Citation:
Yu-Hang Li, Shuai Gao, Lu Zhang, Hanchun Chen, Chong-Chen Wang, Haodong Ji. Insights on selective Pb adsorption via O 2p orbit in UiO-66 containing rich-zirconium vacancies[J]. Chinese Chemical Letters,
2024, 35(8): 109894.
doi:
10.1016/j.cclet.2024.109894
Insights on selective Pb adsorption via O 2p orbit in UiO-66 containing rich-zirconium vacancies
English
Insights on selective Pb adsorption via O 2p orbit in UiO-66 containing rich-zirconium vacancies
Eco-environment and Resource Efficiency Research Laboratory, School of Environment and Energy, Peking University Shenzhen Graduate School, Shenzhen 518055, China
b.
Beijing Key Laboratory of Functional Materials for Building Structure and Environment Remediation, School of Environment and Energy Engineering, Beijing University of Civil Engineering and Architecture, Beijing 100044, China
Received Date:
04 January 2024 Accepted Date:
15 April 2024 Revised Date:
10 April 2024 Available Online:
15 August 2024
Abstract:
Herein, we constructed defective UiO-66 with rich Zr vacancy structure model, in which the defective structure was verified by various characterizations. Also, the Pb adsorption experiments affirmed that defective UiO-66 could display better adsorption and selective adsorption ability than that of perfect UiO-66. The results of partial density of states (PDOS) and Mulliken charge population indicated that the blue shift of O 2p and Zr 4d orbit induced the electron rearrangement of atoms closed to the bonding sites, while the positive charge number of Zr atoms decreased than before. Combining with the expansion of pore size, Pb atom was more inclined to transfer and bond with unsaturated coordination oxygens. More significantly, quantitative structure-activity relationships (QSARs) demonstrated that selective capture of Pb instead of Zn, Cu, Cd and Hg displayed by defective UiO-66 was determined jointly by bond strength, adsorption energy and electron transfer. This work provided some theoretical direction for the purpose of the fabrication of adsorbent and the investigation of mechanism.
Heavy metal ions, as the common pollutants with ultra-high toxicities [1,2], had long been known to the public and extensively existed in the living environment up to this day, resulting in that exploring the removal technologies of heavy metal ions still was a hot study today [3,4]. Adsorption, especially selective adsorption, was a recognized green and facile elimination approach toward heavy metal ions, profiting from that (ⅰ) avoid the interference of other components on the adsorption of target ions [5,6]; (ⅱ) recover high value-added heavy metal ions like Ag, Au, Pt [7,8]; (ⅲ) one-time elimination without any intermediate products and energy input [9,10]. Based on the above superiorities, adsorbents with the abilities of selective sorption were qualified to be the favored environmental functional materials, which could accomplish sustainable development [11].
Our previous study first reported hierarchical porous Zr-MOFs (NH2-UiO-66) with metal vacancies via the modulation of seignette salt, which achieved a breakthrough in selective adsorption capacities from nearly zero to close two hundreds (mg/g) toward lead (Pb(Ⅱ)) [12]. Pb(Ⅱ), as one of the most common heavy metal ions, could be detected in various metal smelting wastewater [13,14], which displayed significant risks to human health due to the inherent toxic for brain tissue, the nervous system and the reproductive system [15]. Therefore, abundant studies selected Pb(Ⅱ) as the target pollutant. We thought that the selective adsorption mechanism insights were not enough from an objective point of view. Beyond that, it was worth noting that most of current studies about selective adsorption utilized adsorption energy and/or distribution coefficient to explain selective removal mechanism [16–18], which could only explain the reason of selective sorption to some extent. In fact, developing and deepening the cognition of selective adsorption mechanism was of great significance for fabrication of efficient adsorbents with adsorption ability in the future.
It was well known that chemical adsorption generally accompanied electron transfer and the formation of new chemical bond [19], in which both the quantity of charge transfer and bond strength between adsorbents and adsorbates were all deemed as significant factors during the selective sorption process. Density functional theory (DFT) calculations had been widely used in related studies including but not limited to the environmental remediation fields [20,21]. DFT calculations could assist researchers to investigate and solve some difficult problems that could not be explained by experimental approaches [22–25]. Thus, to further deepen and enrich the relevant mechanism of selective sorption, we constructed hierarchical porous UiO-66 model with metal vacancies using Materials Studio based on our previous study [12], in which the defective structure was confirmed by X-ray photoelectron spectrometry (XPS), electron paramagnetic resonance (EPR) and thermogravimetric analyses (TGA) analyses. Adsorption energy, electron density, density of states (DOS), electron density difference and molecular orbital theory were adopted to explain the enhancement reason of Pb sorption capacity of defective UiO-66. Moreover, we utilized crystal orbital Hamilton population (COHP), partial density of states (PDOS), electron density difference and Mulliken population to explore selective Pb adsorption mechanism of defective UiO-66, which was also verified by selective adsorption experiment. The quantitative structure-activity relationships (QSARs) confirmed that the adsorption abilities of defective UiO-66 toward different heavy metals were closely related to the strength of formation bond and the electron transfer. As the rare case of exploring selective adsorption mechanism on a theoretical level, this work offered the insight and guidance for researchers into the mutual corroboration between theories and experiments.
All calculation models were constructed by using Materials Studio 2020 [26]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional [27]. The energy cutoff was set as 450 eV (Fig. S1 in Supporting information) and the k-point was 1 × 1 × 1. The convergence standards for optimizing crystal geometry were that: (ⅰ) The energy tolerance was 2.0 × 10−5 eV/atom; (ⅱ) The maximum force tolerance was 0.04 eV/Å; (ⅲ) The maximum stress tolerance was 0.1 GPa; (ⅳ) The maximum displacement tolerance was 0.002 Å. The DOS and electron density were used to analyze the electronic structure of perfect UiO-66 and defective UiO-66. Adsorption energy, PDOS, electron density difference and Mulliken charge population were performed to investigate the interaction between adsorbent and heavy metals. COHP calculations and Mulliken bond population were integrated to characterize the strength of Zr–O–metal bonds. The calculation of COHP was performed in DMol3 module, and other items were all operated in Cambridge Sequential Total Energy Package (CASTEP) module.
Referred to the previous study [18], the unit cell of UiO-66 and defective UiO-66 with linkers missing and partial Zr missing were established using unit cell of UiO-66 (CCDC No. 733458) [12], as well as combined with the characterization results of XPS, EPR and TGA. In this work, the atom number of defective UiO-66 was 167. The different heavy metal adsorption models were formed and discussed. The adsorption energy (Eads) between adsorbent and target heavy metals were calculated as Eads = Ead/sub − Ead − Esub [28], where Ead/sub, Ead, and Esub were the total energies of the optimized adsorbate/substrate system, the adsorbate in the gas phase, and the clean substrate, respectively.
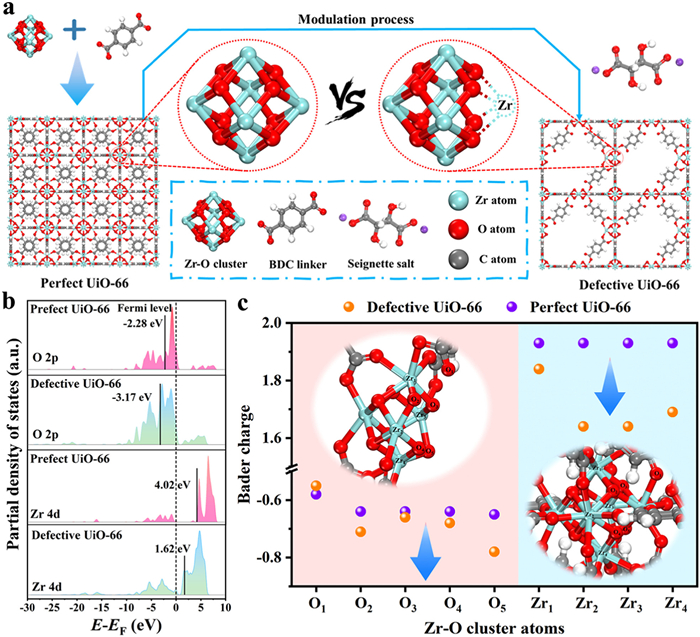
Fig. 1a demonstrated that perfect UiO-66 unit cell was constructed by Zr-O cluster (Zr6O6) and terephthalic acid, in which one Zr-O cluster bridged twelve BDC linkers [29,30]. Compared with perfect UiO-66 unit cell, defective UiO-66 structure model displayed rich defective sites, which were derived from ligand missing and partial Zr in Zr-O cluster missing. The corresponding fabrication methods were displayed in Section S1 (Supporting information). In addition, the powder X-ray diffraction (PXRD) and scanning electron microscope (SEM) confirmed the successful fabrication of adsorbents (Figs. S2 and S3 in Supporting information), in which the defective structure was confirmed by XPS, EPR and TGA analyses (Figs. S4 and S5 in Supporting information). Well known, the distribution of sorption sites and pore structure of adsorbents were two key influence factors during the adsorption process [31,32]. Previous studies had reported that perfect UiO-66 displayed unsatisfactory sorption performances toward cationic heavy metal ions due to the pore diameter and saturated coordination environment [12,29]. Take Pb(Ⅱ) as the example, the ionic radius and hydration radius of Pb(Ⅱ) could respectively reach 1.19 Å and 4.01 Å, signifying that the Pb(Ⅱ) migration process from outside to active sites was difficult in aqueous solution (Table S1 and Fig. S6 in Supporting information). However, defective UiO-66 structure could open the pore and expose the unsaturated coordination sites [32–34], which was in favor to perform adsorption process (Fig. S7 in Supporting information). The corresponding Pb(Ⅱ) capture experiments indicated that defective UiO-66 exhibited superior adsorption capacity and rate to perfect UiO-66 (Tables S2, S3 and Fig. S8 in Supporting information).
Figure 1
Figure 1.
(a) Schematic diagram comparing the unit cell structures of perfect UiO-66 and defective UiO-66 with Zr vacancies. (b) The p-band and d-band center of perfect UiO-66 and defective UiO-66. (c) The Bader charge of different Zr and O atoms in perfect UiO-66 and defective UiO-66.
The electronic distribution near Fermi level (EF) could offer help to understand the electronic properties of perfect and defective UiO-66 [35]. The calculation results of DOS (Fig. S9 in Supporting information) showed that the major electronic distribution near EF were attributed to the contribution of O atom and Zr atom for both perfect UiO-66 and defective UiO-66 (Fig. S9a), in which the O 2p states and Zr 4d states were located in electron occupied area and unoccupied area, respectively (Figs. S9b and c). Interestingly, a blue shift happened to the E − EF of defective UiO-66, indicating that the level position of band center might move down [36,37]. As expected, the energy level of both p-band center and d-band center demonstrated a significant decline after defective modulation (Fig. 1b), signifying that more electrons would be filled in O and Zr orbits, which was in agreement with the results of charge distribution (Fig. 1c). Take a Zr-O cluster with partial Zr missing as the example, the values of charge of all bridging oxygen atoms turned more negative than those of perfect Zr-O cluster and the coordination number became from 3 to 2, which was in favor to form chemical bond with cationic targets. Moreover, the decrease in the values of positive charge on Zr atoms provided favorable conditions for the adsorption of cationic pollutants due to that the repulsive forces turned weak between Zr atoms and adsorbed substance. Based on the above results, defective UiO-66 with partial Zr missing and linker missing could reach effective adsorption toward cationic pollutants.
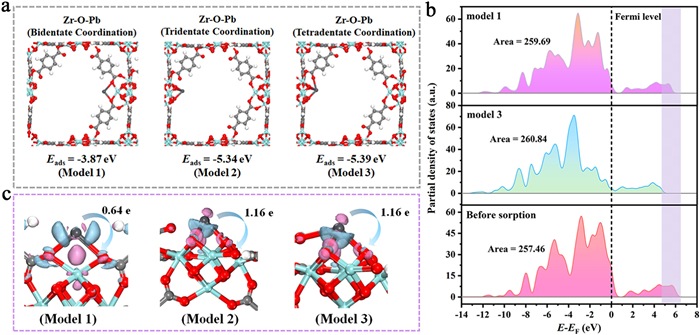
The calculations of adsorption energy could help to estimate whether the sorption behavior could be spontaneous process [38]. Pb with the positive charge preferred to bonding with oxygen atoms due to the electrostatic interaction, and the coordination modes could be change when Pb located at different sorption sites. Thus, all the possible Pb sorption models were constructed and displayed in Fig. 2a, including bidentate coordination (Model 1), tridentate coordination (Model 2) and tetradentate coordination (Model 3). The calculation results showed that all adsorption energy values of the coordination modes were negative, confirming that the Pb sorption behavior was spontaneous. In addition, it was worth noting that the adsorption energy value was the smallest when three bridging oxygens and one carboxyl oxygen coordinated with Pb, illustrating that this coordination mode (Model 3) was the easiest formation and most stable compared with other coordination modes, which could be also certified by results of bond order and bond length. As demonstrated in Fig. S10 and Table S4 (Supporting information), the bond order values of all chemical bonds less than 0.20, illustrating that the composition of ionic bond was dominant [39]. Furthermore, the highest bond order and the shortest bond length signified Model 3 was the most stable coordination structure.
Figure 2
Figure 2.
(a) Different Pb adsorption models and corresponding adsorption energy. (b) The density of states of O 2p orbital analyses before and after Pb adsorption. (c) Electron density difference of Pb adsorption in different models and their corresponding charge transfer. Pale pink and pale blue electronic cloud density represented electron accumulation and electron dissipation, respectively.
To further investigate sorption mechanism from the point of view of electron transfer, the electron density difference, Mulliken charge population and partial density of state (PDOS) were performed to explain the electron transfer between defective UiO-66 and Pb [40,41]. The results of PDOS (Fig. 2b and Fig. S11 in Supporting information) displayed that the distinct changes happened to O 2p orbit of defective UiO-66 before and after Pb adsorption. The area above the Fermi level (electron unoccupied orbits) occurred obvious bule shift, indicating that the additional electrons from Pb atom filled into the unoccupied orbits, further leading to that the number of unoccupied orbits became less. Corresponding to the above conclusion, the area below the Fermi level (electron occupied orbits) after Pb capture turned larger than that of defective UiO-66, demonstrating that electrons had been transferred from Pb to defective UiO-66. More interestingly, the area of electron occupied orbits in Model 3 increased the most, indicating that Model 3 might possess the largest number of charge transfer. Subsequently, the electron density difference on different models (Fig. 2c) showed that the electron cloud overlap appeared near sorption sites, in which the accumulation of electrons happened around oxygen atoms, while the Pb atom presented electrons depletion. Mulliken charges were used to assign to the structure (Table S5 in Supporting information), in which the Mulliken charge of Pb atom increased to 1.16 e for Model 3. Accordingly, the charges of four O atoms bonded by Pb atom increased from −0.55, −0.71, −0.66 and −0.68 to −0.63, −0.85, −0.73 and −0.77, respectively. In addition, it could be observed that the more electrons were transferred, the stronger the adsorption capacity. The above results indicated that the Pb adsorption process over defective UiO-66 was a chemical adsorption with electron transfer rather than a simple physical adsorption, which was also confirmed by the results of XPS analyses (Fig. S12 in Supporting information).
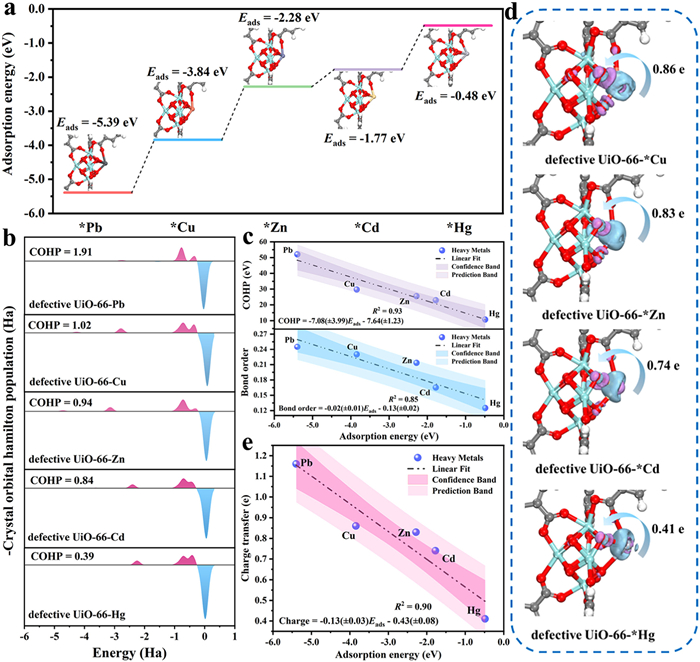
Various other heavy metal atoms like zinc (Zn), cadmium (Cd), cuprum (Cu) and mercury (Hg) were selected to explore selective removal mechanism at the molecule level with the aid DFT calculations. Fig. 3a displayed that the corresponding adsorption energy under the same coordination model followed the order Pb (−5.39 eV) > Cu (−3.84 eV) > Zn (−2.28 eV) > Cd (−1.78 eV) > Hg (−0.48 eV), in which defective UiO-66 displayed lower adsorption energy toward Pb than those of other heavy metals. The result illustrated that defective UiO-66 might possess priority capture ability toward Pb in a solution mixed with multiple cationic heavy metals, which was also in agreement with the results of experiments (Fig. S13 in Supporting information). The COHP curves between metal and O atoms (metal-O bond), composing of positive area (represented bonding components) and negative area (represented antibonding components), were adopted to characterize the strength of metal-O bonds [42,43]. As demonstrated in Fig. 3b, the bond strength formed between Pb and unsaturated coordinated oxygen was calculated to be the strongest, indicating that antibonding orbitals were filled with the fewest electrons. The results of established QSARs exhibited (Fig. 3c) that the bond strength was one significant factor, which determined the adsorption capacity toward different heavy metals. Based on the adsorption models, electron density difference calculations were performed to confirm the amount of charge transfer. It was worth noting that the number of charge transfer from Cu, Zn, Cd and Hg to defective UiO-66 were 0.73, 0.84, 0.78 and 0.43 e, which were much lower than that of defective UiO-66-Pb model (Fig. 3d). Well-known that the number of charge transfers could be used to assess the interaction between adsorbent and heavy metal. Fig. 3e displayed the well-fitted and positive linear relationships of adsorption energy and the amount of charge transfer, demonstrating that the greater the number of charge transfer, the stronger the interaction force between adsorbent and adsorbate. Based on the above analyses results, the heavy metal ions priority capture mechanism was proposed: (ⅰ) The more negative adsorption energy between adsorbent and heavy metal indicated that the target heavy metal could more easily be captured. (ⅱ) The amount of charge transfer determined the strength of the interaction between adsorbent and adsorbate. (ⅲ) The greater the interaction force between adsorbent and adsorbate demonstrated the greater the bond order, also indicating that the antibonding orbitals are less filled with electrons.
Figure 3
Figure 3.
(a) The adsorption energies and (b) -COHP between defective UiO-66 and different heavy metals. (c) The linear relationship of COHP and bond order on adsorption energies over different adsorption models. (d) Electron density difference of different heavy metal adsorption in defective UiO-66 and the corresponding amount of charge transfer. Pale pink and pale blue electronic cloud density represented electron accumulation and electron dissipation, respectively. (e) The influence of charge transfer on adsorption energies over different various heavy metal adsorption models.
In conclusion, this study integrated density functional theoretical calculations and molecular orbital theory to investigate heavy metals adsorption and selective capture mechanism on defective UiO-66 with rich Zr vacancy structure model. Employing molecular orbital theory analyzed the results of PDOS and Mulliken charge population, confirming that the defective structure modulated O 2p orbit and Zr 4d orbit, further leading to that unsaturated coordination sites possessed more negative charge to attract cationic heavy metals due to reconstruction of electronic structure. More importantly, the results of established QSARs displayed that selective capture ability was determined jointly by multiple factors like adsorption energy, the capacity of electron transfer and formation bond strength. As the rare case to explore selective adsorption mechanism in depth via density functional theoretical calculations, this work offered the theoretical direction at a deeper level than before for the purpose of the fabrication of adsorbent and the investigation of mechanism.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial supports from the National Natural Science Foundation of China (NSFC, No. 52100069), the Shenzhen Science and Technology Program (No. JCYJ20220531093205013), and the 2023 Shenzhen Sustainable Supporting Funds for Colleges and Universities (No. 20231121170027002) are greatly acknowledged.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109894.
[1]
J. Li, X. Wang, G. Zhao, et al., Chem. Soc. Rev. 47 (2018) 2322–2356. doi: 10.1039/C7CS00543A
Q. Sun, Z. Li, D.J. Searles, et al., J. Am. Chem. Soc. 135 (2013) 8246–8253. doi: 10.1021/ja400243r
[39]
Y. Sun, S. Sun, H. Yang, et al., Adv. Mater. 32 (2020) e2003297. doi: 10.1002/adma.202003297
[40]
Y. Liu, X. Liu, Z. Lv, et al., Angew. Chem. Int. Ed. 61 (2022) e202117617. doi: 10.1002/anie.202117617
[41]
M. Li, H. Li, C. Ling, et al., Proc. Natl. Acad. Sci. U. S. A. 120 (2023) e2304562120. doi: 10.1073/pnas.2304562120
[42]
V.L. Deringer, A.L. Tchougreeff, R. Dronskowski, J. Phys. Chem. A 115 (2011) 5461–5466. doi: 10.1021/jp202489s
[43]
S. Maintz, V.L. Deringer, A.L. Tchougreeff, R. Dronskowski, J. Comput. Chem. 34 (2013) 2557–2567. 5 doi: 10.1002/jcc.23424
Figure 1
(a) Schematic diagram comparing the unit cell structures of perfect UiO-66 and defective UiO-66 with Zr vacancies. (b) The p-band and d-band center of perfect UiO-66 and defective UiO-66. (c) The Bader charge of different Zr and O atoms in perfect UiO-66 and defective UiO-66.
Figure 2
(a) Different Pb adsorption models and corresponding adsorption energy. (b) The density of states of O 2p orbital analyses before and after Pb adsorption. (c) Electron density difference of Pb adsorption in different models and their corresponding charge transfer. Pale pink and pale blue electronic cloud density represented electron accumulation and electron dissipation, respectively.
Figure 3
(a) The adsorption energies and (b) -COHP between defective UiO-66 and different heavy metals. (c) The linear relationship of COHP and bond order on adsorption energies over different adsorption models. (d) Electron density difference of different heavy metal adsorption in defective UiO-66 and the corresponding amount of charge transfer. Pale pink and pale blue electronic cloud density represented electron accumulation and electron dissipation, respectively. (e) The influence of charge transfer on adsorption energies over different various heavy metal adsorption models.

 DownLoad:
DownLoad:

 下载:
下载:




 下载:
下载:
