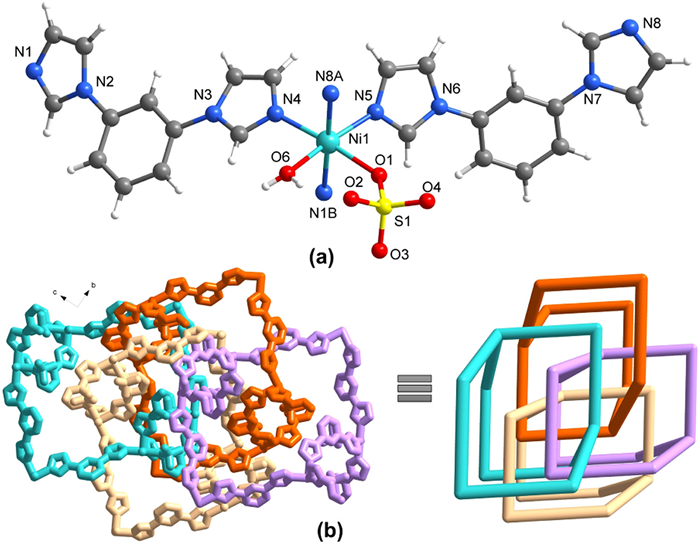
Figure 1.
Crystal structures of TNU-143. (a) Coordination environment of Ni(Ⅱ). (b) Highlight of four-interpenetrated nets and its simplified pattern.
A four-fold interpenetrated MOF for efficient perrhenate/pertechnetate removal from alkaline nuclear effluents
Lei Zhu , Hai-Ruo Li , Yi-Ning Mao , Ruiying Liu , Bo Zhang , Jing Chen , Wengui Xu , Libo Zhang , Cheng-Peng Li

It is of long-term concern to produce clean power with high energy output in addressing the escalating global energy crisis [1]. Nuclear power, as one of the highest density energy sources, supports hundreds of important industrial manufactures in many countries. Despite its advantages, the risk of nuclear leaks from power plants poses a serious threat to global security, especially in the face of extreme climatic disasters. In the event of a leak, significant amounts of radioactive species could enter the environment, leading to unforeseeable consequences for human beings. Among the various radioisotope contaminants, technetium-99 (99Tc) has accumulated in considerable amounts (150 t), primarily from the nuclear fission of 235U in reactors [2]. 99Tc has a long half-life (t1/2 = 2.13 × 105 years) and releases β-rays during decay [3]. As the main form of 99Tc, pertechnetate anion (99TcO4–) poses a challenge due to its high solubility (11.3 mol/L at 20 ℃) and non-complexing nature. This makes it difficult to immobilize, allowing it to easily enter local groundwater and ecological systems [4]. In light of these challenges, the management of 99Tc is crucial for ensuring the safety of nuclear power plants, facilitating environmental remediation, and safeguarding human health.
To address this unmet challenge, solid-phase sorbents are considered promising anion exchange materials for the sequestration of 99TcO4–, given their ease of implementation and superior performances [5–8]. Due to the radioactive nature of 99Tc, ReO4– is typically used as a nonradioactive surrogate in common research, owing to their similar charge density and thermodynamic parameters [9]. In the large-scale treatment process of 99TcO4–, commercialized polymeric anion exchange resins (such as superLig-639 or Purolite A520E) have been applied; however, they exhibit slow anion exchange kinetics and/or poor radiation resistance [10,11]. On the other hand, inorganic cationic materials, such as layered double hydroxide (LDH) [12] and NDTB-1 [13], usually lack specific exchange sites, resulting in low sorption capacity and poor selectivity. Recently, advanced porous materials such as metal-organic frameworks (MOFs) [14–17], covalent organic frameworks (COFs) [18–20] and porous aromatic frameworks (PAFs) [21–23] have been considered as potential game-changing candidates for TcO4–/ReO4– sorption.
In our previous study, we utilized the single-crystallinity property of MOFs as a crucial tool to elucidate the structure-mechanism correlation during the anion exchange process [24,25]. However, these MOFs face challenges in maintaining their structures and activities under highly alkaline conditions, which are necessary for spent nuclear fuel reprocessing with poor regeneration. To address this limitation, the development of more stable MOF sorbents for sequestrating TcO4–/ReO4– becomes crucial for nuclear waste management, particularly for the alkaline high-level waste (HLW) streams, e.g., the Savannah River Site (SRS) effluents [26]. Currently, only a few MOFs have demonstrated effective TcO4–/ReO4– sequestration form alkaline solutions [27–30]. However, none of the anion exchanged products from these MOFs can maintain their single-crystallinities, hindering a convincing interpretation of their highly efficient capture ability toward TcO4–/ReO4–. Consequently, the systematic design and development of MOFs with good alkaline resistance and single-crystallinity during the anion exchange process are still in their early stages. To achieve decent alkaline stability, it is highly desirable to consider the appropriate softness of metal ions and organic ligands in MOFs, considering the principles of the hard and soft acids and bases theory. In this context, the coordination bonds between the metal ions and ligand are robust enough to be destroyed by OH–, thus preventing the formation of metal-hydroxide bonds. Therefore, the strategy by choosing transition metal ions and N-heterocyclic ligand with high pKa values is applicable to construct alkaline-stable MOFs [31].
Furthermore, the anion components within MOFs can undergo stoichiometric anion exchange with TcO4–/ReO4–, to give rise to high uptake capacity [32]. Specifically, the hydrophobic channels or cavities in MOFs based on neutral organic linkers prove to be more advantageous for enhancing sorption selectivity [25,26]. In this study, we chose a bidentate N-donor ligand, 1,3-di(1H-imidazol-1-yl)benzene (bib), to assemble with NiSO4, resulting in a four-fold interpenetrated MOF named TNU-143. It shows good alkaline resistance, high sorption capacity and exceptional ReO4– capture selectivity, even in simulated legacy nuclear wastes. Impressively, TNU-143 can maintain the single-crystallinity during the anion exchange process to yield TNU-143(Re). This distinctive single-crystal-to-single-crystal (SC-SC) transformation explicitly demonstrates the anion exchange mechanism, wherein the SO42– anions in TNU-143 readily exchange with ReO4–, accompanied by changes in the coordination geometry of Ni2+ and network structure in TNU-143(Re). This process is a thermodynamically favourable reactions as proved by density functional theory (DFT) calculations.
Single-crystal X-ray diffraction analysis shows that TNU-143 crystallizes in an orthorhombic phase with space group Pbca (Table S1 in Supporting information). The asymmetric unit consists of one Ni(Ⅱ) atom, two bib ligand, one water molecule, and one SO42− anion (Fig. 1a). Each Ni(Ⅱ) adopts a six-coordinated octahedral geometry, and binds to four nitrogen atoms from bib ligands, and two oxygen atoms from H2O and SO42− in the cis-location, featuring a distorted octahedral geometry (Table S2 in Supporting information). As a result, the Ni(Ⅱ) atoms are linked by bib ligands to form a 3D coordination network (Fig. S1 in Supporting information). In this structure, 1D channels are observed along the a axis, providing enough voids (19 × 17 Å2) for network entanglement. Specifically, four coordination nets intertwine to form a four-interpenetrated framework (Fig. 1b and Fig. S2 in Supporting information). From a topological perspective, each 3D net can be regarded as a uninodal 4-connected dia network.
Anion exchange of ReO4– was performed by soaking TNU-143 materials (30 mg, 0.05 mmol) into the water solution (20 mL) of KReO4 (29 mg, 0.1 mmol). The progress of anion exchange was monitored through FTIR, PXRD, XPS and STEM-EDS mapping analyses. As shown in Fig. 2a, the reduced intensity of the SO42– peak at 1032 cm–1 and 601 cm–1, and a new strong IR peak at 909 cm–1 arises after one day of anion exchange, indicating the formation of ReO4–-loaded materials, TNU-143(Re). Simultaneously, PXRD patterns of TNU-143 and TNU-143(Re) confirmed the well crystallinity of materials but revealed completely different crystalline structures (Fig. 2b). In the high-resolution XPS spectra (Fig. 2c), TNU-143(Re) displayed peaks of Re 4f (ca. 46.0 eV) compared to TNU-143, along with the disappearances of S 2p (ca. 168.2 eV) peaks, demonstrating the complete anion exchange of SO42– by ReO4–. Furthermore, the Re 4f core-level spectra in TNU-143(Re) are different from that of KReO4, which indicates that the ReO4– species may form interactions with Ni(Ⅱ) during the ion-exchange process. As probed by the STEM-EDS mapping of TNU-143 and TNU-143(Re), the block crystal morphology was well-maintained during the anion-exchange process, with tiny crystals adhering to the crystal surfaces (Fig. 2d). The similar distribution of Ni and C elements in TNU-143 and TNU-143(Re) revealed the intact Ni-bib frameworks (Fig. S3 in Supporting information). In the STEM-EDS mapping of TNU-143(Re), Re elements were evenly distributed in the adsorbents, further confirming the successful uptake of ReO4–.

To evaluate the effectiveness of TNU-143 in capturing ReO4–, kinetics sorption experiments were carried out by soaking 15 mg of TNU-143 sample in 5 mL of aqueous ReO4– solution containing 72 ppm Re. As shown in Fig. 3a, TNU-143 exhibits an exceptionally rapid sorption rate. Within the first minute, the relative amount of ReO4– removal reaches 96.39%, and TNU-143 achieves adsorption equilibrium within 10 min. Correspondingly, the residual Re(Ⅶ) concentration decreases to 0.64 ppm, resulting in a trapping efficiency of 99.11%. The kinetic data can be well fitted by the pseudo-second-order model with a high correlation coefficient of > 0.9999 (Table S3 in Supporting information). Notably, the rate constant (k2) for TNU-143 (0.72 g mg–1 min–1) is much higher than that of commercial Purolite A532E (4.60 × 10–3 g mg–1 min–1) and A530E (6.75 × 10–3 g mg–1 min–1) [33]. Furthermore, these commercial anion exchange resins reportedly take up to 120 min to reach adsorption equilibrium [34]. The rapid sorption kinetics exhibited by TNU-143 is crucial for a sorbent dealing with TcO4–, as an immediate response to radioactive waste can significantly reduce the potential risk of sorbent decomposition and facilitate the swift disposal of accidental 99TcO4– spills. To further assess the performance of TNU-143 as an anion scavenger, the parameter of distribution coefficient (Kd) is used. The Kd value is calculated based on the equation Kd = [(C0 − Ce)V/Ce]/m, in which C0 and Ce are the initial and equilibrium concentrations of ReO4– (mg/L), V is the volume of the treated solution (mL), m is the mass of TNU-143 (g). As a result, the Kd value of TNU-143 toward ReO4– is 5.18 × 105 mL/g, which is considered to be excellent sorbent [35].

To further evaluate the adsorption capacity of TNU-143 toward ReO4–, anion exchange isotherm experiments were conducted under ambient conditions. TNU-143 (10 mg) was immersed in a 10 mL ReO4– solution with stepwise concentrations ranging from 100 ppm to 2000 ppm. As shown in Fig. 3b, sorption of ReO4– by TNU-143 is fitted well with the Langmuir isotherm model, with a high correlation coefficient of > 0.99 (Fig. S4 and Table S4 in Supporting information). As a result, the calculated uptake capacity of ReO4– by TNU-143 is 844 mg/g, corresponding to 628 mg of Re per gram of sorbent. This value is slightly lower than the theoretically maximum ReO4– capacity (846 mg/g), which is calculated according to the hypothesis that all of the SO42– in TNU-143 are completely exchanged by ReO4–. Notably, this value is higher than most of reported MOF materials [36], other lab-made sorbents [37], and commercial products [38].
It is very important to assess the sorbent performance in harsh chemical conditions, since most of nuclear wastes are either strongly acidic or alkaline. Therefore, sorption of ReO4– (50 ppm) at various pH conditions (pH 0–14) was investigated. In the pH range of 3–12, TNU-143 showed > 80% ReO4− removal (Fig. 3c). If in more acidic solution, ReO4− removal percentage obviously declined, possibly attributed to framework collapse (Fig. 3d). Interestingly, TNU-143 shows remarkable stability even at pH 14, and can remove 61% of ReO4− (OH–: ReO4– = 10,000:1).
The presence of a large excess of coexisting anions poses a significant challenge to the practical application of TcO4– decontamination in radioactive waste streams. Therefore, anion exchange selectivity experiments were conducted assess TNU-143′s capability to remove ReO4− in the presence of competing anions, including NO3–, NO2–, Cl–, PO43–, ClO4–, SO42–, and CO32–. As shown in Fig. 4a, TNU-143 exhibited remarkable selectivity, removing ReO4− with nearly 100% efficiency in the presence of equimolar competing anions. In particular, the amounts of NO3–, CO32– and Cl– in specific high-level liquid waste were far more than that of ReO4– anion. These anions, known for their high charge density, typically present significant challenges in waste stream treatment due to strong electrostatic interactions with adsorption resins. Sorption experiments involving varying molar ratios of interfering anions to ReO4– (ranging from 1:1 to 500:1) were conducted. For NO3– and CO32–, the removal efficiencies consistently exceeded 95% at a ratio of 100:1 and remained around 90% even at the ratio of 500:1 (Figs. 4b and c). In the case of Cl–, TNU-143 maintained high removal percentages of 93% for ReO4–, even when the concentration of Cl− was 500 times in excess (Fig. 4d).
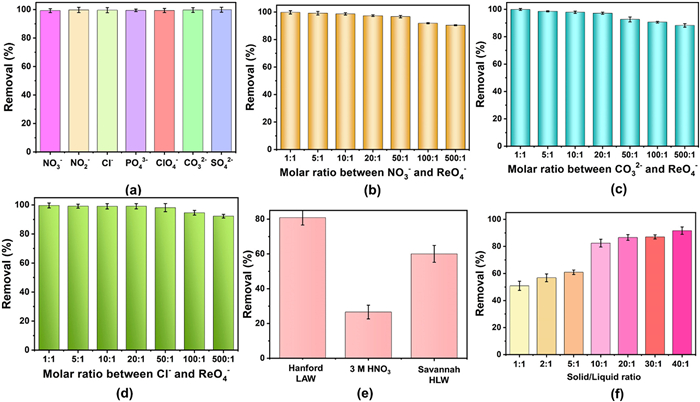
Encouraged by the aforementioned results, we proceeded to investigate the uptake ability of TNU-143 for ReO4− in typical simulated nuclear waste streams, including Hanford LAW Melter Recycle Stream, highly acidic spent nuclear fuel solution (3 mol/L HNO3, 200 ppm Re) and SRS HLW Stream (Tables S5 and S6 in Supporting information). In the simulated Hanford LAW recycle stream and 3 mol/L HNO3 solution, more than 80.2% and 27.6% of ReO4− could be removed by TNU-143 in a solid/liquid ratio of 5 g/L (Fig. 4e). Notably, TNU-143 achieved a remarkable 60.8% removal of ReO4− from the simulated SRS HLW Stream in just one hour (Fig. 4e). Additionally, this accomplishment is particularly challenging for MOF sorbents, given their inherent instability in alkaline solutions. Very few materials have been reported to effectively sequester TcO4− under such harsh conditions. Among them, only three MOFs showed promising performance (Table S7 in Supporting information). The PAQ series materials can remove 64%–77% of TcO4− from the simulated SRS LAW at 10 g/L solid/liquid ratio [39]. Outstandingly, SCU-CPN-4 [40] and SCU-103 [27] can extract 94% and 90% TcO4− at the solid/liquid ratio of 20 and 40 g/L, respectively. At the solid/liquid ratio of 40 g/L, TNU-143 shows 91.6% of ReO4– removal (Fig. 4f).
To elucidate the underlying reason for the superior ReO4– uptake performances by TNU-143, we employed single-crystal to single-crystal (SC-SC) transformations, providing compelling evidence for the heterogeneous anion exchange process [32]. After many attempts, single crystals of anion exchange product TNU-143(Re) were successfully obtained from the SC-SC transformation method and structurally determined by single-crystal X-ray diffraction. A structural comparison for TNU-143 and TNU-143(Re) would help us to underderstand the sorption mechanism. As shown in Fig. 5a, the paired SO42− ions ligands in TNU-143 are exchanged by two ReO4− ion ligands in TNU-143(Re). As a result, the Ni central atoms are connected by four bib ligands to form a 2D coordination layer (Fig. 5b). These adjacent layers are arranged in a parallel manner, creating a 3D stacking pattern (Fig. S5 in Supporting information). The structural divergences between TNU-143 and TNU-143(Re) can be attributed to the positions of coordination ions. Specifically, two SO42− ions in TNU-143 are situated at cis positions, whereas two ReO4− ions in TNU-143(Re) occupy trans sites. Considering steric and electrostatic effects, the bulky negatively charged ligands tend to be distanced from each other and positioned at trans sites, following the principle of lowest energy.
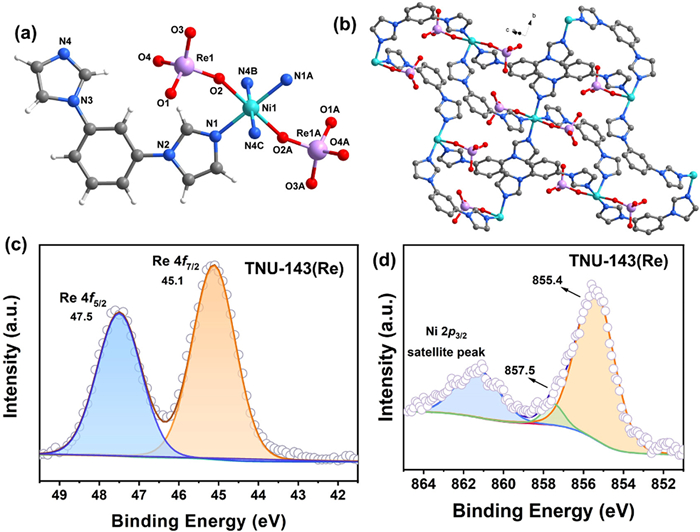
In the XPS survey of TNU-143(Re), the high-resolution Re 4f spectra show two peaks at 47.5 eV of Re 4f5/2 and 45.1 eV of Re 4f7/2 (Fig. 5c). In comparison, the XPS signals of Re 4f5/2 and Re 4f7/2 in free ReO4– are reported at 48.3 and 45.9 eV [41]. These results clearly reveal a decrease of electron density of ReO4– in TNU-143(Re), caused by the coordination bonding between Ni and ReO4–. Meanwhile, the slight difference in the Ni 2p3/2 peaks observed in high-resolution XPS survey may be attributed to the different coordination environment of Ni in TNU-143 and TNU-143(Re) (Fig. 5d and Fig. S6 in Supporting information). Moreover, the morphology of the single crystals does not obviously change between TNU-143 and TNU-143(Re) (Fig. S7 in Supporting information).
Density function theory (DFT) calculations provide a comprehensive understanding of the anion-exchange process. We selected a typical fragment of TNU-143 as a theoretical model. Unlike the use of electrostatic adsorption of imidazole ring in the MOF material of SCU-102 reported by Wang [16], the present model is generated from the anion exchange of SO42– by ReO4– to form coordination interactions with Ni2+. In addition, the stability of adsorption is enhanced by the steric effect of macrocycles. The electrostatic potential (ESP) drawn based on the crystal structure (Fig. 6a) clearly shows that the Ni2+ is at the maximum value of the ESP, while the imidazole ring is located at the ESP minimum, further proving that the adsorption site is located at the Ni2+ and the imidazole rings are uncharged. Furthermore, the binding energy (∆BE) between TNU-143 and ReO4– is –23.38 kcal/mol, indicating a thermodynamically favorable anion exchange reaction (Fig. 6b), corresponding to the heated synthesis conditions of TNU-143.

This work presents a rare example of alkaline-resistant four-fold interpenetrated MOF, TNU-143, demonstrating excellent ReO4–/TcO4– segregation capabilities even in the simulated Hanford LAW and SRS HLW streams. The unique intertangled polymeric nets containing sterically crowded Ni2+ coordination geometry in TNU-143 create hydrophobic channels and specific trapping sites for the selective capture of ReO4–/TcO4–. Consequently, TNU-143 exhibits outstanding resistance to OH− attack in highly alkaline solutions. The underlying sorption mechanism was further elucidated by the single crystal structures of TNU-143(Re) and DFT calculation. These analyses clearly reveal that, coordination interactions between Ni2+ and ReO4–/TcO4−, instead of SO42–, results in the formation of more stable Ni–O bonds. This process follows a thermodynamically favorable anion exchange mechanism. This work not only reports a rare example of MOF sorbent showing superior performance in ReO4–/TcO4− sequestration under extreme conditions of high alkalinity and a large excess of competing anions, but also elucidates the sorption mechanism using SC-SC transformation methods, successfully opening up new insights into designing solid sorbent materials for pollutant remediation.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Lei Zhu: Methodology, Investigation. Hai-Ruo Li: Validation, Data curation. Yi-Ning Mao: Investigation, Data curation. Ruiying Liu: Software, Formal analysis. Bo Zhang: Investigation. Jing Chen: Visualization. Wengui Xu: Resources. Libo Zhang: Resources. Cheng-Peng Li: Writing – review & editing, Writing – original draft, Supervision, Conceptualization.
This work was supported by National Natural Science Foundation of China (No. 22171210), Research Project of Tianjin Education Commission (No. 2023KJ182), and Tianjin Research Innovation Project for Postgraduate Students (No. 2022BKY200).
Supplementary material associated with this article can be found, in the online version, at doi:
I.E.A., World Energy Outlook, 2022, p. 2022.
J. Li, X. Wang, G. Zhao, et al., Chem. Soc. Rev. 47 (2018) 2322–2356. doi: 10.1039/c7cs00543a
D. Banerjee, D. Kim, M.J. Schweiger, A.A. Kruger, P.K. Thallapally, Chem. Soc. Rev. 45 (2016) 2724–2739. doi: 10.1039/C5CS00330J
H. Ji, Y. Zhu, J. Duan, W. Liu, D. Zhao, Chin. Chem. Lett. 30 (2019) 2163–2168. doi: 10.1016/j.cclet.2019.06.004
Z.Y. Di, Z.F. Liu, H.R. Li, Z. Liu, C.P. Li, Inorg. Chem. Front. 10 (2023) 952–958. doi: 10.1039/d2qi02147a
J.X. Qi, C.R. Zhang, X.J. Chen, et al., Anal. Chem. 94 (2022) 10850–10856. doi: 10.1021/acs.analchem.2c01932
W. Zhou, A. Li, P.A. Gale, Q. He, Cell Rep. Phys. Sci. 3 (2022) 100875. doi: 10.1016/j.xcrp.2022.100875
C. Liu, J. Lan, Q. Yan, et al., Chin. Chem. Lett. 33 (2022) 3561–3564. doi: 10.3390/ma15103561
J. Li, L. Chen, N. Shen, et al., Sci. China Chem. 64 (2021) 1251–1260. doi: 10.1007/s11426-020-9962-9
L. Zhu, H.R. Li, Z.F. Liu, et al., Chem. Eur. J. 29 (2023) e202302168. doi: 10.1002/chem.202302168
W.R. Wilmarth, G.J. Lumetta, M.E. Johnson, et al., Solvent Extr. Ion Exch. 29 (2011) 1–48. doi: 10.1080/07366299.2011.539134
H.H. Fei, M.R. Bresler, S.R.J. Oliver, J. Am. Chem. Soc. 133 (2011) 11110–11113. doi: 10.1021/ja204577p
S. Wang, P. Yu, B.A. Purse, et al., Adv. Funct. Mater. 22 (2012) 2241–2250. doi: 10.1002/adfm.201103081
Y. Song, J. Phipps, C. Zhu, S. Ma, Angew. Chem. Int. Ed. 62 (2023) e202216724. doi: 10.1002/anie.202216724
C.P. Li, H.R. Li, J.Y. Ai, J. Chen, M. Du, ACS Cent. Sci. 6 (2020) 2354–2361. doi: 10.1021/acscentsci.0c01342
D. Sheng, L. Zhu, X. Dai, et al., Angew. Chem. Int. Ed. 58 (2019) 4968–4972. doi: 10.1002/anie.201814640
H. Xu, C.S. Cao, H.S. Hu, et al., Angew. Chem. Int. Ed. 58 (2019) 6022–6027. doi: 10.1002/anie.201901786
Z.F. Liu, K. Liu, X.J. Zheng, et al., Chem. Mater. 34 (2022) 5452–5460. doi: 10.1021/acs.chemmater.2c00377
Y. Wang, M. Xie, J. Lan, et al., Chem 6 (2020) 2796–2809. doi: 10.1016/j.chempr.2020.08.005
P. Zhang, Z. Wang, S. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202213247. doi: 10.1002/anie.202213247
Y. Huang, M. Ding, J. Ding, et al., Chem. Eng. J. 435 (2022) 134785. doi: 10.1016/j.cej.2022.134785
R. Zhao, D. Chen, N. Gao, et al., Adv. Funct. Mater. 32 (2022) 2200618. doi: 10.1002/adfm.202200618
D. Chen, Z. Liu, S. Li, et al., Chem. Eng. J. 452 (2023) 139148. doi: 10.1016/j.cej.2022.139148
C.P. Li, H. Zhou, J.J. Wang, et al., ACS Appl. Mater. Interfaces 11 (2019) 42375–42384. doi: 10.1021/acsami.9b16386
C.P. Li, H. Zhou, J. Chen, et al., ACS Appl. Mater. Interfaces 12 (2020) 15246–15254. doi: 10.1021/acsami.0c00775
A. Khayambashi, L. Chen, X. Dong, et al., Chin. Chem. Lett. 33 (2022) 3429–3434. doi: 10.1016/j.cclet.2022.02.011
N. Shen, Z. Yang, S. Liu, et al., Nat. Commun. 11 (2020) 5571. doi: 10.1038/s41467-020-19374-9
K. Kang, S. Liu, M. Zhang, et al., Adv. Funct. Mater. 32 (2022) 2208148. doi: 10.1002/adfm.202208148
Q.H. Hu, Y.Z. Shi, X. Gao, et al., Environ. Sci. Pollut. Res. 29 (2022) 86815–86824. doi: 10.1007/s11356-022-21870-y
A.V. Desai, A. Roy, P. Samanta, B. Manna, S.K. Ghosh, iScience 3 (2018) 21–30. doi: 10.1016/j.isci.2018.04.004
V. Colombo, S. Galli, H.J. Choi, et al., Chem. Sci. 2 (2011) 1311–1319. doi: 10.1039/c1sc00136a
A.V. Desai, B. Manna, A. Karmakar, A. Sahu, S.K. Ghosh, Angew. Chem. Int. Ed. 55 (2016) 7811–7815. doi: 10.1002/anie.201600185
K.M. Long, G.S. Goff, S.D. Ware, G.D. Jarvinen, W.H. Runde, Ind. Eng. Chem. Res. 51 (2012) 10445–10450. doi: 10.1021/ie300534e
J. Li, L. Zhu, C. Xiao, L. Chen, Z. Chai, S. Wang, Radiochim. Acta 106 (2018) 581–591. doi: 10.1515/ract-2017-2829
C. Xiao, A. Khayambashi, S. Wang, Chem. Mater. 31 (2019) 3863–3877. doi: 10.1021/acs.chemmater.9b00329
X. Wang, L. Chen, L. Wang, et al., Sci. China Chem. 62 (2019) 933–967. doi: 10.1007/s11426-019-9492-4
B. Aguila, D. Banerjee, Z. Nie, et al., Chem. Commun. 52 (2016) 5940–5942. doi: 10.1039/C6CC00843G
Q. Sun, B. Aguila, S. Ma, Trends Chem. 1 (2019) 292–303. doi: 10.1016/j.trechm.2019.02.010
Q. Sun, L. Zhu, B. Aguila, et al., Nat. Commun. 10 (2019) 1646. doi: 10.1038/s41467-019-09630-y
J. Li, B. Li, N. Shen, et al., ACS Cent. Sci. 7 (2021) 1441–1450. doi: 10.1021/acscentsci.1c00847
W. Liu, B.H. Han, Environ. Sci. Technol. 54 (2020) 216–224. doi: 10.1021/acs.est.9b05308

Figure 1 Crystal structures of TNU-143. (a) Coordination environment of Ni(Ⅱ). (b) Highlight of four-interpenetrated nets and its simplified pattern.

Figure 2 (a) FT-IR spectra, (b) PXRD patterns, (c) XPS surveys, and (d) STEM-EDS mapping of TNU-143 and TNU-143(Re).

Figure 3 (a) ReO4– sorption curve vs. time in TNU-143 suspension. Inset: Pseudo-second-order kinetic plot for sorption. (b) ReO4– sorption curve vs. concentration in water suspension of TNU-143. (c) Removal percentages of ReO4– by TNU-143 in various pH solutions. (d) PXRD patterns of TNU-143 after anion exchange of ReO4– in pH 0 and 14 for 12 h.

Figure 4 (a) Removal efficiency of ReO4− by TNU-143 in the presence of equimolar competing anions. (b) Removal efficiency of ReO4− by TNU-143 in the presence of excessive NO3−, (c) CO32− and (d) Cl–. (e) Comparison of the removal efficiency of ReO4− by TNU-143 after being exposed to Hanford LAW melter recycle stream, the solution of 3 mol/L HNO3 with 200 ppm Re, and simulated SRS HLW melter recycle streams with solid/liquid ratio of 5 g/L. (f) Removal efficiency of ReO4− by TNU-143 after being exposed to simulated SRS HLW melter recycle streams with various solid/liquid ratios.

Figure 5 (a) Coordination environment of Ni(Ⅱ) in TNU-143(Re). (b) 2D coordination layer of TNU-143(Re). (c) XPS survey spectra of Re and (d) Ni peaks in TNU-143(Re).

Figure 6 (a) Electrostatic potential (ESP) distribution on the van der Waals surfaces of TNU-143 (get rid of SO42– anion and H2O molecular). (b) Crystal models of the typical fragments of TNU-143 and TNU-143(Re). Corresponding color: yellow = Ni, orange = S, pink = Re, red = O, blue = N, gray = C and white = H.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: