Received Date:
24 January 2024 Accepted Date:
18 April 2024 Revised Date:
17 April 2024 Available Online:
15 December 2024
Abstract:
Temperature plays a crucial role in regulating polymorphism in supramolecular polymers. Understanding the mechanism behind temperature-dependent supramolecular polymorphism is crucial as it provides an opportunity to tailor polymorphs for specific properties and applications. In this study, we present our findings on a naphthalimide-substituted benzene-1,3,5-tricarboxamide derivative, R-Nap-1, which exhibits two distinct polymerization pathways at varying temperatures. At 313 K, polymerization results in the formation of an M-chiral polymorph, whereas at 253 K, a P-chiral polymorph is formed. Both polymorphs are notably stable, remaining unchanged for over six months under ambient conditions. Theoretical calculations and experimental investigations allowed us to elucidate the mechanisms underlying these polymorphic transformations. The formation of the M-chiral polymorph at 313 K is attributed to the nucleation and growth of R-Nap-1 monomers once their concentration surpasses a critical threshold. Conversely, at lower temperatures (e.g., 253 K), the monomers undergo facile transformation into dimers due to a lower energy barrier and reduced Gibbs energy compared to the monomeric state. Subsequently, these dimers undergo nucleation-elongation to form the P-chiral polymorph when their concentration exceeds the critical polymerization concentration. The stability and lack of interconversion between the two polymorphs can be attributed to their close thermodynamic stabilities, as evidenced by variable-temperature CD spectra and DFT calculations. These findings highlight the importance of accurate temperature control in supramolecular polymerization processes, making a significant contribution to the understanding of supramolecular polymorphism, thus advancing the field of supramolecular chemistry.
Polymorphism is a widely observed phenomenon in crystalline materials, wherein a single substance can adopt different arrangements within the unit cells, resulting in the formation of multiple crystalline phases with distinct performances [1–3]. For example, there are several species of calcium carbonate crystalline polymorphs found in nature, including aragonite, calcite, vaterite, monohydrocalcite, and ikaite, each showcasing specific physicochemical attributes [4,5]. In the pharmaceutical industry, polymorphism also carries significant implications for drug dissolution rate, solubility, bioavailability, and efficacy [6–8]. Consequently, substantial efforts have been dedicated to comprehending and controlling the formation of crystal polymorphs [9–11].
On the other hand, supramolecular polymers have gained significant attention within the scientific community due to their versatile design [12–25], reversible properties [26–28], responsiveness to stimuli [29–33], and tunable characteristics [34–43]. The investigation of supramolecular polymerization mechanism has unveiled the existence of "pathway complexity" [44] and "on/off pathway" [45–61] phenomena, occurring during the transition from monomers to kinetically and thermodynamically stable aggregates. Additionally, supramolecular polymorphism, characterized by the formation of distinct stable polymers from identical monomers under different conditions, has been observed [62–70]. By manipulating external factors such as temperature, solvent, and concentration, it becomes possible to achieve diverse supramolecular polymorphs with unique properties. Notably, the effect of temperature on supramolecular polymorphism has been extensively studied. For instance, Wan et al. reported the formation of right-handed ribbons when a D-glucoside-based low-molecular-mass gelator was rapidly cooled, whereas left-handed ribbons were observed during slow cooling, the ribbons were further employerd as templates for constructing an opposite chiral arrangement of silver nanoparticles [71,72]. Additionally, Würthner and colleagues discovered three distinct polymorphs of a perylene bisimide (PBI)-based molecule by manipulating temperature and sonication time, with these polymorphs exhibiting long-term stability at room temperature [69]. Recently, Liu et al. found a 3-(4-pyridyl)acrylic acid glutamide derivative present distinct chiral nanotwist and nanotubes depending on the cooling rate from hot monomer solutions [73]. Therefore, it is highly desirable to develop temperature-dependent supramolecular polymorphs and unravel the underlying mechanisms.
In this study, we investigated the supramolecular polymorphic behavior of R-Nap-1, a derivative of naphthalimide-substituted benzene-1,3,5-tricarboxamide. The R-Nap-1 system demonstrates temperature-dependent supramolecular polymorphism behavior, which should be caused by the temperature-dependent molecular conformations, as depicted in Fig. 1. At room temperature, R-Nap-1 monomers adopt a folded conformation with intramolecular hydrogen bonding. However, with increasing temperature, the predominant conformation of R-Nap-1 transitions to an extended form without hydrogen bonding. Conversely, decreasing the temperature to 253 K may encourage the preferential formation of energy-favorable dimers. Remarkably, when the concentration of R-Nap-1 in the solvent exceeds a critical threshold, two distinct supramolecular polymerization pathways are observed at different temperatures. Polymerization conducted at 313 K results in the formation of a thermally stable M-chiral polymorph. Conversely, conducting polymerization at 253 K under the same solvent and concentration conditions generates a P-chiral polymorph, as depicted in Fig. 1b. Notably, both of two polymorphs have demonstrated long-term stability, lasting over six months under ambient conditions. The combination of theoretical calculations and experimental results provides compelling evidence that the M-chiral supramolecular polymorph arises from the nucleation and growth of R-Nap-1 monomers at 313 K. Conversely, the P-chiral polymorph originates from the cooperative polymerization of R-Nap-1 dimers at 253 K. These findings provide valuable insights into the governance of supramolecular polymorphism by distinct molecular units during polymerization under different temperature conditions.
Figure 1
Figure 1.
(a) Chemical structures of R-Nap-1 and multiple molecular conformations based on the organization of intramolecular hydrogen bonding. (b) The diagram of supramolecular polymorphism of R-Nap-1 under different temperatures, where M-helical polymorph Agg313 was obtained under 313 K (pathway Ⅰ), and P-helical polymorph Agg253 was obtained under 253 K (pathway Ⅱ).
R-Nap-1 and S-Nap-1 were synthesized according to a multistep procedure, as depicted in Scheme S1 (Supporting information). Firstly, trimesoyl chloride was reacted with either 2 N of R-2-aminooctane or S-2-aminooctane, resulting in the formation of a benzene dicarboxamide intermediate. Subsequently, this intermediate was further reacted with (2-aminoethyl)naphthalimide to obtain R-Nap-1 or S-Nap-1. The synthesis and characterization details can be found in Supporting information.
R-Nap-1 displays high solubility in chloroform (CHCl3) but low solubility in toluene (Tol). However, when R-Nap-1 was heated and dissolved in a series of solvent mixtures consisting of CHCl3 and Tol, followed by cooling at 293 K, the formation of a transparent gel was observed (Fig. S1 in Supporting information, cT = 1 × 10−3 mol/L), indicating supramolecular polymerization. Furthermore, the optimal medium for studying the supramolecular polymerization of R-Nap-1 was determined to be a solvent mixture with a volumetric ratio of CHCl3: Tol = 5:95, based on the highest intensity of the CD signal observed in this specific solvent mixture at polymerization temperatures of 313 K and 253 K (Fig. S2 in Supporting information).
To investigate the temperature-dependent supramolecular polymerization pathway, the following experiments were conducted. Initially, R-Nap-1 was fully dissolved in a CHCl3/Tol solvent at 353 K at the concentration of 1 × 10−3 mol/L (Fig. S3 in Supporting information, CD silence). The resulting hot solution was quickly transferred to a pre-cooled chamber maintained at a specific temperature for approximately 2 h to facilitate sufficient supramolecular polymerization. Subsequently, the sample was removed from the chamber and allowed to equilibrate to room temperature for further characterization (Fig. 2a). Remarkably, supramolecular polymerization occurred within the temperature range of 318 K to 278 K, leading to the formation of a transparent gel (Fig. 2a and Fig. S4a in Supporting information). However, when the temperature for supramolecular polymerization ranged from 273 K to 248 K, only a suspension was observed (Fig. 2a and Fig. S4a). These findings strongly indicate the presence of distinct self-assembly pathways depending on the temperature of supramolecular polymerization.
Figure 2
Figure 2.
(a) The supramolecular polymerization procedure of R-Nap-1 in a CHCl3/Tol solvent (cT = 1 × 10−3 mol/L), R-Nap-1 is initially dissolved in the solvent mixture at 353 K and then quickly transferred to a pre-cooled chamber maintained at a specific temperature for supramolecular polymerization. At 313 K, a transparent gel consisting of thin nanofibers is formed, while at 253 K, a suspension consisting of nanofiber bundles is observed. (b) UV–vis absorption (ABS) and (c) fluorescence (FL) spectra of R-Nap-1 in a diluted CHCl3/Tol solvent (cT = 1 × 10−5 mol/L), as well as the aggregated states Agg313 and Agg253 (cT = 1 × 10−3 mol/L). (d) The circular dichroism (CD) and (e) circularly polarized luminescence (CPL) spectra of R/S-Agg313 and R/S-Agg253 obtained from R-Nap-1 and S-Nap-1 respectively (cT = 1 × 10−3 mol/L). In all cases, the solvent mixture of CHCl3 and Tol was with a volumetric ratio of 5:95.
Distinct microstructures were observed when analyzing the morphology of supramolecular polymers obtained at different temperatures. Polymerization carried out within the temperature range of 318 K to 298 K yielded well-defined nanofibers with widths ranging from 50 nm to 100 nm and lengths reaching several hundred micrometers. On the other hand, polymerizations conducted at temperature ranging from 273 K to 248 K resulted in thicker nanofiber bundles with widths of 200–500 nm and lengths of several tens micrometers (Fig. 2a and Fig. S4b in Supporting information). At temperature ranging from 293 K to 278 K, a combination of nanofibers and nanofiber bundles was observed (Fig. S4b). These results suggest that the preferential formation of a gel-like structure occurs through an entangled network composed of thin nanofibers, while the presence of bundle-like nanostructures tends to result in a suspension.
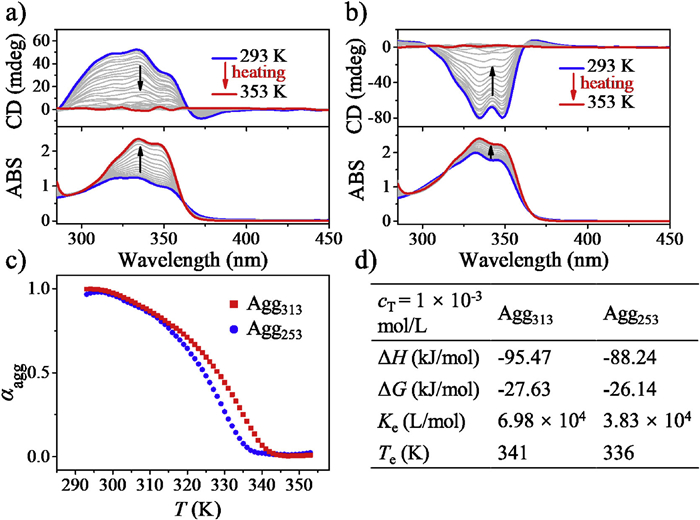
Distinct circular dichroism (CD) signals were observed for polymers obtained at various temperatures, as illustrated in Fig. 2d and Fig. S5 (Supporting information). Polymers obtained between 318 K and 303 K exhibited a bisignated Cotton effect with a crossover at approximately 365 nm. Negative maxima were observed at 370 nm, while positive maxima appeared at 334 nm, indicating the presence of M-chiral naphthalimide-naphthalimide (Nap-Nap) packing in the polymers. The CD signal intensity gradually increased with higher supramolecular polymerization temperatures, peaking at 313 K, as shown in Fig. S5. Subsequently, as the temperature continued to rise, there was a reduction in the CD signal intensity. Interestingly, supramolecular polymers formed between 298 K and 248 K displayed an inverse bisignated Cotton effect, with negative maxima around 334 nm and positive maxima at 365 nm, suggesting a preference for P-chiral packing of the Nap chromophores in these polymers (Fig. 2d and Fig. S5). Notably, as the supramolecular polymerization temperature decreased, the CD signal intensity gradually increased, reaching its maximum at 253 K (Fig. S5). To further elucidate the underlying mechanism of supramolecular polymorphism, we selected two model polymers with the most prominent chiral signals: Agg313 and Agg253, obtained at 313 K and 253 K, respectively. These model polymers will serve as representatives for investigating the underlying phenomena in detail.
The absorption and emission spectra of R-Nap-1 monomer, Agg313 and Agg253 were investigated. In a diluted CHCl3/Tol solvent, the absorption spectrum of R-Nap-1 showed an absorption band at 335.5 nm, corresponding to the π→π* electronic transition of the Nap moiety (Fig. 2b, cT = 1 × 10−5 mol/L). However, the absorption bands of both Agg313 and Agg253 exhibited hypsochromic shifts which were 333.1 nm and 332.6 nm, respectively, indicating the formation of H-type aggregation in both polymorphs (Fig. 2b). The fluorescence spectrum of R-Nap-1 in dilute solution exhibited a broad emission with a maximum at around 402 nm. However, Agg313 and Agg253 exhibited red-shifted emission maxima at approximately 448 nm and 440 nm respectively, suggesting the presence of intermolecular π-π stacking among the Nap chromophores (Fig. 2c). Moreover, the excited states of Agg313 and Agg253 polymorphs exhibited distinct chiral emission behavior. Agg313 exhibited a negative circularly polarized luminescence (CPL) signal at approximately 450 nm, while Agg253 exhibited a stronger positive CPL signal at around 440 nm (Fig. 2e). These results indicated significant differences in the molecular packing structures between Agg313 and Agg253 polymorphs. As expected, the polymorphs obtained from S-Nap-1 exhibited nearly mirrored CD and CPL characteristics compared to R-Nap-1 polymorphs, respectively (Figs. 2d and e). It is worth noting that both CD and CPL signals of Agg313 and Agg253 are only slightly changed after aging for six months under ambient condition. This finding indicated that the polymorphs maintain long-term stability without undergoing interconversion (Fig. S6 in Supporting information).
The concentration-dependent supramolecular polymerization behavior of R-Nap-1 was investigated at temperatures of 313 K and 253 K using their CD and FL spectra. The critical concentrations for polymerization were determined by analyzing the concentration-dependent CD spectra (Figs. S7a and c in Supporting information). At 313 K, the critical concentration was found to be 3 × 10−4 mol/L, while at 253 K, it was 2 × 10−4 mol/L. The degree of polymerization gradually increased and reached its maximum when the concentration of R-Nap-1 exceeded 7 × 10−4 mol/L at both temperatures (Figs. S7a, S7c and S8 in Supporting information). Analysis of FL spectra revealed the similar trends. At 313 K, as the concentration of R-Nap-1 increased from 1 × 10−4 mol/L to 3 × 10−4 mol/L, there was a red shift in the emission maximum from 402 nm to approximately 439 nm. Further increasing the concentration beyond 7 × 10−4 mol/L caused an additional red shift, resulting in an emission maximum around 448 nm (Fig. S7b in Supporting information). A similar behavior was observed for polymerization at 253 K, where the emission maximum red-shifted to 436 nm at a concentration of 2 × 10−4 mol/L and reached equilibrium beyond a concentration of 7 × 10−4 mol/L. These findings indicate that polymorphs can form once the concentration of R-Nap-1 surpasses a critical value, with Agg253 being obtained at a relatively lower concentration compared to Agg313.
Variable-temperature CD spectra were employed to investigate the supramolecular polymerization mechanism of the Agg313 and Agg253 polymorphs (Figs. 3a and b, Fig. S9 in Supporting information). However, due to the considerable difficulties in monitoring the polymerization process for Agg313 and Agg253, only the dissociation process was considered for understanding the polymerization mechanism. The degree of aggregation (αagg) was determined by analyzing the maximum Cotton effect at 334 nm and plotted against temperature (Fig. 3c and Fig. S10 in Supporting information). For Agg313, the αaggversus temperature curve exhibited a nonsigmoidal shape with a sharp transition at the critical temperature (Te), indicating a cooperative nucleation-elongation model. Increasing the concentration from 8 × 10−4 mol/L to 1.2 × 10−3 mol/L resulted in an increase in Te from 336 K to 346 K (Table S1 in Supporting information). Similarly, the temperature-dependent αagg for Agg253 also followed a cooperative polymerization mechanism (Fig. 3c and Fig. S10 in Supporting information). Increasing the concentration led to elevated Te values, ranging from 331 K to 341 K. The corresponding thermodynamic parameters ΔH, ΔG, and Ke of both Agg313 and Agg253 were calculated using a nucleation-elongation model (Fig. 3d, Tables S1 and S2 in Supporting information). Notably, for each concentration, the ΔG value of both polymorphs approximated closely, while the Agg313 polymorph consistently displayed slightly negative values compared to that of the Agg253 polymorph. Thus, from the thermodynamic analysis, it can be concluded that R-Nap-1 formed a more thermodynamically stable aggregate at 313 K than at 253 K. In addition, a thermal hysteresis between cooling and heating curves of αagg was observed under a cooling/heating rate of 1 K/min, indicating a kinetically controlled cooling process and the existence of kinetic metastable state (Fig. S11 in Supporting information).
Figure 3
Figure 3.
Variable-temperature CD spectra of (a) Agg313 and (b) Agg253 obtained from heating process (1 K/min), respectively. (c) Plot of the αaggversus temperature for Agg313 and Agg253, cT = 1 × 10−3 mol/L. (d) Thermodynamic parameters of Agg313 and Agg253 were calculated using a nucleation-elongation model.
To comprehensively investigate the supramolecular polymorphism mechanism of R-Nap-1, several crucial steps were undertaken. Initially, the molecular conformation of the R-Nap-1 monomer was thoroughly examined. Subsequently, the packing arrangement of R-Nap-1 in both aggregates and dimeric forms was meticulously analyzed. Finally, the supramolecular polymerization pathway and energy landscape were elucidated to facilitate a deeper understanding of the mechanisms underlying the supramolecular polymorphism observed in R-Nap-1.
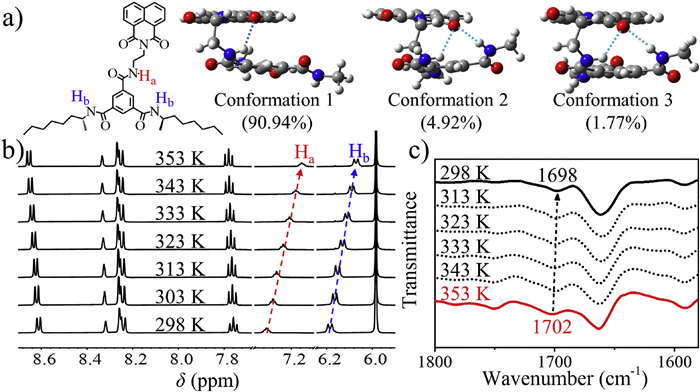
To investigate the molecular conformation of monomeric R-Nap-1, variable-temperature 1H NMR spectroscopy was conducted in a soluble solvent, specifically 1,1,2,2-tetrachloroethane-d2 (TCE-d2), at a concentration of 1 × 10−3 mol/L. The obtained results revealed temperature-dependent changes in two distinct types of protons. One type originated from the amide group directly linked to the Nap moiety, exhibiting a triplet signal split. The other type arose from the adjacent symmetrical amide on the benzene ring, displaying a doublet signal split. Remarkably, as the temperature increased from 298 K to 353 K (Fig. 4b), both types of protons exhibited up-field shifts. These observed shifts can be possibly attributed to the intramolecular hydrogen bonds in monomeric R-Nap-1 at 298 K. However, elevating temperature disrupts these hydrogen bonding interactions due to increased molecular vibrational energy. Furthermore, theoretical calculations were performed to simulate the molecular conformation of monomeric R-Nap-1. MD simulations based on the xTB method [74] and QM calculations employing B3LYP-D3/6-31G(d) and wB97XD/aug-cc-pvtz levels within the Gaussian16 program [75] were combined to conduct a conformational search. The optimized most stable molecular structures in toluene, as shown in Fig. 4a, supported the formation of a foldable structure through intramolecular hydrogen bonding interactions involving the N—H group of the amide and the C═O group of Nap. In addition, the stretching vibration of the C═O band of the Nap moiety shifted from 1702 cm−1 to 1698 cm−1 as the temperature decreased from 353 K to 298 K (Fig. 4c), providing additional evidence supporting the presence of intramolecular hydrogen bonding between the amide hydrogen and the carbonyl oxygen within Nap at 298 K. Consequently, based on these findings, it can be concluded that R-Nap-1 molecule tends to adopt a folded conformation at room temperature, facilitated by the presence of intramolecular hydrogen bonds. This conformational arrangement hinders the formation of intermolecular hydrogen bonding among R-Nap-1 molecules, thereby impeding polymerization at low concentrations (e.g., cT < 2 × 10−4 mol/L at 253 K).
Figure 4
Figure 4.
(a) The most stable molecular structures of R-Nap-1 monomer in toluene obtained by conformational search calculations, the calculated Boltzmann polulation at 298 K of each conformation is listed in parentheses. (b) Variable-temperature 1H NMR spectroscopy of R-Nap-1 dissolved in TCE-d2 (cT = 1 × 10−3 mol/L). (c) Variable-temperature Fourier-transform infrared (FT-IR) spectrum of the R-Nap-1 dissolved in TCE (cT = 1 × 10−3 mol/L).
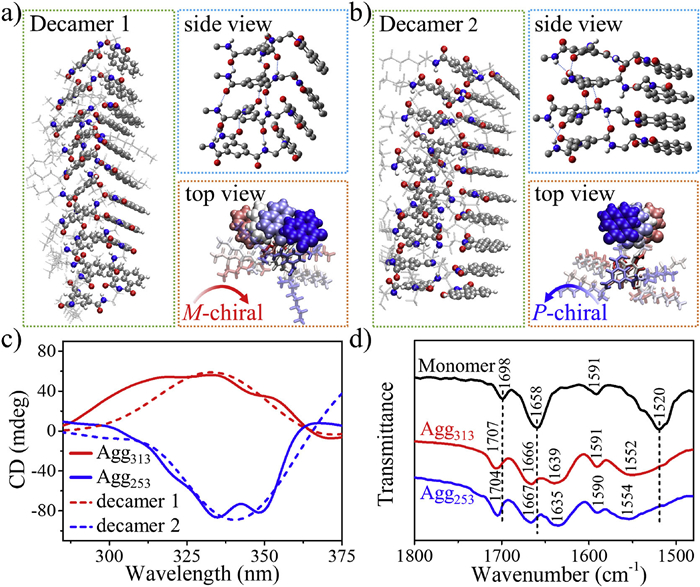
To study the self-assembling process and understand the packing structure of Agg313 and Agg253, conformational search computation has also been done on R-Nap-1 decamers. The Molclus program's "gentor" [76] feature was utilized to generate a series of initial structures for R-Nap-1 decamers. The large systems were optimized using the semiempirical GFN2-xTB method, known for its similarity to DFTB3 [77,78]. Two most stable decamer structures were identified, as depicted in Fig. 5. Decamer 1 was found to be the global energy minimum with a Boltzmann population of 88.44% at 298 K. In this structure, three hydrogen bonds existed between the amide protons and amide oxygen, and the R-Nap-1 monomeric repeating units exhibited M-chiral packing. Decamer 2 is the second lowest energy state, slightly higher than decamer 1 (ΔEtot= 1.22 kcal/mol), possessed a Boltzmann population of 11.23%. Detailed examination revealed that decamer 2 consisted of dimers serving as basic units, featuring three hydrogen bonds within each dimer and two additional hydrogen bonds connecting the dimers. Consequently, a P-chirality packing arrangement was observed. To further analyze the structures, we further calculated the CD spectra of the selected tetramers of decamer 1 and decamer 2 by using (TD)B3LYP-D3/6-31G(d, p). Remarkably, the theoretical CD spectrum of decamer 1 closely matched the experimental data of Agg313, while the theoretical CD spectrum of decamer 2 resembled the experimental data of Agg253 (Fig. 5c). Therefore, combining these findings with the fitted thermodynamic parameter ΔG of Agg313 and Agg253, it can be inferred that Agg313 adopts the packing structure of decamer 1, while Agg253 corresponds to decamer 2. Notably, the stretching frequency of C═O group in Nap for both Agg313 and Agg253 was around 1706 cm−1 (Fig. 5d), indicating that the carbonyl groups within Nap do not participate in hydrogen bonding in polymorphs, which is consistent with the proposed model.
Figure 5
Figure 5.
The theoretical two most stable R-Nap-1 decamer configurations (a) decamer 1 and (b) decamer 2, where the selected tetramers were particularly shown in sideview with hydrogen bonds and in topview with π-π stacking Naps. (c) Experimental CD spectra of Agg313 and Agg253 and theoretical CD spectra of the selected tetramers of decamer 1 and decamer 2. (d) FT-IR spectrum of R-Nap-1 monomer, Agg313 and Agg253.
In order to assess the potential formation of a dimer in the supramolecular polymerization process of Agg253, theoretical calculations were conducted. The aggregation behavior of two optimized R-Nap-1 molecules at different temperatures (253 K and 313 K) in toluene were studied by MD combined with GFN2-xTB method respectively. A starting configuration consisting of two folded monomers of R-Nap-1 in a face-to-face stacking structure was employed for the MD simulations, where a total simulation time 3 ns with a time step of 2 fs was set. Fig. S12 (Supporting information) presents selected MD snapshots of the two R-Nap-1 molecules at 253 K and 313 K. At 253 K, the simulations demonstrated a gradual unfolding of the folded molecules. Due to the π-π stacking interaction between Naps and intermolecular hydrogen bonding forces, a stable dimer structure with π-π stacked Naps formed within 100 ps and remained stable thereafter. However, at 313 K, despite the unfolding of the two folded molecules within 10 ps, the dimer exhibited significant changes throughout the simulation due to enhanced molecular vibrations resulting from the higher temperature. The histograms of the distances between two Naps of all MD snapshots helped us visualize the good stability of π-π stacking dimer structure at 253 K, while the instability of dimer at 313 K (Fig. S13 in Supporting information). These observations suggest that dimer formation is favored at 253 K but hindered at 313 K. Additionally, a conformational search calculation was conducted to determine the R-Nap-1 dimer structure as applied to the R-Nap-1 monomer. The resulting optimized structures, depicted in Fig. S14 (Supporting information), revealed the presence of both intramolecular and intermolecular hydrogen bonds in the R-Nap-1 dimer. Moreover, the binding energy of the dimer, calculated as Ebind = (Gdimer – 2Gmonomer)/2, was found to be −4.168 kcal/mol, indicating R-Nap-1 molecules are energetically favorable for the formation of dimer.
The formation of the dimer species at 253 K can be verified by the following experiments. Firstly, the variable temperature 1H NMR spectroscopy was conducted on R-Nap-1 in a deuterated toluene and CDCl3 mixture solution with a concentration of 1 × 10−4 mol/L, which is below the critical polymerization concentration, allowing for the capture of dimer species. The results revealed pronounced broadening and reduced intensity of proton signals corresponding to Phenyl and Nap moieties as the temperature decreased from 298 K to 253 K, indicating the occurrence of π-π stacking between Naps and Phenyls. In contrast, no broadening was observed for the proton signals these moieties in the case of monomeric R-Nap-1 dissolved in TCE-d (Fig. S15 in Supporting information). This significant contrast further supports the proposition that the observed changes are specific to the formation of dimeric structures at 253 K. Moreover, the proton signal spectrum of R-Nap-1 in the deuterated toluene and CDCl3 mixture solution returned to its initial state upon returning to 298 K, indicating the reversible nature of the dimerization process, which is well consistent with the theoretical calculation (Fig. S15). In addition, fluorescence spectra of R-Nap-1 in CHCl3/Tol solvent (cT = 1 × 10−4 mol/L) exhibited a red-shifted emission maximum from 402 nm to 406 nm as the temperature decreased from 298 K to 253 K, which reverted to 402 nm upon recovery to 298 K. Conversely, no obvious red-shift was observed for monomeric R-Nap-1 in TCE as the temperature decreased (Fig. S16 in Supporting information). These results further support the formation of dimeric structure of R-Nap-1 in CHCl3/Tol solvent at 253 K. Moreover, matrix-assisted laser desorption/ionization (MALDI) mass spectrometry demonstrated distinct signals for the dimeric and monomeric forms of R-Nap-1. Specifically, an observable signal at m/z = 1331.7 corresponding to the Na+ adduct of dimer, along with a peak representing the Na+ adduct of the monomer at m/z = 677, were detected for Agg253 (Fig. S17 in Supporting information). In contrast, only a monomeric signal could be observed for Agg313. These results indicated the existence of dimer species in the case of Agg253. Therefore, the spectral and mass spectrometry data demonstrated the formation of the R-Nap-1 dimer species at a temperature of 253 K.
Based on theoretical calculations and experimental results, the energy landscape was illustrated in Fig. 6, revealing two distinct pathways dependent on the polymerization temperature. R-Nap-1 monomer exists as folded conformation due to the intramolecular hydrogen bonding between amides and Nap moiety at room temperature. At lower temperatures (e.g., 253 K), the monomers readily undergo transformation into dimers, facilitated by a low energy barrier and lower Gibbs free energy compared to that of the monomeric state. However, the dimer lacks stability at higher temperatures (e.g., 313 K) due to increased molecular vibrational energy. Supramolecular polymerization leads to the formation of Agg253 when the concentration of R-Nap-1 surpasses a critical threshold (2 × 10−4 mol/L) at 253 K. The presence of sufficient dimers in the system initiates nucleation, which subsequently elongates into the P-chiral Agg253 aggregates. Conversely, at 313 K, the dominant species are the monomeric R-Nap-1 molecules, and nucleation proceeds towards the thermodynamically stable M-chiral Agg313 as the concentration reaches 3 × 10−4 mol/L. An additional experiment demonstrated that polymerization does not occur with R-Nap-1 at a concentration of 2 × 10−4 mol/L at 313 K, while the Agg253 polymorph could be obtained upon cooling the solution to 253 K (Fig. S18 in Supporting information). Therefore, the formation of stable dimers and their energetic preference for aggregation towards Agg253 is crucial factors for achieving polymorphism. It is noteworthy that both Agg253 and Agg313 exhibit similarly low energies, contributing to the exceptional stability observed for these two distinct polymorphs.
Figure 6
Figure 6.
The energy landscape of R-Nap-1 supramolecular polymerization pathways at 253 K and 313 K respectively.
Furthermore, the supramolecular polymerization behavior of a similar derivative R-Nap-2 was investigated. In R-Nap-2, the alkyl chain connecting the Nap and benzene ring was elongated from two methylene units to three methylene units (Fig. S19 in Supporting information). The results revealed that at 313 K, R-Nap-2 exhibited a CD signal pattern comparable to that observed in R-Nap-1 based Agg313, indicating the formation of M-chiral aggregates. However, when the polymerization process was conducted at a lower temperature of 253 K, a similar CD signal pattern was observed, albeit with diminished intensity (Fig. S19b). This observation suggests that M-chiral aggregates were also formed at 253 K, which is different from that of R-Nap-1, and there is a lack of polymorphism in the R-Nap-2 system. The CPL spectra of R-Nap-2 presented similar behavior (Fig. S19c). Additionally, variable temperature 1H NMR analysis and Theoretical calculations further demonstrated the formation of a folded structure for R-Nap-2 monomer under ambient condition, similar to R-Nap-1 (Figs. S19a and S20 in Supporting information). However, MD snapshots revealed that the formation of dimers is unfavorable for R-Nap-2 at both 313 K and 253 K (Fig. S21 in Supporting information). As a result, R-Nap-2 could only undergo monomeric nucleation polymerization to form M-chiral aggregates at both temperatures. These findings provide further confirmation that the formation of a stable dimer, as observed in R-Nap-1 at low temperatures, plays a pivotal role in the phenomenon of polymorphism.
In conclusion, the supramolecular polymorphism of a naphthalimide-substituted benzene-1,3,5-tricarboxamide derivative R-Nap-1 was carefully investigated. The study provides insights into the formation of two distinct temperature-dependent polymorphs. At 253 K, the P-chiral Agg253 polymorph was observed, while at 313 K, the M-chiral Agg313 polymorph was formed. Both the Agg253 and Agg313 polymorphs exhibited exceptional stability, demonstrated by minimal changes in their CD and CPL signals over six months. Theoretical calculations and experimental findings supported the hypothesis that the R-Nap-1 monomers actively engaged in cooperative polymerization, leading to the formation of the M-chiral Agg313 polymorph at 313 K. Conversely, at 253 K, R-Nap-1 monomers favored the formation of energetically favorable dimers, thereby promoting the nucleation elongation of dimers and resulting in the formation of the P-chiral Agg253 polymorph. These findings significantly contribute to our understanding of the relationship between supramolecular polymorphism and polymerization temperature, thereby highlighting the potential to selectively obtain supramolecular polymorphs with unique properties through meticulous temperature control.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was supported by the Chinese Academy of Sciences CAS Project for Young Scientists in Basic Research (No. YSBR-027).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109909.
J. Matern, Y. Dorca, L. Sánchez, G. Fernández, Angew. Chem. Int. Ed. 58 (2019) 16730–16740. doi: 10.1002/anie.201905724
[49]
A.T. Haedler, S.C.J. Meskers, R.H. Zha, et al., J. Am. Chem. Soc. 138 (2016) 10539–10545. doi: 10.1021/jacs.6b05184
[50]
J. Kang, D. Miyajima, T. Mori, et al., Science 347 (2015) 646–651. doi: 10.1126/science.aaa4249
[51]
J.S. Valera, R. Gómez, L. Sánchez, Small 14 (2017) 1702437.
[52]
S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu, M. Takeuchi, Nat. Chem. 6 (2014) 188–195. doi: 10.1038/nchem.1849
[53]
M. Wehner, M.I.S. Röhr, V. Stepanenko, F. Würthner, Nat. Commun. 11 (2020) 5460. doi: 10.1038/s41467-020-19189-8
[54]
C. Naranjo, S. Adalid, R. Gómez, L. Sánchez, Angew. Chem. Int. Ed. 135 (2023) e202218572. doi: 10.1002/ange.202218572
[55]
I. Helmers, G. Ghosh, R.Q. Albuquerque, G. Fernández, Angew. Chem. Int. Ed. 60 (2020) 4368–4376.
[56]
C.A. Shen, D. Bialas, M. Hecht, et al., Angew. Chem. Int. Ed. 60 (2021) 11949–11958. doi: 10.1002/anie.202102183
[57]
T. Fukui, S. Kawai, S. Fujinuma, et al., Nat. Chem. 9 (2017) 493–499. doi: 10.1038/nchem.2684
[58]
S. Ogi, V. Stepanenko, J. Theirs, F. Würthner, J. Am. Chem. Soc. 138 (2016) 670–678. doi: 10.1021/jacs.5b11674
[59]
H. Choi, S. Ogi, N. Ando, S. Yamaguchi, J. Am. Chem. Soc. 143 (2021) 2953–2961. doi: 10.1021/jacs.0c13353
[60]
E.E. Greciano, J. Calbo, E. Ortí, L. Sanchez, Angew. Chem. Int. Ed. 59 (2020) 17517–17524. doi: 10.1002/anie.202005837
[61]
F. Wang, R. Liao, F. Wang, Angew. Chem. Int. Ed. 62 (2023) e202305827. doi: 10.1002/anie.202305827
[62]
M. Hecht, P. Leowanawat, T. Gerlach, et al., Angew. Chem. Int. Ed. 59 (2020) 17084–17090. doi: 10.1002/anie.202006744
[63]
Z. Fernandez, B. Fernandez, E. Quinoa, F. Freire, Angew. Chem. Int. Ed. 60 (2021) 9919–9924. doi: 10.1002/anie.202100162
[64]
S. Datta, D. Chaudhuri, Angew. Chem. Int. Ed. 61 (2022) e202201956. doi: 10.1002/anie.202201956
[65]
N. Suda, T. Saito, H. Arima, S. Yagai, Chem. Sci. 13 (2022) 3249–3255. doi: 10.1039/d2sc00690a
[66]
B. Bartolec, A. Kiani, M.A. Beatty, et al., Chem. Sci. 13 (2022) 14300–14304. doi: 10.1039/d2sc03909e
[67]
N.M. Matsumoto, R.P.M. Lafleur, X.W. Lou, et al., J. Am. Chem. Soc. 140 (2018) 13308–13316. doi: 10.1021/jacs.8b07697
[68]
A. Langenstroer, K.K. Kartha, Y. Dorca, et al., J. Am. Chem. Soc. 141 (2019) 5192–5200. doi: 10.1021/jacs.8b11011
[69]
M. Wehner, M.I.S. Röhr, M. Bühler, et al., J. Am. Chem. Soc. 141 (2019) 6092–6107. doi: 10.1021/jacs.9b02046
[70]
S. Bujosa, A. Doncel-Giménez, N. Bäumer, et al., Angew. Chem. Int. Ed. 61 (2022) e202213345. doi: 10.1002/anie.202213345
[71]
R. Wang, J. Cui, X. Wan, J. Zhang, Chem. Commun. 55 (2019) 4949–4952. doi: 10.1039/c9cc01015g
[72]
J. Cui, A. Liu, Y. Guan, et al., Langmuir 26 (2010) 3615–3622. doi: 10.1021/la903064n
[73]
S. Du, Y. Jiang, H. Jiang, L. Zhang, M. Liu, Angew. Chem. Int. Ed. 63 (2023) e202316863.
[74]
C. Bannwarth, S. Ehlert, S. Grimme, J. Chem. Theory Comput. 15 (2019) 1652–1671. doi: 10.1021/acs.jctc.8b01176
[75]
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 16 Rev. C. 01, Wallingford, CT, 2016.
[76]
T. Lu, Molclus program, Version 1, 9.9.7, http://www.keinsci.com/research/molclus.html (accessed 17 February 2022).
[77]
Y. Yang, H.B. Yu, D. York, Q. Cui, M. Elstner, J. Phys. Chem. A 111 (2007) 10861–10873. doi: 10.1021/jp074167r
[78]
M. Gaus, A. Goez, M. Elstner, J. Chem. Theory Comput. 9 (2013) 338–354. doi: 10.1021/ct300849w
Figure 1
(a) Chemical structures of R-Nap-1 and multiple molecular conformations based on the organization of intramolecular hydrogen bonding. (b) The diagram of supramolecular polymorphism of R-Nap-1 under different temperatures, where M-helical polymorph Agg313 was obtained under 313 K (pathway Ⅰ), and P-helical polymorph Agg253 was obtained under 253 K (pathway Ⅱ).
Figure 2
(a) The supramolecular polymerization procedure of R-Nap-1 in a CHCl3/Tol solvent (cT = 1 × 10−3 mol/L), R-Nap-1 is initially dissolved in the solvent mixture at 353 K and then quickly transferred to a pre-cooled chamber maintained at a specific temperature for supramolecular polymerization. At 313 K, a transparent gel consisting of thin nanofibers is formed, while at 253 K, a suspension consisting of nanofiber bundles is observed. (b) UV–vis absorption (ABS) and (c) fluorescence (FL) spectra of R-Nap-1 in a diluted CHCl3/Tol solvent (cT = 1 × 10−5 mol/L), as well as the aggregated states Agg313 and Agg253 (cT = 1 × 10−3 mol/L). (d) The circular dichroism (CD) and (e) circularly polarized luminescence (CPL) spectra of R/S-Agg313 and R/S-Agg253 obtained from R-Nap-1 and S-Nap-1 respectively (cT = 1 × 10−3 mol/L). In all cases, the solvent mixture of CHCl3 and Tol was with a volumetric ratio of 5:95.
Figure 3
Variable-temperature CD spectra of (a) Agg313 and (b) Agg253 obtained from heating process (1 K/min), respectively. (c) Plot of the αaggversus temperature for Agg313 and Agg253, cT = 1 × 10−3 mol/L. (d) Thermodynamic parameters of Agg313 and Agg253 were calculated using a nucleation-elongation model.
Figure 4
(a) The most stable molecular structures of R-Nap-1 monomer in toluene obtained by conformational search calculations, the calculated Boltzmann polulation at 298 K of each conformation is listed in parentheses. (b) Variable-temperature 1H NMR spectroscopy of R-Nap-1 dissolved in TCE-d2 (cT = 1 × 10−3 mol/L). (c) Variable-temperature Fourier-transform infrared (FT-IR) spectrum of the R-Nap-1 dissolved in TCE (cT = 1 × 10−3 mol/L).
Figure 5
The theoretical two most stable R-Nap-1 decamer configurations (a) decamer 1 and (b) decamer 2, where the selected tetramers were particularly shown in sideview with hydrogen bonds and in topview with π-π stacking Naps. (c) Experimental CD spectra of Agg313 and Agg253 and theoretical CD spectra of the selected tetramers of decamer 1 and decamer 2. (d) FT-IR spectrum of R-Nap-1 monomer, Agg313 and Agg253.


 DownLoad:
DownLoad:



 下载:
下载:







 下载:
下载:
