Citation:
Liangliang Jia, Ye Hong, Xinyu He, Ying Zhou, Liujiao Ren, Hongjun Du, Bin Zhao, Bin Qin, Zhe Yang, Di Gao. Fighting hypoxia to improve photodynamic therapy-driven immunotherapy: Alleviating, exploiting and disregarding[J]. Chinese Chemical Letters,
2025, 36(2): 109957.
doi:
10.1016/j.cclet.2024.109957
Fighting hypoxia to improve photodynamic therapy-driven immunotherapy: Alleviating, exploiting and disregarding
English
Fighting hypoxia to improve photodynamic therapy-driven immunotherapy: Alleviating, exploiting and disregarding
The Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology, Xi’an Jiaotong University, Xi’an 710049, China
b.
Center of Digestive Endoscopy, Shaanxi Provincial Cancer Hospital, Xi’an 710061, China
c.
Research Institute of Xi’an Jiaotong University, Hangzhou 311200, China
d.
Department of Gastroenterology, Second Affiliated Hospital, Xi’an Jiaotong University, Xi’an 710004, China
e.
Department of Ophthalmology, Xijing Hospital, Xi’an 710032, China
f.
Department of Epidemiology, Shaanxi Provincial Cancer Hospital, Xi’an 710061, China
gaodi422@xjtu.edu.cn (D. Gao). 1 These authors contributed equally to this work.
Received Date:
28 February 2024 Accepted Date:
30 April 2024 Revised Date:
28 April 2024 Available Online:
15 February 2025
Abstract:
Innovative anti-cancer therapies that activate the immune system show promise in combating cancers resistant to conventional treatments. Photodynamic therapy (PDT) is one such treatment, which not only directly eliminates tumor cells but also functions as an in situ tumor vaccine by enhancing tumor immunogenicity and triggering anti-tumor immune responses through immunogenic cell death (ICD). However, the effectiveness of PDT in enhancing immune responses is influenced by factors, such as photosensitizers and the tumor microenvironment, particularly hypoxia. Current clinically used PDT heavily relies on oxygen (O2) availability and can be limited by tumor hypoxia. Additionally, the tumor immunosuppressive microenvironment induced by hypoxia affects the anti-tumor immunity of tumor-infiltrating effector T cells. Meanwhile, the immunosuppressive myeloid-lineage cells are recruited to the hypoxic tumor tissue and exhibit higher immunosuppressive capabilities under hypoxia conditions. Consequently, numerous strategies have been developed to modulate tumor hypoxia or to create hypoxia-compatible PDT, aiming to reduce the effects of tumor hypoxia on PDT-driven immunotherapy. This review investigates these strategies, including approaches to alleviate, exploit, and disregard tumor hypoxia within the context of PDT/immunotherapy. It also emphasizes the role of advanced nanomedicine and its benefits in these strategies, while outlining current challenges and future prospects in the field.
Photodynamic therapy (PDT) is an innovative medical treatment that utilizes light to activate photosensitizers (PSs), leading to the production of highly toxic reactive oxygen species (ROS) that cause damage to target cells [1,2]. Owing to its minimally invasive characteristic, precise targeting, low systemic toxicity, and potential for repeated administration, PDT has found extensive application in diverse medical fields, including dermatology [3], ophthalmology [4], dentistry [5], vascular diseases [6], and oncology [7–9]. In the field of cancer treatment, PDT is utilized not only for superficial tumors (e.g., basal cell carcinoma and squamous cell carcinoma), but also employs endoscopic examination or intervention techniques to directly deliver optical fibers to tumor sites for treating other types of cancer such as esophagus cancer [10], biliary duct cancer [11], breast cancer [12]. This approach enables precise tumor localization, neglects drug resistance, reduces off-target toxicity to surrounding tissues, and mitigates the pain associated with traditional surgery for cancer patients. Consequently, PDT has emerged as a highly promising method for cancer treatment.
In recent years, evidence has emerged indicating that PDT not only directly eliminates tumor cells and damages microvascular endothelium, but also acts as an in situ tumor vaccine by enhancing tumor immunogenicity and triggering anti-tumor immune responses [13]. This process occurs through the induction of effective immunogenic cell death (ICD) in tumor cells, leading to the exposure or release of tumor-associated antigens (TAAs) and damage-associated molecular patterns (DAMPs) such as adenosine-triphosphate (ATP), calreticulin (CRT), high mobility group protein B1 (HMGB1), and heat shock protein (HSP). DAMPs can facilitate the maturation of antigen-presenting cells, including dendritic cells (DCs), induce the activation of antigen-specific T cells, promote the infiltration of immune cells into the tumor, and thus enhance the immune response within solid tumors [14–16]. Furthermore, mature DCs release cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α), which contribute to the proliferation and activation of natural killer (NK) cells and T cells, ultimately promoting adaptive immune responses [17]. Actually, not all PDT process are capable of inducing ICD. The effectiveness of these therapies relies on various factors related to the PSs, such as its intracellular sub-localization, photodynamic activity, dosage, and light conditions (e.g., light power density and light illumination duration) [18,19]. Additionally, the unfavorable tumor microenvironment (TME), characterized by features like hypoxia, significantly influences the efficacy of PDT-induced ICD effect and its impact on the anti-tumor immune response [20–22].
Currently, the clinically used PDT is mainly type Ⅱ PDT [23]. In this process, the excited PSs in the triplet state preferentially transfer their energy to adjacent oxygen molecules (O2), resulting in the production of highly toxic singlet oxygen (1O2). This leads to a significant cytotoxic effect, as specific mechanisms for PDT are detailed in Section 2. Therefore, type Ⅱ PDT is critically dependent on O2 availability [24]. However, the rapid proliferation of tumor cells and inadequate blood supply within the tumor often result in low O2 concentration, leading to hypoxia in the internal regions of solid tumors. Furthermore, the ROS generated by PDT can impact the tumor microvessels, causing damage to the endothelial cells and resulting in microvessel constriction and thrombosis. Consequently, this deprives the tumor cells of O2 and nutrients, rapidly inducing ischemic cell death while aggravating O2 consumption [25,26]. Ultimately, this exacerbates tumor hypoxia, hindering the production of ROS by the photosensitizer (PS) upon light irradiation and limiting the effectiveness of PDT-induced ICD.
Furthermore, the tumor immunosuppressive microenvironment, directly or indirectly induced by hypoxia [27,28], further influences the survival of tumor-infiltrating effector T cells and the exertion of anti-tumor immune effects, thereby impacting the efficacy of PDT-driven immunotherapy [29]. For example, hypoxic tumor cells tend to metabolize glucose through the glycolytic pathway, resulting in the accumulation of lactate, leading to decreased pH within the tumor tissue and the creation of an acidic environment [30]. This acidic environment alters the metabolism of tumor cells in turn, enhancing their invasive and migratory properties [31]. Moreover, the acidic TME impedes T cell expansion within the tissue and hinders cytotoxic effects [32]. Additionally, immunosuppressive myeloid-lineage cells (tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs)) are recruited to the hypoxic tumor tissue to a greater extent and undergo expansion [33]. Moreover, their phenotype is programmed with higher immunosuppressive capabilities under hypoxia condition [34]. The findings demonstrate that tumor hypoxia not only hampers the successful induction of ICD by PDT, but also diminishes the efficacy of PDT-driven immunotherapy by promoting an immunosuppressive microenvironment within the tumor. Hence, the development of regulatory strategies to tackle tumor hypoxia is crucial for improving treatment outcomes.
Moreover, the groundbreaking advancements in immunotherapy have led to the establishment of several clinically effective cancer immunotherapy strategies, encompassing immune checkpoint inhibition [35,36], vaccination [37–39], and adoptive T cell transfer, which includes chimeric antigen receptor (CAR)-engineered [40] and T cell receptor (TCR)-engineered T cell therapies [41]. These strategies not only impede the growth of primary tumors but also have the potential to impede tumor metastasis and recurrence through the patient's innate immune system, demonstrating significant utility in clinical practice. The integration of PDT with the aforementioned immunotherapeutic approaches as part of combination therapy can further disrupt immune suppression, reprogram the TME, and amplify the therapeutic effects of PDT-based cancer treatment [42].
In this review, we will examine recent advancements in strategies aimed at addressing tumor hypoxia in the context of PDT and PDT-driven immunotherapy (Fig. 1 and Table S1 in Supporting information). We will approach this examination from three angles: alleviating, exploiting, and disregarding tumor hypoxia, respectively. The content encompasses the optimization of the structure of PSs or the development of comprehensive strategies for regulating hypoxia. Furthermore, due to the important and unique advantages demonstrated by advanced nanomedicine in the aforementioned strategies, such as achieving targeted drug delivery and controlled release [43], improving drug bioavailability [44], and providing a multi-drug nanoplatform [45,46], this review will also focus on PDT-driven immunotherapy against hypoxic tumors mediated by nanomedicine. Lastly, we will summarize the current challenges and future prospects of photodynamic immunotherapy, including further clinical development of nanomedicine in this filed.
