ for 12 h (Fig. 3a). Thermogravimetric (TG) analysis of HIF showed an initial loss of about 4.09% before 80 ℃ owing to the loss of the accommodated solvent molecules (heating at 80 ℃ for 6 h). The remaining solvent molecules are removed (2.14%) as the temperature continues to rise, followed by framework decomposition after 300 ℃ (Fig. 3b). The solid-state 13C NMR (SSNMR) spectrum of HIF that after activation at 80 ℃ for 12 h revealed the solvent molecules in HIF channel are ethyl alcohol and diethyl ether. As shown in Fig. S1 (Supporting information), the peak at 158.64 ppm is assigned to carbocation in the guanidinium. The peaks at 66.21 and 14.93 ppm belong to ethyl alcohol. In addition, the peak at 58.54 and 17.15 ppm can attribute to diethyl ether, which is produced by dehydration of ethanol catalyzed by acid during HIF preparation. However, further heating to 120 ℃ leads to the samples partially changed, as verified by two new tiny peaks at 9.54° and 18.19° in PXRD pattern (Fig. S2 in Supporting information). X-ray photoelectron spectroscopy (XPS) spectra provide information on chemical composition of HIF. The results agree with the composition of {(CN3H6)2[Ti(μ2O)(SO4)2]}n (Fig. S3 in Supporting information).
Figure 3
Figure 3.
The stability and adsorption isotherms of the HIF. (a) Comparison of PXRD patterns of the simulated pattern from single-crystal structure determination, fresh HIF and samples after activation at 80 ℃ for 12 h. (b) Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) curves of HIF at a heating rate of 10 ℃/min. (c) N2 at 77 K, CO2 at 263 K and Ar at 87 K sorption isotherms for activated HIF. (d) Optical image of the HIF crystals.
To examine the porosity of HIF, we measured gas adsorption isotherms of N2 at 77 K and CO2 at 263 K after degassing the sample at 80 ℃ under vacuum for 12 h Comparison of TG curves of HIF before and after activation suggests that part of the solvent molecules in the channel are removed (Fig. S4 in Supporting information). N2 adsorption did not occur, while trace CO2 uptake could be ascribed to surface adsorption (Fig. 3c). The results seem to suggest that the framework is non-porous. In sharp contrast and unexpectedly, Ar sorption measurements at 87 K clearly show a S-shaped adsorption isotherm with a remarkable Ar uptake (Fig. 3c).
Based on the nature of adsorption isotherms, the International Union of Pure and Applied Chemistry (IUPAC) classifies adsorption behaviors into eight different types [40]. The Ar adsorption isotherm is similar to the curve of type Ⅱ, which is caused by the physisorption of most gasses on nonporous or macroporous adsorbents. However, there is a curious paradox since no adsorption was observed for N2 and CO2. Moreover, it is generally believed that the appearance of type Ⅱ isotherms is resulted from the adsorption in the macro-voids between adjacent small particles or the pileup of nanoplates in the samples. The samples used in sorption measurements are needle-like large crystals with diameters and length at micrometer and millimeter scale, respectively (Fig. 3d). Therefore, we exclude the possibility that the Ar uptake originates from the packing of sample particles. If so, the N2 and CO2 will give rise to the similar adsorption behaviors. All the dynamic molecular sizes of the three gas molecules are smaller than the window opening of the channel as listed in Table S1 (Supporting information), so that we could exclude the effect of aperture screening. The HIF structure has 1D channels with positively-charged pore walls encircled by Gua+, which is unusual in porous materials as discussed above. This special pore property may be responsible for the anomalous and unique adsorption phenomenon of HIF.
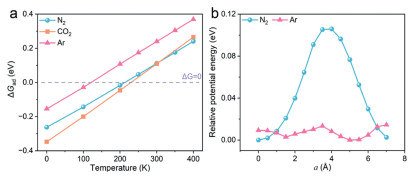
In order to understand the mechanism of abnormal adsorption and selectivity, we resort to the DFT simulations implemented in the Vienna Ab-initio Simulation Package (VASP) (see details in Supporting information). The binding energies between HIF structure and different gas molecules (N2, CO2, Ar) were calculated and then corresponding adsorption free energies (ΔGad) were estimated utilizing harmonic frequencies of adsorbates. The correlation between ΔGad and temperature is plotted in Fig. 4a. Although Ead for all gasses are negative, at experimental temperatures (N2-77 K, CO2-263 K, Ar-87 K), ΔGad for N2, Ar and CO2 are −0.17, −0.05 and 0.11 eV, respectively. It means that CO2 cannot be spontaneously adsorbed at 263 K and standard pressure. This explains why HIF exhibits almost no adsorption for CO2 in our experimental conditions. On the other hand, N2 adsorption at 77 K and Ar adsorption at 87 K seem thermodynamically favorable thanks to their negative ΔGad. Kinetically, however, to enable continuous adsorption of these gasses, the adsorbates necessarily diffuse along the 1D channel. To this end, the relative potential energy of Ar and N2 at varied depth when they move straightly in the channel from the surface adsorption site to the other end is displayed in Fig. 4b. It displays clear differences between Ar and N2, where the diffusion of Ar is almost barrierless, while N2 experiences a much larger energy variation. The free energy variations along this diffusion path at respective experimental temperatures of Ar and N2 are ~0.01 eV versus ~0.11 eV. It is obviously more difficult for N2 molecules to pass through the channel and continuously enter the adsorption site. Instead, Ar molecules is almost unimpeded during diffusion in the HIF channel and thus more easily fill the specific surface area of the porous framework. This qualitatively explains the selectivity for Ar adsorption.
Figure 4
Figure 4.
DFT calculations. (a) ΔGad and temperature for N2, CO2 and Ar. (b) Relative potential energy of species in HIF when they traverse the channel along a direction with other coordinates fixed at their most stable adsorption geometries, noted that a = 0 is defined as the center of mass of the molecule being at the same plane with one pair Gua+ in the (b × c) plane.
The interaction between gas molecules and HIF arises from both dispersion forces and Coulomb repulsions. The repulsive force increases when the gas molecule gets close to Gua+, because Gua+ features a strong positive charge (+0.86 e). In this regard, N2 is more significantly influenced by the presence of a positive charge due to its larger polarizable size and tends to apart from Gua+. Since Gua+ appear in staggered positions in the framework (Fig. S5 in Supporting information), it is difficult for N2 to find a facile diffusion path that is not too close to Gua+ from an adsorption site to the other. By contrast, Ar in the HIF channel feels a much weaker repulsion by Gua+, which experiences more gentle energy variation and more flexible diffusion route.
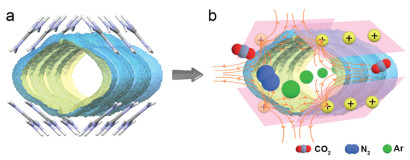
The theoretical results suggest that the selective adsorption of HIF to Ar results from the low polarizability of Ar molecules which are less influenced by the positive electric field in the channel. As conceptually shown in Fig. 5a, the 1D channel is surrounded by four ribbons made up of Gua+ with a positive charge and it seems like a bunch of point charges around the channel (Fig. 5). In turn, it can be imaged the positive electric field pointing towards the center of the channel (Fig. 5b), so that the gas molecules are exposed to a positive electric field when approaching or enter the channel. For molecules with varied polarizabilities, they experience different repulsive forces during the diffusion in the channel, resulting in HIF adsorption selectivity. In the helical track of CPOS-5, specific host-guest interactions are responsible for a unique screwing dynamic of CO2 traveling along ultra-micropores. In contrast to CPOS-5, HIF exhibits a net positive electric field inside the channel. The large diffusion barrier for N2 at 77 K is caused by the net positive electric field.
Figure 5
Figure 5.
The positive electric field in HIF. (a) The channel encircled by Gua+ are simplified as positive point charges with the positive electric field indicating using yellow arrow lines (b). In the channel, CO2 molecules are not adsorbable owing to the positive adsorption energy, N2 molecules can be adsorbed but not be able to diffuse owing to the positive energy of diffusion barrier, and Ar molecules are allowed for adsorption and subsequent diffuses.
In conclusion, a unique adsorption phenomenon was observed for HIF of (CN3H6)2n[Ti(μ2O)(SO4)2]n, which exhibits a negligible N2 and CO2 adsorption but show a remarkable uptake for Ar. The density functional theory simulations indicated that the adsorption free energy is positive at 263 K for CO2. In addition, it is difficult for N2 to diffuse in the channel because of the relatively large barrier. The unusual selective adsorption for Ar is related to the lower polarizability and special channel property in HIF structure with positive charge field. This study helps us understand the relationship between pore/channel structure and adsorption property and provides a new strategy for building charged porous materials towards specific selective adsorption. Inspired by HIF, using the positive electric field within the framework channel to control gas molecules' diffusion in the channel to separate gas molecules with different polarizabilities is a feasible strategy. Cationic building blocks of various sizes, charges, and shapes can be selected to control the channel diameter, shape, and charge distribution on the channel wall of HIF. Additionally, the framework with a negative electric field in the channel may exhibit incredible adsorption properties. Moreover, tuning the charge distribution and the subsequent electric field inside ionic porous materials might be important for catalytic application as the electric field could polarize the reagent molecules in the confined space and thus promote the catalytic reactions.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Congyan Liu: Data curation, Writing – original draft. Xueyao Zhou: Methodology, Writing – original draft. Fei Ye: Validation. Bin Jiang: Writing – review & editing. Bo Liu: Writing – review & editing.
Acknowledgments
We acknowledge support from the Chinese Academy of Sciences and University of Science and Technology of China, National Key Research and Development Program of China (No. 2021YFA1500402), National Natural Science Foundation of China (Nos. 21571167, 51502282 and 22075266), Fundamental Research Funds for the Central Universities (Nos. WK2060190053 and WK2060190100).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109969.
M. Thommes, K. Kaneko, A.V. Neimark, et al., Pure Appl. Chem. 87 (2015) 1051–1069. doi: 10.1515/pac-2014-1117
Figure 1
Classification of ionic porous materials. Type Ⅰ, backbones are charged, and pores are filled with counterions. (a) Anionic skeleton with counter cations in the pores. (b) Cationic skeleton with counter anions and (c) electroneutral skeleton modified with organic salt. Type Ⅱ, Zwitterionic channels. Frameworks assembled from (d) regular and alternating arrangement of cationic and anionic building units and (e) double helices with opposites charges. Type Ⅲ, (f) the ionic porous frameworks with channels bearing net electric fields.
Figure 2
The channel structure of {(CN3H6)2[Ti(μ2—O)(SO4)2]}n. (a) View of HIF along the a-axis. Space encircled by yellow represents empty channels (a rolling sphere of 1.4 Å was used). (b) The channel wall structure of HIF, Gua+ assembles into the channel wall (blue part in (a)). (c) The 1D chain structure of [Ti(μ2—O)(SO4)2]}n and arrangement of Gua+, which assembles into the channel wall (orange part in (b)). The dash line represents the hydrogen bonds.
Figure 3
The stability and adsorption isotherms of the HIF. (a) Comparison of PXRD patterns of the simulated pattern from single-crystal structure determination, fresh HIF and samples after activation at 80 ℃ for 12 h. (b) Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) curves of HIF at a heating rate of 10 ℃/min. (c) N2 at 77 K, CO2 at 263 K and Ar at 87 K sorption isotherms for activated HIF. (d) Optical image of the HIF crystals.
Figure 4
DFT calculations. (a) ΔGad and temperature for N2, CO2 and Ar. (b) Relative potential energy of species in HIF when they traverse the channel along a direction with other coordinates fixed at their most stable adsorption geometries, noted that a = 0 is defined as the center of mass of the molecule being at the same plane with one pair Gua+ in the (b × c) plane.
Figure 5
The positive electric field in HIF. (a) The channel encircled by Gua+ are simplified as positive point charges with the positive electric field indicating using yellow arrow lines (b). In the channel, CO2 molecules are not adsorbable owing to the positive adsorption energy, N2 molecules can be adsorbed but not be able to diffuse owing to the positive energy of diffusion barrier, and Ar molecules are allowed for adsorption and subsequent diffuses.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