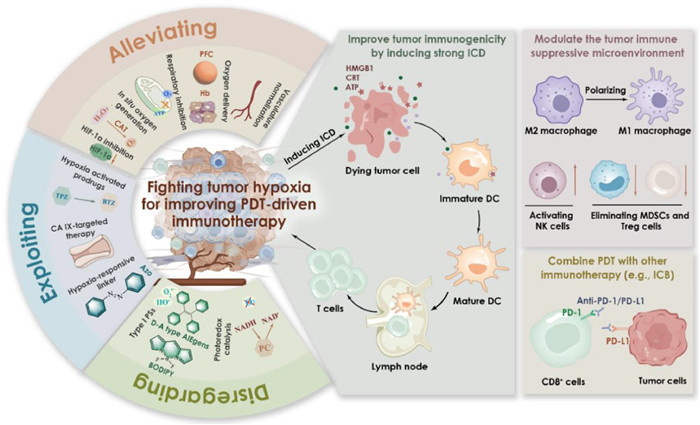
Figure 1
Figure 1.
Schematic illustration of strategies for fighting tumor hypoxia to improve PDT-driven immunotherapy. These strategies can be categorized into three sections: (Ⅰ) direct or indirect alleviation of tumor hypoxia, (Ⅱ) exploitation of tumor hypoxia, and (Ⅲ) disregard of tumor hypoxia. Fighting tumor hypoxia can amplify the therapeutic effects of PDT, and thus improving tumor immunogenicity by inducing strong ICD. The effectiveness of this process can be enhanced by modulating the tumor immunosuppressive microenvironment and combining PDT with other immunotherapies.
PDT is commonly classified into two types, known as type Ⅰ and type Ⅱ, based on their different photophysical and photochemical mechanisms [47]. Put simply, PSs in their ground singlet state (S0) can be excited to the unstable excited singlet state (Sn) when subjected to light irradiation. Subsequently, they transition to the relatively stable excited triplet state (Tn) through intersystem crossing (ISC). Following this, they participate in electron transfer or energy transfer with surrounding substrates or O2, thereby generating ROS through type Ⅰ and/or type Ⅱ mechanisms (Fig. S1 in Supporting information) [48]. In more common type Ⅱ PDT, a PS in the Tn state (3PS*) transfers energy to the triplet state of 3O2, directly yielding highly cytotoxic 1O2 [49]. On the other hand, in type Ⅰ PDT, 3PS* initially interacts with intracellular substrates (such as reduced coenzymes, amino acids, vitamins, and nitrogenous bases) via electron transfer. This interaction subsequently triggers a cascade of reactions that culminate in the generation of various ROS [50]. Notably, these reactions serve to generate additional O2, effectively recycling O2 during PDT treatment, ultimately leading to low O2-dependent PDT and enhancing the therapeutic efficacy of PDT against hypoxic tumors.
3.
Alleviating tumor hypoxia
Elevating the O2 levels in the TME could potentially reduce tumor hypoxia and enhance the effectiveness of PDT [51]. In addition, hindering the hypoxia-inducible factor-1α (HIF-1α) signaling pathway and disrupting tumor cell respiration to indirectly counteract the impact of tumor hypoxia on cancer treatment may also enhance the synergistic therapeutic outcomes [52,53]. Several review articles have thoroughly introduced various strategies for alleviating hypoxia in tumors [24,54,55]. This article will commence with recent relevant research and provide a comprehensive review, with a heightened focus on the utilization of tumor oxygenation strategies within the research field of photodynamic immunotherapy.
The poor solubility of O2 in the blood makes direct intravenous O2 delivery unfeasible, necessitating alternative strategies to augment tumor oxygenation [56,57]. On one hand, normalizing the tumor vasculature can enhance blood flow to facilitate tumor oxygenation [58,59]. On the other hand, using external materials with high O2 affinity can enable targeted O2 delivery to the tumor or initiate in situ O2 generation through chemical reactions, thereby ameliorating the hypoxic TME [60–62]. Furthermore, the rapid advancement of nanotechnology has positioned nanoplatforms as supportive tools for the previously mentioned strategies. The following sections will elaborate on these strategies in detail.
3.1.1
Normalizing the tumor vasculature
Tumor tissues upregulate the expression of erythropoietin (EPO) and vascular endothelial growth factor (VEGF) to facilitate neovascularization in response to hypoxia, thereby promoting the proliferation of endothelial cells [63]. Consequently, this process culminates in the formation of disorganized and dysfunctional tumor vasculature, exacerbating the hypoxic conditions. Hence, medications that either anti-VEGF antibodies [64] or VEGF receptors-targeted inhibitors with anti-angiogenic properties, such as small molecule tyrosine kinase inhibitors (e.g., regorafenib (Reg) [65], sunitinib (SU) [66], and erlotinib [67]), can normalize the structure and function of blood vessels to alleviate tumor hypoxia. However, long-term clinical use of high-dose aforementioned drugs can lead to many adverse reactions (e.g., emesis, diarrhea and hand-foot skin reaction) and increase the risk of cardiovascular diseases [68]. In response to these issues, researchers have developed multifunctional nano-drug delivery systems to effectively load drugs, enabling targeted delivery to tumor tissues [69–71]. Moreover, in studies related to PDT, vascular normalization additionally enhances the O2 levels in tumor tissues, ultimately improving the therapeutic outcomes of PDT [72]. This strategy has also been applied in X-ray-excited PDT (XPDT) recently, an innovative PDT-based treatment developed to overcome the limitation of light penetration depth in biological tissues. Unlike traditional PDT, XPDT involves the use of a scintillator to convert X-rays into ultraviolet or visible light, which then activates PSs to generate ROS. Jiang and co-workers developed a nanoplatform named CCT-DPRS, featuring a dual-core-satellite structure, to allow the separate and sequential loading of a nanoscintillator (CaF2), the PS Rose Bengal (RB), and the anti-angiogenic tyrosine kinase inhibitor SU. The results demonstrated that the combination of XPDT with antiangiogenic drugs resulted in significant tumor regression, surpassing the efficacy of XPDT alone [73].
In the context of immunotherapy, anti-angiogenesis via inhibiting the VEGF/VEGFR pathway has the potential to promote the maturation of DCs and facilitate the presentation of antigens, as well as the immune response of T cells. Furthermore, anti-angiogenesis can normalize tumor blood vessels, augmenting the infiltration of T cells into the tumor, while diminishing the presence and activity of immune suppressor cells (e.g., MDSCs, TAMs, regulatory T cells (Tregs)), thereby promoting the transition of TME from an immunosuppressive state to an immune-active state [74]. A preclinical study has demonstrated that low-dose VEGF-targeting drugs, such as Reg, have the capability to improve the immunosuppressive TME [75]. Therefore, it is evident that the concurrent administration of antiangiogenic drugs and PDT may further enhance the efficacy of photodynamic immunotherapy. Wan and co-workers developed a nanomedicine referred to as NP-PDT@Reg. This formulation utilized a ROS-responsive diselenide-containing polymer as a carrier to encapsulate the degradable pseudo-conjugate polymer-based PS PSPbodipy and Reg. Under 808 nm laser irradiation, NP-PDT@Reg demonstrated the ability to produce abundant ROS and effectively initiate the release of Reg for alleviating tumor hypoxia, thus augmenting the efficacy of PDT and inducing ICD to stimulate anti-tumor immune responses (Fig. S2A in Supporting information). More importantly, Reg was found to have the capability to reprogram TAMs from a pro-tumor M2 phenotype to a tumor-killing M1 phenotype, consequently reversing the immunosuppressive TME and enhancing the potential of photodynamic immunotherapy (Fig. S2B in Supporting information) [23].
Mild hyperthermia (≤45 ℃) also offers an additional effective approach for promoting vascular normalization. Elevated solid stress within solid tumors, caused by activation of cancer-associated fibroblasts (CAFs) and increased extracellular matrix (ECM), can diminish blood flow perfusion, reduce external O2 supply, and exacerbate tumor hypoxia [63]. Hyperthermia has the potential to address these issues by depleting tumor CAFs and degrading ECM, thus facilitating the normalization of tumor blood vessels [76]. It is essential to consider that the treatment parameters of thermal therapy, including light fluence and fluence rate, can significantly impact the treatment temperature, which plays a crucial role in determining the vascular effects of thermal therapy. Coagulative thermal therapy (50–100 ℃) may lead to vascular collapse, consequently reducing tumor oxygenation and diminishing the effectiveness of PDT. In contrast, subcoagulative thermal therapy doses or mild hyperthermia can enhance tumor oxygenation [77]. Therefore, precise temperature control during thermal therapy is paramount for practical applications.
Additionally, dexamethasone (DXM), a corticosteroid frequently used in clinical practice, demonstrates the capability to normalize vessels and the tumor ECM, consequently lowering interstitial fluid pressure, tissue rigidity, and solid stress to enhance vascular permeability and alleviate tumor hypoxia [78,79]. Furthermore, low-dose DXM can effectively decrease the expression of programmed death ligand 1 (PD-L1) and indoleamine 2,3-dioxygenase 1 (IDO1) in tumor cells, thus inhibiting tumor immune evasion [80]. Li and co-workers pre-treated 4T1 tumor-bearing mice with DXM to enhance tumor micro-vessel density, a process referred to as “delivery unlocking”. This aimed to restore O2 supply and improve the delivery efficiency of ZnPc@FOM-Pt, a hybrid nanoparticle for PDT and Pt-mediated catalysis of tumor-overexpressed H2O2 to generate O2 (Fig. S2C in Supporting information). Immunofluorescence imaging revealed increased expression of CD31 and α-SMA, as well as decreased levels of HIF-1α in 4T1 tumors from the DXM group compared to the PBS group, revealing the alleviation of tumor hypoxia by DXM. Therapeutic outcomes also showed increased levels of cytotoxic T lymphocyte infiltration and extended survival rate in the group treated with ZnPc@FOM-Pt in combination with anti-PD-L1 therapy and laser irradiation (Figs. S2D and E in Supporting information) [81].
3.1.2
Delivering exogenous oxygen
Hyperbaric oxygen (HBO) therapy has emerged as a promising approach to address tumor hypoxia and the immunosuppressive microenvironment by directly delivering exogenous O2 to solid tumors [82,83]. Inspired by this, the development of effective, safe, and convenient O2 delivery strategies is vital in this context. Researchers are currently exploring the use of nanotechnology to create a variety of nano-O2 carriers to dissolve O2 in O2-rich environments and, upon reaching the tumor site, release O2 using specific mechanisms in hypoxic conditions, thereby facilitating tumor oxygenation [84–86].
Hemoglobin (Hb) molecules comprise four heme groups, each containing a Fe2+ capable of binding to an O2 [87]. Although Hb exhibits efficient O2-carrying and releasing capabilities, free Hb displays poor stability and has a shorter half-life, rendering it an inadequate option as an O2 carrier for tumors. Moreover, the oxidation of the Fe2+ in heme leads to a significant decrease in Hb's O2-carrying capacity [88]. Current studies focus on the use of carriers such as polymers [89], liposomes [90], albumin [91], or red blood cells (RBCs) [92] to protectively deliver Hb to tumor tissues, ultimately enhancing the effectiveness of O2-dependent PDT. Recently, researchers have employed hybrid proteins to develop a new type of Hb delivery system, resulting in enhanced stability and tumor-targeting capabilities for Hb. Wu and co-workers developed the nanoplatform ODP-TH, co-loaded with doxorubicin (Dox) and protoporphyrin Ⅸ (PpⅨ). It employed a hybrid protein, transferrin (TFR)-Hb, linked by disulfide bonds, as a carrier for improved O2 storage and controlled release. Additionally, the nanoplatform crossed the blood-brain barrier (BBB) via TFR receptor-mediated transport, enabling O2 delivery to alleviate tumor hypoxia [93]. In the context of immunoregulation, Chen and co-workers developed a hybrid protein O2 nanocarrier, denoted as C@HPOC, by combining human serum albumin (HSA) and Hb through disulfide bonds and loading it with Ce6. Apart from its favorable biosafety profile and O2 delivery capacity, C@HPOC exhibited the ability to augment the ICD effect triggered by PDT by mitigating tumor hypoxia [94].
In addition to Hb with inherent O2 delivery capacity, perfluorocarbons (PFCs), with their carbon chain skeleton fully fluorinated, also demonstrate a strong affinity for O2. It has been demonstrated that 100 mL of PFC solution can dissolve 40–50 mL of O2, equivalent to the O2 content in 200 mL of blood [95]. The outstanding O2-carrying capacity and biocompatibility of PFCs make them widely employed as O2 carriers. However, owing to their poor solubility in water, PFCs require modification through liposomes, polymers, or proteins [96–98]. Fluorocarbon chains, similar to PFCs droplets, also exhibit excellent O2-carrying capability, which have been introduced into nanocarrier materials for assisting PDT-driven immunotherapy [99]. In a recent work, Zhang and co-workers incorporated fluorocarbon chains and PS into the same polymer backbone to develop a fluorinated polysensitizer, named PolyFBODIPY, which can self-assemble into nanoparticles NP@PolyFBODIPY. This nanophotosensitizer achieves O2 delivery, efficient ROS generation, and ROS-responsive degradation (Fig. S3A in Supporting information). Moreover, the infiltration of immune-active cells was elevated in the oxygenated PDT group (Figs. S3B and C in Supporting information). Furthermore, the metabolism of lipids, amino acids, and D-glutamine was significantly influenced under a strong oxidizing condition, thereby facilitating autophagy and apoptosis of cancer cells (Figs. S3D and E in Supporting information) [100].
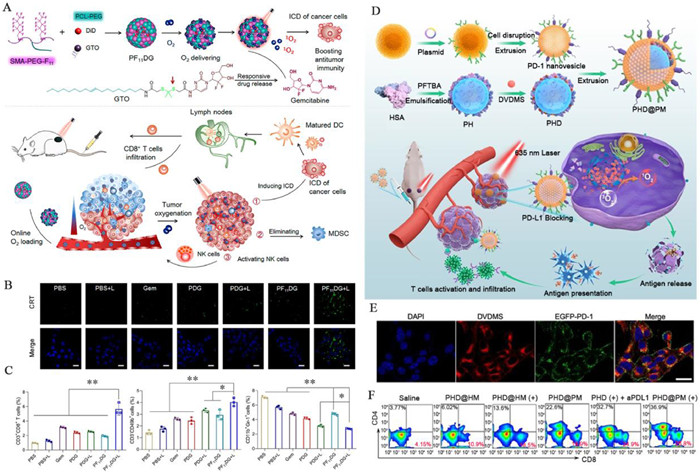
In addition to the aforementioned strategy, incorporating immunomodulatory drugs may improve the immunosuppressive TME and enhance the anti-tumor immune effects of PDT. Wang and co-workers developed PF11DG, a PFC-modified nanoparticle that co-loaded the DiD (as PS) and the chemo-immunomodulatory agent, gemcitabine prodrug. Gemcitabine, capable of generating ROS, synergized with PDT to promote ICD (Figs. 2A and B). Moreover, gemcitabine activated NK cells and diminished immunosuppressive MDSCs, thereby bolstering the anti-tumor immune response. The results demonstrated a 1.8-fold increase in the frequency of NK cells in tumors treated with PF11DG and laser irradiation compared to those treated with PBS, while the frequency of MDSCs decreased by 61.09% (Fig. 2C) [101]. Furthermore, the PDT-induced ICD effect can be utilized as an in situ vaccine in combination with anti-programmed cell death protein-1 (PD-1)/PD-L1 therapy, aiming to enhance the response rate of PD-1/PD-L1 antibodies. Simultaneously, the co-administration of exogenous O2 to mitigate tumor hypoxia can improve the cooperative efficacy of PDT and anti-PD-1/PD-L1 therapy. Zhang and co-workers developed a biomimetic nanoliposome called PHD@PM, which was camouflaged with cell membranes expressing PD-1 and contained the DVDMS (as PS) and an HSA-binding perfluorotributylamine nanoemulsion (PHD). PHD facilitated the delivery of exogenous O2, thereby enhancing anti-tumor PDT and inducing efficient ICD. Additionally, the PD-1 proteins on the external cell membrane layer of PHD@PM exerted an anti-PD-1 effect, further enhancing the effectiveness of immunotherapy via immune checkpoint blockade (ICB) (Figs. 2D–F) [102]. It is worth noting that the aforementioned O2-carrying substances (e.g., Hb or PFCs) can only carry a single dose of O2, making it challenging to effectively address long-term tumor hypoxia. Furthermore, achieving controlled release of O2 presents a significant challenge.
Figure 2
Figure 2.
The co-administration of immunomodulatory drugs or anti-PD-1/PD-L1 therapy enhances the anti-tumor immune effects of PDT. (A) Schematic illustration of preparation of PF11DG and antitumor immune responses elicited by PF11DG. (B) CLSM images of CRT exposed on cell surface. Scale bar: 25 µm. (C) Frequency of CD3+CD8+ T cells, NK cells and MDSCs in tumors of different treatment groups. Reproduced with permission [101]. Copyright 2021, American Chemical Society. (D) Schematic illustration of preparation of PHD@PM and PHD@PM-based photodynamic immunotherapy. (E) CLSM images of 4T1 cells after a 2 h-incubation with PHD@PM. Scale bar: 10 µm. (F) The flow cytometry plots of the percentages of tumor-infiltrating CD4+ and CD8+ T cells in tumors of different treatment groups. Reproduced with permission [102]. Copyright 2021, Wiley Publishing Group.
The in situ generation of O2 within tumors represents a significant strategy for alleviating tumor hypoxia. Tumor tissues exhibit elevated levels of H2O2 relative to normal tissues. The concentration of H2O2 in normal tissues is around 0.02 µmol/L, while it is up to 100 µmol/L in tumor tissues [103]. Converting H2O2 into O2 can effectively facilitate in situ O2 production. Catalase (CAT) is a ubiquitous enzyme, which facilitates the catalysis of H2O2 into water and O2. However, CAT is susceptible to physiological conditions and prone to deactivation and degradation [104]. Recently, researchers have devoted themselves to developing various metal and carbon-based nanoenzymes to mimic the enzymatic activity of CAT in generating O2in situ, thus mitigating tumor hypoxia [105–107]. To further increase H2O2 levels within tumors, researchers are endeavoring to increase H2O2 levels within tumors by administering exogenous H2O2 [108], reducing H2O2 elimination [109], or enhancing H2O2 generation [110]. Luo and co-workers developed MnZ, a hybrid material comprising Mn-doped carbon dots (Mn-CDs) as the core and zeolitic imidazolate framework-8 (ZIF-8) as the shell. They further functionalized MnZ by anchoring ultrasmall gold nanoparticles (AuNPs) onto its surface via ion exchange and in-situ reduction techniques, leading to the formation of the nanozyme MnZ@Au (Figs. S4A and B in Supporting information). MnZ@Au acted as a catalyst, initially converting glucose into gluconic acid and H2O2 through AuNPs activity resembling glucose oxidase (GOx), followed by Mn-CDs-mediated catalysis to produce O2 akin to CAT [111]. Even so, nanoenzymes still exhibit certain limitations, such as the time-consuming and energy-intensive processes involved in their synthesis, purification, and storage before administration [112].
In order to further simplify the construction and management process of the O2 generator, researchers have recently turned their attention to potassium permanganate (KMnO4), a cost-effective, commercially available, stable, and clinically utilized chemical reagent. Pan and co-workers proposed an innovative approach of directly injecting KMnO4 into tumors to create an in situ and minimalist O2 generator. The primary oxidation product of KMnO4 is manganese dioxide (MnO2) instead of Mn2+, attributed to the limited presence of H2O2 and H+ in TME. MnO2 then facilitates the decomposition of H2O2 to generate O2, thereby mitigating PDT resistance caused by tumor hypoxia. Furthermore, KMnO4 possesses inherent strong oxidizing capabilities, enabling it to directly eliminate tumor cells. Moreover, a histopathological examination of major organs uncovered no visible organ lesions or irregularities during treatment, indicating that the developed KMnO4Ce6 system is a safe and efficacious approach for tumor treatment [113]. In another work, Wang and co-workers developed a three-in-one oncolytic adenovirus system p-Adv-CAT-KR. This system utilized KillerRed as a genetically-engineered PS, CAT as an in situ O2 supply medium, and adenovirus as a bio-renewable immune-stimulating carrier. In tumor cells, the virus successfully replicated, leading to the generation of more viral progeny and ultimately inducing strong ICD (Fig. S4C in Supporting information). This system concurrently initiated and amplified immune responses, resulting in an increase in antigen-presenting cells and enhanced T-cell infiltration (Figs. S4D–F in Supporting information) [114]. Tumor in situ O2 production can be achieved not only by converting H2O2 to O2 but also through other exogenous chemical substances (e.g., H2O [115], CaO2 [116]) or photo-controlled photosynthesis [117]. Qiao and co-workers delivered RBC membrane (RBCM)-engineered algae to hypoxic tumor regions, aiming to increase local O2 levels through microalgae-mediated photosynthesis [118]. This in situ O2 generation strategy presented here opens up a new avenue for anti-tumor PDT-immunotherapy.
3.2
Indirectly ameliorating tumor hypoxia
Clinical trials have demonstrated that the O2-rich environment paradoxically fosters tumor growth and metastasis [119]. Thus, precise regulation of tissue O2 levels is imperative for directly tumor oxygenation. Alternatively, researchers have developed indirect approaches to alleviate tumor hypoxia, including inhibiting tumor cell respiration and suppressing the HIF-1α signaling pathway.
3.2.1
Decreasing oxygen consumption
One of the primary causes of tumor hypoxia is the substantial physiological O2 consumption resulting from the cellular mitochondrial-associated oxidative phosphorylation (OXPHOS) [120]. Thus, blocking OXPHOS to reduce endogenous O2 consumption can be an effective approach to alleviate tumor hypoxia. Inhibitors of OXPHOS can be classified into three categories according to their mechanisms of action: mitochondrial respiratory inhibitors that impede electron transport in the respiratory chain, phosphorylation inhibitors that hinder ATP synthesis, and uncouplers. Certain inhibitors not only inhibit OXPHOS but also modulate the tumor immune microenvironment. For instance, atovaquone (ATO) functions as a mitochondrial respiratory inhibitor by impeding mitochondrial complex Ⅰ-Ⅲ. Simultaneously, ATO can be involved in regulating immune cells by suppressing the Wnt/β-catenin signaling pathway [121]. Feng and co-workers synthesized ATO/PpIX-SMN nanocubes using a modified nanoprecipitation method to co-encapsulate the PS PpIX and ATO for promoting PDT-induced ICD (Fig. S5A in Supporting information). The findings indicated that the mitochondrial O2 consumption rate (OCR) was substantially lower in 4T1 cells treated with ATO/PpIX-SMN, and the exposure of CRT was highest for the same group. Furthermore, ATO could augment the secretion of the chemokine (C—C motif) ligands 4 (CCL4), which induced the maturation of DCs by downregulating the Wnt/β-catenin signaling pathway. In addition, the coupling with anti-PD-L1 therapy increased cytotoxic T lymphocytes (CTLs) infiltration, decreased Tregs, and enhanced the immune response in triple-negative breast cancer (TNBC) (Fig. S5B in Supporting information) [122].
HIF-1α is activated and stabilized under hypoxic conditions. It acts as a key transcription factor for numerous hypoxia-related genes, promoting tumor invasion, proliferation, angiogenesis, and metastasis [123]. Consequently, inhibiting the HIF-1α signaling pathway can synergistically enhance the anti-tumor effects of PDT. Thereinto, RNA interference technology can be employed to precisely and efficiently suppress the target HIF-1α protein using HIF-1α small interfering RNA (siRNA) [124]. Additionally, HIF-1α inhibitor, such as YC-1, can impede the expression of HIF-1α at the post-transcriptional stage, resulting in enhanced antitumor effects when combined with PDT [125]. HIF-1α is also involved in the regulation of tumor immune evasion. Studies have shown that under hypoxic conditions, HIF-1α upregulates the expression of PD-L1 in glioblastoma and influences the expression of CD47 by upregulating VEGF to evade immune surveillance [126]. Therefore, suppressing the expression of HIF-1α can impede the immune evasion of hypoxic tumors. Zhou and co-workers chemically modified TiO2 nanoparticles with a ruthenium-based PS (Ru) and loaded HIF-1α siRNA through electrostatic adsorption to create a novel nanosystem TiO2@Ru@siRNA (Fig. S5C in Supporting information). Upon laser irradiation, TiO2@Ru@siRNA induced lysosomal damage via photodynamic effects, leading to the release of HIF-1α siRNA for HIF-1α knockdown. The study demonstrated that inhibition of HIF-1α was concentration-dependent (Fig. S5D in Supporting information). Interestingly, the key proteins of pyroptosis (gasdermin-D, abbreviated as GSDMD) were upregulated in the group treated with TiO2@Ru@siRNA with laser irradiation, indicating the promotion of cellular pyroptosis by TiO2@Ru@siRNA-mediated PDT (Fig. S5E in Supporting information). Pyroptosis further induced ICD and triggered an anti-tumor immune response. Furthermore, TiO2@Ru@siRNA-mediated PDT downregulated PD-L1 levels in tumor cells through the HMGB1/nuclear factor-κB (NF-κB)/PD-L1 axis (Fig. S5F in Supporting information), increased the secretion of the anti-tumor cytokine IL-24, and activated CD4+ and CD8+ T cells [127].
4.
Exploiting tumor hypoxia
In addition to the tumor oxygenation-based PDT enhancement strategies, the exploitation of tumor hypoxia and considering the hypoxic microenvironment as a therapeutic target for cancer treatment has introduced new perspectives. Among these, hypoxia activated prodrugs (HAPs) are a type of anticancer drug that are designed to be activated specifically in the hypoxic regions of tumors. Besides, carbonic anhydrase Ⅸ (CA Ⅸ) is frequently overexpressed in hypoxic environments, particularly within various solid tumors, making it a potential target for anticancer strategies. Additionally, integrating hypoxia-cleavable linkers into the drug system offers opportunities for controlled drug release during PDT. This section explores recent advancements in combining these approaches with PDT and their potential to enhance PDT-driven immunotherapy.
4.1
Hypoxia activated prodrugs
HAPs depend on reductases that are overexpressed in hypoxic tumors to convert initially non-toxic prodrugs into highly toxic active therapeutic agents, thereby efficiently targeting and eliminating hypoxic tumor cells [128]. Currently, the class of HAPs include various compounds such as banoxantrone (AQ4N) [129,130], tirapazamine (TPZ) [131], apaziquone (EO9) [132]. It is well known that type Ⅱ PDT is a process that continuously consumes O2, ultimately inducing aggravated hypoxia in tumors. This serves as a stimulus to enhance the bioreduction of HAPs and further improve PDT efficacy [133,134]. However, facilitating HAPs into the interior of tumor is crucial for enhancing the synergistic therapeutic effects of HAPs/PDT especially for certain types of cancer with greater penetration barriers. Gliomas often exhibit a pronounced hypoxic environment. Additionally, the BBB and blood-tumor barrier restrictions hinder many prodrugs from reaching the brain or tumor lesions. Moreover, the disorder and complexity of the para-tumor vascular system lead to dynamic circulation of hypoxia and reoxygenation, resulting in unstable degree hypoxia and making it difficult to fully activate HAPs [135,136]. To address this issue, Zhang and co-workers encapsulated ZnPc and TPZ in liposomes that were subsequently modified with penetrating peptide iRGD, which proved to actively transport TPZ to gliomas [137].
The role of HAPs in enhancing the immunogenic effects of PDT has been demonstrated in recent years. Combining them with immune checkpoint inhibitors or adjuvants can strengthen the anti-tumor effects in hypoxic environments, providing potential for eliminating primary tumors and their metastases [138,139]. Shao and co-workers undertook a study in which they developed a specific type of metal-organic framework (MOFs) as nPSs for encapsulating TPZ within their nano-pores. The results demonstrated remarkable anti-tumor treatment effects in tumor immunotherapy by this nanosystem. Moreover, when exploited in combination with PD-L1 checkpoint inhibitors, it completely inhibited the untreated distant tumor growth by generating specific tumor-infiltrating cytotoxic T cells [140]. More recently, Wang and co-workers confirmed that HAPs can also induce ICD, further explaining the reasons behind the enhanced anti-tumor immune effects in PDT/HAPs synergistic therapy (Fig. S6A in Supporting information). Specifically, a multifunctional TME-responsive nanoplatform, denoted as IR 780-NLG919-TPZ NPs, was firstly developed using bovine serum albumin (BSA) as carrier material to encapsulate IR780 as a PS and Di-NLG919 as a glutathione-sensitive IDO inhibitor prodrug. Additionally, TPZ was conjugated on the surface of the nanoparticles. Upon laser irradiation, the generated ROS facilitated both PDT and release of TPZ from the nanosystem via the cleavage of thioketal (TK) linkers. The results indicate that ICD induced during this process stemmed from both PDT and TPZ-based chemotherapy, which is supported by the upregulation of CRT exposure, ATP secretion, and HMGB1 release in the IR780-TPZ NPs + L group compared to the IR780 NPs + L group. Furthermore, the enrichment of glutathione (GSH) in the TME could promote the activation of the NLG919 prodrug, leading to the specific inhibition of IDO1 and alleviating the immunosuppressive TME [141].
4.2
CA Ⅸ-targeted immunotherapy
CA Ⅸ is a transmembrane protein that is solely overexpressed exclusively on the cell membrane of hypoxic tumors [142]. Its expression is regulated by the HIF-1α in an O2-dependent manner, and it is not expressed in normal tissues [143,144]. In recent years, studies have suggested that targeted inhibitors of CA Ⅸ can be utilized for specific immunotherapy, in collaboration with PDT, to overcome hypoxia limitations in cancer treatment [55]. Based on the aforementioned characteristics of CA Ⅸ, researchers have developed strategies for synergistic treatments. Su and co-workers developed CA-Re, a PS consisting of rhenium(Ⅰ)-based PSs (Re) and a CA Ⅸ anchoring group (CA). The CA Ⅸ anchoring property enables CA-Re to remain fixed on the cell membrane even after extended incubation, promoting on-site production of ROS and potent lipid peroxidation along with disrupting the cell membrane. Interestingly, during CA-Re-mediated PDT, pyroptotic cell death was induced due to extensive lipid oxidation, leading to the depletion of glutathione peroxidase 4 (GPX4) (Fig. S6B in Supporting information). GPX4 negatively regulates the cleavage of GSDMD, a critical marker of pyroptosis, and the process of pyroptosis. Decreased expression of GPX4 and increased level of GSDMD-N and cleaved caspase-1 were observed in CA-Re treated cells upon light irradiation (Fig. S6C in Supporting information). Furthermore, the pyroptosis induced by CA-Re-mediated PDT may additionally enhance ICD and provoke T cell-dependent adaptive immune responses in vivo [145].
4.3
Hypoxia-responsive drug release
Hypoxia-responsive labile linkers, such as azobenzene (Azo), and chemical groups like the nitroaromatic group, have been utilized to develop a hypoxia-sensitive drug delivery system for PDT-driven immunotherapy, aiming to achieve precise and controlled drug release within the TME. For instance, Im and co-workers developed a nanocomposite material called CAGE, in which the immune adjuvant CpG oligonucleotides/glycol chitosan (GC) complex and PEG were conjugated to Ce6-predoped mesoporous silica nanoparticles (MSN) using hypoxia-cleavable Azo bonds. During PDT, as the tumor became more hypoxic, the Azo bonds would cleave, causing PEG to detach and release the CpG/GC complex from the cage. The immune adjuvant (CpG) could bind to toll-like receptor 9 (TLR9), modulating DCs activity and recruiting DCs, thereby effectively presenting antigens and enhancing immunotherapy [138]. This strategy avoided the kidney clearance and enzymatic degradation of CpG oligonucleotides when administered alone. In other cases, polymers used in the fabrication of nanocarriers containing Azo bonds can withstand normal O2 conditions, but they degrade quickly in the hypoxic TME [146,147]. This degradation leads to the release of encapsulated drugs, offering opportunities to develop polymeric nano-platforms for combined PDT/immunotherapy.
More recently, Kang and co-workers synthesized a nanocomposite, M1-MPNP, consisting of the aggregation-induced emission luminogen (AIEgen) MTPE-TT as PS, alongside the hypoxia-responsive paclitaxel (PTX) prodrug PTX-NB, which was further incorporated into the membrane of M1 macrophages (Fig. S6E in Supporting information). PTX-NB was anticipated to transform into the active drug in a hypoxic environment through hypoxic bioreduction, generating the unstable amine intermediate, which would subsequently undergo spontaneous self-elimination to release PTX. M1-MPNP not only exhibited effective tumor-killing capabilities under light exposure but also functioned as an inducer of ICD, enhancing tumor immunogenicity to inhibit both primary and distant tumor growth (Figs. S6F–I in Supporting information). Furthermore, due to improved tumor cell targeting mediated by the M1 macrophage membrane, M1-MPNPs exhibited exceptional tumor penetration abilities [148]. Besides nitroaromatic group, various hypoxia-responsive groups are also utilized for the formulation of nanosystems. For example, the reduction of hydrophobic 2-nitroimidazole to hydrophilic 2-aminoimidazole can be achieved through biological reductases, such as nicotinamide adenine dinucleotide phosphate (NADPH) [149]. This modification alters the solubility of the materials in an aqueous solution, providing additional avenues for developing hypoxia-sensitive drug delivery systems for PDT-driven immunotherapy.
5.
Disregarding tumor hypoxia
To decrease the O2-dependency and even disregard tumor hypoxia, researchers have developed a range of innovative type Ⅰ PSs and applied the principle of photoredox catalysis to enhance the production of ROS during PDT. This approach has resulted in a substantial improvement in the therapeutic efficacy of PDT for hypoxic tumors. Various reviews have systematically introduced type Ⅰ PSs [150,151]. Herein, we provide a comprehensive summary of two prevalent categories of organic type Ⅰ PSs from recent studies: type Ⅰ AIE PSs based on donor-acceptor (D-A) structure and type Ⅰ PSs based on boron dipyrromethene (BODIPY). Furthermore, we examine the utilization of type Ⅰ PSs in PDT-immunotherapy and introduce O2-independent PDT mechanisms, such as photoredox catalysis.
5.1
Type Ⅰ PDT and photosensitizers
In recent years, researchers have been dedicated to design various type Ⅰ PSs for hypoxic tumors boron dipyrromethene [152]. Based on the mechanism of type Ⅰ PDT, the following conditions should be satisfied: (1) A low energy gap ΔEST between the S1 and T1 states for the convenience of ISC process and enhance the T1 yield of PSs (Fig. S1); (2) long lifetime of the T1 state for PSs to facilitate the subsequent generation of ROS; (3) a suitable redox potential or electron-hole separation capability (typical in inorganic metal oxides) to facilitate electron transfer, or a lower energy level difference between the T1 and S0 states of PSs than the energy required for producing 1O2 by 3O2 (1.12 eV) to impede energy transfer [153,154]. The stringent conditions and the absence of a comprehensive theoretical framework for guiding the design and synthesis of type Ⅰ PSs have led to a scarcity of reports on type Ⅰ PSs compared to type Ⅱ ones. At present, the most extensively researched type Ⅰ PSs primarily comprise inorganic metal oxides (e.g., TiO2 [155], Fe2O3 [156] and ZnO [157]) or carbon [158] and small organic molecules based PSs (e.g., methylene blue (MB) [159] and BODIPY derivatives [160]). As compared with inorganic type Ⅰ PSs, organic ones exhibit superior biocompatibility, reduced side effects, and facile metabolism.
5.1.1
D-A type AIEgens based type Ⅰ photosensitizer
Traditional organic small molecule-based PSs, possess a hydrophobic planar molecular structure that readily aggregates in physiological environments, resulting in fluorescence quenching (ACQ) and a significant reduction in their photodynamic activity [161]. In 2001, Tang made a significant discovery while researching silole, uncovering the phenomenon of AIE [162]. Unlike ACQ dyes, AIE dyes (known as AIEgens) dissipate energy in the excited state through non-radiative pathways when in their single molecule state, which is attributed to molecular freedom of movement. However, in the aggregated state, non-radiative decay pathways are impeded by restricted molecular movement, leading to the activation of radiative decay and ISC pathways. This activation results in a concurrent enhancement in fluorescence and efficiency of ROS generation upon light irradiation [163,164].
In recent years, researchers have focused on developing type Ⅰ AIE PSs specifically tailored for hypoxic tumors to enhance the therapeutic effectiveness of PDT. This involves promoting the ISC process, where the separation of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) by the donor-acceptor (D-A) is crucial for reducing ΔEST and facilitating ISC [165]. Additionally, creating an electron-rich environment is advantageous for the triplet PS to efficiently capture external electrons under light conditions and execute the type Ⅰ PDT process [166]. Consequently, researchers have developed a series of type Ⅰ AIE PSs based on the D-A structure. Wang and co-workers developed a near-infrared (NIR) anion-π+ AIEgen termed Pys-QM-TT, as PS for effective inhibition of bacterial infections and ablation of tumor tissue. The construction of Pys-QM-TT involved the incorporation of an electron donor (triphenylamine (TPA) unit), a π bridge (thiophene), and an electron-withdrawing group (pyridinium salt unit) into the AIE building block quinoline-malononitrile (QM), resulting in enhanced D-π-A behavior and a strong intramolecular charge transfer (ICT) effect. The ICT effect led to an increased Stokes shift (254 nm for Pys-QM-TT), extending the emission wavelength, and effectively reduced the ∆EST, thereby enhancing ISC and the generation of ROS. Additionally, the pyridinium salt created an electron-rich environment and facilitated electron transfer, promoting type Ⅰ PDT [167]. The previously mentioned PS has been shown to effectively produce ROS to induce apoptosis in tumor cells within hypoxic tumors. However, its constant activation state may cause potential phototoxic effects in normal tissues, leading to symptoms such as burning sensations, skin redness, and scabbing [168]. To address the issue, Tian and co-workers developed a type Ⅰ PS responsive to hypoxia-normoxia cycling, denoted as TPFN-AzoCF3. This PS was constructed based on the reversibly redox-responsive arylazo group and the type Ⅰ PS TPFN, which features a typical D-A structure. TPFN-AzoCF3 was optimized on the basis of common OFF-ON PSs and achieved quenched photosensitization pre and post PDT, which could be activated only in tumor hypoxic environment, thereby minimizing side effects [169].
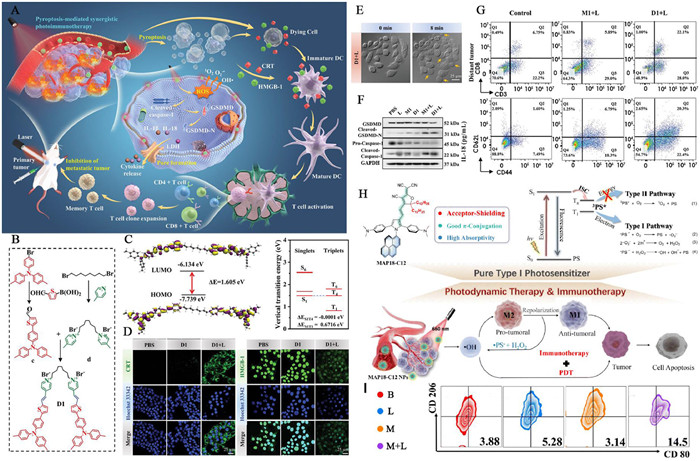
In the context of immunoregulation, type Ⅰ PDT can also induce ICD and enhance tumor immunogenicity. Furthermore, type Ⅰ PDT-induced ICD presents a more attractive option than type Ⅱ PDT due to its low O2 dependency and the highly cytotoxic OH• it generates [170]. Tang and co-workers developed a photosensitive dimer, denoted as D1, based on D-π-A structured AIEgens, consisting of two π-conjugated photosensitive chromophores (M1) and a flexible octyl group linker. Each chromophore comprised an electron donor methyl-substituted triphenylamino thiophene unit, an electron acceptor pyridinium, and a π-bridge carbon-carbon double bond (Figs. 3A–C). Compared to the monomer M1, D1 exhibited enhanced aggregation capability, thereby boosting AIE-mediated ROS generation efficiency and PTT effects. Under light irradiation, D1 effectively induced type Ⅰ PDT and PTT, thereby triggering and enhancing tumor pyroptosis, leading to ICD, and activating the antitumor immune response (Figs. 3D and E). The results indicated a significant increase in the concentrations of GSDMD-N and caspase-1, key markers of pyroptosis, in D1-treated tumor cells under light irradiation (Fig. 3F). In bilateral tumor models, the proportion of CD8+ T cells in distant tumors treated with D1 plus light was obviously higher than in the control group, reaching 22.1% (Fig. 3G) [171].
Figure 3
Figure 3.
photodynamic immunotherapy mediated by D-A type AIEgens based type Ⅰ photosensitizer. (A) Schematic illustration of pyroptosis-mediated photothermal/photodynamic immunotherapy enabled by photosensitive dimer D1. (B) Synthesis of D1. (C) HOMO and LUMO wave functions in the geometrical structure and singlet and triplet energy levels and ΔEST values of D1. (D) The immunofluorescent images of CRT on 4T1 cells and the CLSM analysis of HMGB-1 release from 4T1 cells after treatment with D1. (E) Real-time observation of cell membrane expansion and content release in pyroptosis of D1-treated cells. (F) Quantitative analysis of Western blot was obtained to determine the relative intensity of GSDMD and caspase-1. (G) The flow cytometry plots of the proportion of infiltrating CD8+ T cells and CD44 and CD62L expression on CD3+CD8+ T cells of distant tumor. Reproduced with permission [171]. Copyright 2023, Wiley Publishing Group. (H) Schematic illustration of chemical structure of MAP18-C12 and its immunotherapy processes. (I) The flow cytometry plots of the proportion of the M1 phenotype in tumor tissues after treatment. Reproduced with permission [173]. Copyright 2022, Royal Society of Chemistry.
In addition to inducing ICD, the HO• generated by type Ⅰ PSs can degrading NF-κB inhibitors to activate NF-κB factors, thereby stimulating protumoral M2 macrophages to transform into antitumoral M1 macrophages [172]. Recently, Qu and co-workers developed a multiaryl-pyrrole (MAP) derivative MAP18-C12, comprises of a MAPs core, an N,N-dimethylaniline electron donor group, and a 2-(3-cyano-5,5-dialkylfuran-2-ylidene)propane-dinitrile (FE) electron acceptor. Notably, an acceptor-shielding strategy was incorporated into the chemical structure design of the PS to enhance PDT efficacy. This involved the introduction of a dodecyl group into MAP18-C12, yielding improved D-A electron transfer, a higher molar absorption coefficient, a longer triplet lifetime, and a narrower singlet-triplet energy gap (Fig. 3H). The researchers further encapsulated MAP18-C12 into distearoyl phosphoethanolamine-polyethylene glycol (DSPE-PEG2000) to produce MAP18-C12 NPs. HO• produced by type Ⅰ PDT can convert M2 macrophages to M1. Results indicated that the proportion of M1 cells in the tumor treated with MAP18-C12 NPs under light irradiation was 3.7 times higher than that of the control group, demonstrating the synergistic effects of PDT and immunotherapy (Fig. 3I) [173].
5.1.2
BODIPY-based type Ⅰ photosensitizer
BODIPY is a commonly used commercial organic fluorescent dye, widely utilized in biomedical research owing to its substantial molar absorption coefficients, excellent photostability, highly adjustable photophysical properties, and numerous chemical reaction sites [174]. Recent research has demonstrated that the distinctive π-conjugated system of the BODIPY unit facilitates the generation of supramolecular interactions more effectively, offering a fresh perspective for the manipulation of photophysical properties of BODIPY [175]. Yang's research group has conducted intriguing research in this field. Firstly, a series of α,β-linked BODIPY dimers and a trimer were designed as PSs that exclusively generate O2•− through the type Ⅰ process under NIR light irradiation (Fig. S7A in Supporting information). The efficient formation of triplet states originates from the transition of the initially populated S1 to T1 states, mediated by an intermediate triplet (T2) state. The ultralong lifetime of the T1 state, extending to the microsecond timescale, and the low reduction potential facilitate efficient O2•− generation. Additionally, the energy gaps between the T1 state and the ground S0 state of these PSs were narrower than the gap between3O2 and 1O2, effectively impeding the energy transfer process from the PSs to O2, which is beneficial for type Ⅰ PDT (Figs. S7B and C in Supporting information) [154]. However, challenges arise in the design of type Ⅰ PSs guided by the aforementioned strategy, primarily attributable to the unpredictable nature of the triplet state energy of PSs. Yang and co-workers further proposed a supramolecular strategy to enhance electron transfer through host-guest interactions, effectively converting conventional type Ⅱ PS to type Ⅰ PS. They designed a supramolecular polymer, HG, comprising a guest molecule and electron acceptor, iodide BODIPY (G), and a macrocyclic host and electron donor, bispillar[5]arene (BP5A). The host-guest interaction promoted electron cloud overlap between the electron-rich substrates and the PSs, facilitating efficient electron transfer from BP5A to G and generating G•−. Subsequently, G•− reacted with O2 to produce O2•−via the type Ⅰ PDT mechanism (Figs. S7D and E in Supporting information) [176].
The mechanism of both type Ⅰ PSs aforementioned involves 3PS* initially abstracting an electron from surrounding substrates, generating PS•−. This PS•− then transfers an electron to O2 to produce O2•−. However, the efficiency of electron transfer between PS and surrounding substrates is limited, resulting in lower O2•− generation efficiency. Theoretically, 3PS* can also transfer an electron to an electron-deficient substrate, which subsequently transfers an electron to O2 to produce O2•− [177]. Yang and co-workers proposed another strategy to convert existing type Ⅱ PS to a type Ⅰ PS by introducing an electron acceptor to existing type Ⅱ PSs. They designed three different kinds of electron acceptors and co-assembled with conventional type Ⅱ PS to form quadruple hydrogen-bonded supramolecular nPSs. Since the PS was close to the electron acceptor with a matching redox potential, under light irradiation, electrons transferred from PS to the electron acceptor, and then to O2, thereby efficiently generating O2•−. It is remarkable that this process was accompanied by the generation of PS•+, which can also oxidize nicotinamide adenine dinucleotide (NADH), thereby exacerbating the biological damage to tumor cells (Figs. S7F and G in Supporting information) [178]. In another study, the same group employed an analogous approach to develop an O2-independent supramolecular PS to produce OH• by oxidizing water in the presence of intracellularly abundant pyruvic acid under O2-free conditions [179].
5.2
Photoredox catalysis
The most of reported type Ⅰ PSs are low O2-dependent and show enhanced therapeutic effects against hypoxic tumors. However, O2 is still indispensable to type Ⅰ PDT. Additionally, the intracellular antioxidant systems of tumor cells such as high GSH expression can resist PDT. To address these challenges, a novel form of phototherapy has emerged that functions independently of O2 and does not rely on ROS, utilizing the mechanism of photoredox catalysis. When subjected to light irradiation, single electron transfer (SET) occurs, facilitating the transfer of electrons from substrates like NAD(P)H to photoredox catalysts (PCs). This process leads to the oxidation of these substrates, subsequently influencing the cellular metabolism in which they are involved. Simultaneously, the intermediate radicals of the PCs engage in reduction reactions with other molecules, such as oxidative cytochrome c under hypoxic conditions, enabling PCs to revert to their original active state and complete the photocatalytic cycle [180,181].
NADH serves as the electron source in the respiratory electron transport chain (ETC) and acts as a coenzyme in oxidoreductases to maintain the intracellular redox balance [182]. Consequently, photoredox catalysis has the potential to upset the intracellular redox equilibrium by depleting NADH and disrupting the ETC, thus leading to the efficient eradication of cancer cells under hypoxic conditions. In 2019, Hang and co-workers pioneered the use of photoredox catalysis in hypoxic tumor phototherapy. They developed an iridium complex, [Ir(ttpy)(pq)Cl]PF6 (1(IrⅢ)), which exhibited high photocatalytic activity for NADH oxidation and nearly equivalent photocytotoxicity under both normoxia and hypoxia. Furthermore, the release of HMGB1 and transfer of CRT indicated that 1(IrⅢ) could induce ICD and stimulate the body's anti-tumor immune response. Regarding the 1(IrⅡ) species, under hypoxia, Fe3+-cyt c served as a terminal electron acceptor and was reduced by 1(IrⅡ), yielding Fe2+-cyt c and 1(IrⅢ) (Figs. S8A and B in Supporting information) [183].
PCs are primarily abiotic transition-metal complexes, known for their long excited-state lifetime and large redox window. However, the biomedical application of these PCs is hindered by unpredicted metal toxicity and limited tissue penetration of the light required for excitation [184]. Organometallic complexes also possess the capacity to interfere with cellular biological processes, including enzyme activity and gene expression, which can result in oxidative stress and DNA damage. Moreover, the in vivo application of photoredox catalysis faces challenges such as “off-target” and nonspecific photoactivation. Therefore, the development of organic, metal-free PCs with controlled photoredox catalytic activity is essential to enhance the therapeutic effects of photoredox catalysis on hypoxic tumors. Li and co-workers have introduced the concept of “conditionally activatable photoredox catalysis” and developed a nitroreductase (NTR)-triggered PC called Se-NO2, which is based on classic self-immolative chemistry (Fig. S8C in Supporting information). Se-NO2 was synthesized by combining a metal-free PC (Se-NH2) with a nitrobenzene group. The fluorescence of Se-NO2 was minimal due to electron transfer caused by the strong electron-withdrawing effect of the nitro group, and its extinction coefficient decreased ten-fold at 660 nm, indicating a reduced light-harvesting capacity. These photophysical properties rendered Se-NO2 incapable of inducing NIR photoredox catalysis. However, under hypoxic conditions, NTR catalyzed the departure of the nitrobenzene group from Se-NO2, releasing the photoredox catalytic activity of Se-NH2 [185]. More recently, Li and co-workers hypothesized that photoredox catalysis played a significant role in traditional PDT. To validate their hypothesis, they conducted experiments to demonstrate that well-established PDT PSs could cause depletion of NADH, acting as PCs. They also developed a BODIPY-derived molecular targeting PC named CatER, comprising a BODIPY-type PS and erlotinib (ER), a tyrosine kinase inhibitor [186]. Additionally, Deng and co-workers developed NIR-activated Pt(Ⅳ) photooxidants based on the concept of the metal-enhanced photooxidation effect (Fig. S8D in Supporting information). Pt(Ⅳ) was capable of directly photooxidizing intracellular biomolecules essential for cell survival in an O2-independent behavior. Mechanistic studies revealed that the cell death induced by Pt(Ⅳ) differed from typical forms of cell death such as autophagy and necrosis, and was instead characterized by the intensive intracellular oxidative stress and acidosis. Furthermore, Pt(Ⅳ) was found to induce ICD in tumor cells and activate immune cells, thereby augmenting its anti-tumor effects (Figs. S8E and F in Supporting information) [187].
6.
Challenge and future prospects
PDT is effective in eliminating tumor cells and inducing ICD, which boosts immune response. Yet, the hypoxic tumor microenvironment reduces PDT efficacy and promotes immune suppression, hampering PDT-based immunotherapy. Strategies to modulate tumor hypoxia or develop hypoxia-compatible PDT hold promise for enhancing PDT-driven immunotherapy. Despite the satisfactory outcomes of these strategies, there are still some underlying challenges that remain to be resolved. Firstly, the limited light penetration in tissue hinders PDT due to most photosensitizers being activated by visible or NIR Ⅰ light that can only reach less than 1 cm beneath the skin. To address this challenge and broaden PDT applications, developing PSs with longer excitation wavelengths (NIR Ⅱ or Ⅲ), improving light source selectivity, and advancing optical technologies such as endoscopes are crucial. Additionally, XPDT offers a solution by converting X-rays to ultraviolet (UV) or visible light via a scintillator to activate photosensitizers. This advancement can enhance PDT efficacy by improving light delivery to tumor tissues, necessitating increased focus on organic scintillators with enhanced biocompatibility and safety.
Secondly, there is an imperative to develop general methods for designing novel PSs that are either low or not dependent on O2. While a molecular design principle for exclusive type Ⅰ PSs has been proposed and several type Ⅰ PSs have been constructed based on this principle, it is challenging to generalize such a principle due to its lack of specificity and universal applicability. Existing pure type Ⅰ PSs have largely been developed empirically. The recent introduction of photoredox catalysis as a component of common PDT has shown promising results, providing a photo-controlled O2-independent immunogenic apoptotic mechanism. Hence, the development of O2-independent PSs based on photoredox catalysis holds significant potential.
Thirdly, enhancing the synergy between PDT and immunotherapy is essential. While PDT-driven immunotherapy typically relies on photodynamic ICD, overcoming the immunosuppressive tumor environment is crucial for better treatment outcomes. Strategies to reverse tumor immunosuppression, such as tumor oxygenation, need further exploration for their potential to improve treatment efficacy. The emerging concept of pyroptosis in cancer research offers promising prospects in tumor immunotherapy. Developing photosensitizers capable of inducing tumor immunity through photo-induced pyroptosis activation could lead to positive outcomes in antitumor immunotherapy.
In addition, nanomedicine plays a crucial role in enhancing the effectiveness of photodynamic immunotherapy. However, the clinical progress of nanomedicine is challenged by significant biological obstacles. While the permeability and retention (EPR) effect has been key in nanoparticle accumulation in tumors, its applicability in human solid tumors remains debated. Recent findings suggest that nanoparticles may enter solid tumors through complex mechanisms beyond EPR effect [188]. It is essential to develop innovative strategies to enhance tumor-specific nanoparticle accumulation for successful clinical translation. Light exposure can increase the accumulation of nanomedicines in tumors by altering the tumor microenvironment, but further research is needed to fully comprehend this phenomenon [189,190]. Simultaneously, identifying a standardized animal experimental model system can enhance the precision in assessing the therapeutic efficacy of nanomedicine during the preclinical phase, potentially expediting the translation of nanomedicine from the laboratory to clinical applications. Furthermore, the swift advancements in artificial intelligence (AI) applications offer invaluable support in constructing predictive models for nano-bio interactions, targeted delivery efficiency, and the safety and effectiveness of nanomedicine. While still in the nascent stage of exploration, these tools and methods possess significant potential to revolutionize cancer nanomedicine and redefine the cancer treatment paradigm.
Overall, PDT-driven immunotherapy, as an innovative approach in cancer treatment, has been demonstrated to leverage immune activation in the fight against conventional therapy-resistant cancers. Furthermore, a heightened focus on understanding the influence of tumor hypoxia on the therapeutic outcomes, coupled with the development of robust strategies to alleviate, exploit or disregard tumor hypoxia, is anticipated to enhance the overall effectiveness of this treatment modality.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the Qin Chuangyuan Traditional Chinese Medicine (TCM) and Innovation Research and Development Project of Shaanxi Provincial Administration of TCM (No. 2022-QCYZH-017), Natural Science Foundation of Zhejiang Province (No. LY24E030010), Natural Science Foundation of Shaanxi Province (Nos. 2022JM183, 2024JC-YBMS-272), the Shaanxi Fundamental Science Research Project for Chemistry & Biology (No. 22JHQ072), Shaanxi Provincial Key R&D Program (No. 2022SF-342HZ), the Fundamental Research Funds for the Central Universities (Nos. xzy012022037, xzy012023002), the Postdoctoral Science Foundation of Shaanxi Province (No. 2023BSHYDZZ05), Foundation by Shaanxi Provincial Administration of TCM (No. 2021-ZZ-JC032).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109957.
M. Izci, C. Maksoudian, B.B. Manshian, S.J. Soenen, Chem. Rev. 121 (2021) 1746–1803. doi: 10.1021/acs.chemrev.0c00779
[189]
Z. Yang, D. Gao, J. Zhao, et al., Nat. Rev. Clin. Oncol. 20 (2023) 116–134. doi: 10.1038/s41571-022-00717-y
[190]
W. Gao, Z. Wang, L. Lv, et al., Theranostics 6 (2016) 1131. doi: 10.7150/thno.15262
Figure 1
Schematic illustration of strategies for fighting tumor hypoxia to improve PDT-driven immunotherapy. These strategies can be categorized into three sections: (Ⅰ) direct or indirect alleviation of tumor hypoxia, (Ⅱ) exploitation of tumor hypoxia, and (Ⅲ) disregard of tumor hypoxia. Fighting tumor hypoxia can amplify the therapeutic effects of PDT, and thus improving tumor immunogenicity by inducing strong ICD. The effectiveness of this process can be enhanced by modulating the tumor immunosuppressive microenvironment and combining PDT with other immunotherapies.
Figure 2
The co-administration of immunomodulatory drugs or anti-PD-1/PD-L1 therapy enhances the anti-tumor immune effects of PDT. (A) Schematic illustration of preparation of PF11DG and antitumor immune responses elicited by PF11DG. (B) CLSM images of CRT exposed on cell surface. Scale bar: 25 µm. (C) Frequency of CD3+CD8+ T cells, NK cells and MDSCs in tumors of different treatment groups. Reproduced with permission [101]. Copyright 2021, American Chemical Society. (D) Schematic illustration of preparation of PHD@PM and PHD@PM-based photodynamic immunotherapy. (E) CLSM images of 4T1 cells after a 2 h-incubation with PHD@PM. Scale bar: 10 µm. (F) The flow cytometry plots of the percentages of tumor-infiltrating CD4+ and CD8+ T cells in tumors of different treatment groups. Reproduced with permission [102]. Copyright 2021, Wiley Publishing Group.
Figure 3
photodynamic immunotherapy mediated by D-A type AIEgens based type Ⅰ photosensitizer. (A) Schematic illustration of pyroptosis-mediated photothermal/photodynamic immunotherapy enabled by photosensitive dimer D1. (B) Synthesis of D1. (C) HOMO and LUMO wave functions in the geometrical structure and singlet and triplet energy levels and ΔEST values of D1. (D) The immunofluorescent images of CRT on 4T1 cells and the CLSM analysis of HMGB-1 release from 4T1 cells after treatment with D1. (E) Real-time observation of cell membrane expansion and content release in pyroptosis of D1-treated cells. (F) Quantitative analysis of Western blot was obtained to determine the relative intensity of GSDMD and caspase-1. (G) The flow cytometry plots of the proportion of infiltrating CD8+ T cells and CD44 and CD62L expression on CD3+CD8+ T cells of distant tumor. Reproduced with permission [171]. Copyright 2023, Wiley Publishing Group. (H) Schematic illustration of chemical structure of MAP18-C12 and its immunotherapy processes. (I) The flow cytometry plots of the proportion of the M1 phenotype in tumor tissues after treatment. Reproduced with permission [173]. Copyright 2022, Royal Society of Chemistry.

 DownLoad:
DownLoad:

 下载:
下载:




 下载:
下载:
