Scheme 1.
Representative synthesis for axially chiral binaphthol.

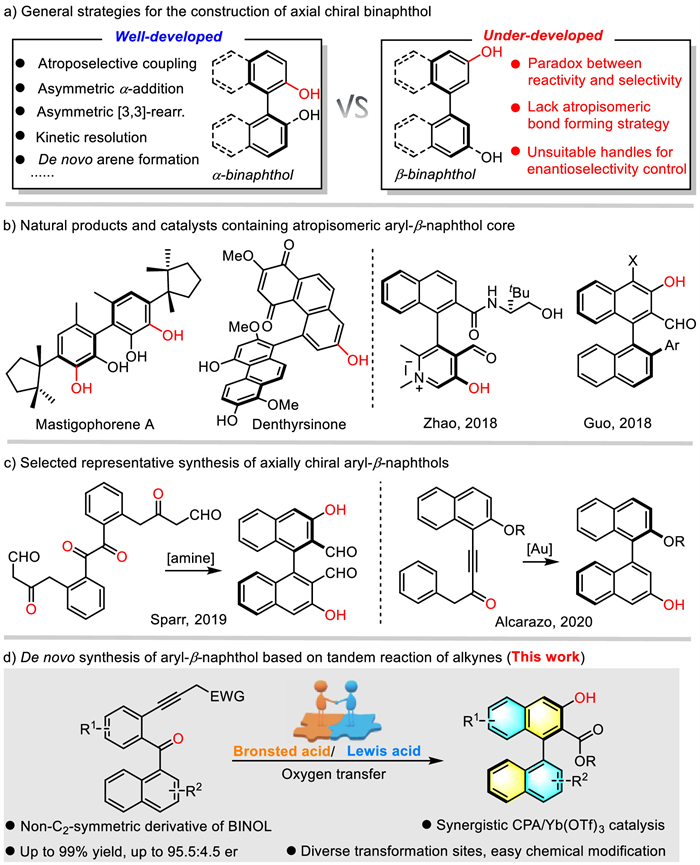
Atropisomerism is a phenomenon whereby the constrained rotation of σ bond leads to the formation of stereoisomers. Atropisomers are considered to be useful frameworks because of their unique structure and function [1–3], which is also key component of catalysts [4], natural products and medicines [5,6]. Among them, binaphthol, a representative privileged architecture of chiral ligand, has played an important role in transition-metal catalysis. Thus the practical synthetic strategies of axially chiral α-binaphthol and its analogs have been extensively investigated over the past decades, including (1) oxidative coupling of two units [7–11], (2) asymmetric addition or rearrangement followed by chirality transfer [12–19], (3) stereoselective functionalization of racemic or prochiral precursors [20–26] and (4) de novo arene formation (Scheme 1a) [27–29]. In sharp contrast, successful synthesis and application of non-α-binaphthol remain limited [30,31], probably due to the conflicts between the inherent reactivity and desired regioselectivity for β-binaphthol synthesis.
Aryl-β-naphthol is also a prominent skeleton of many natural products and bioactive compounds, such as Mastigophorene A [32], Denthyrsinone (Scheme 1b) [33]. Recently, aryl-β-naphthol derivatives have received more attention especially in organic catalysis. For example, in 2018, Zhao and coworkers [34] reported an elegant biomimetic asymmetric Mannich reaction via carbonyl catalysis. What's more, a new contribution by the group of Guo [35] using axially chiral β-naphthol derivatives has been developed (Scheme 1b). However, in these reports, relative lengthy synthetic routes (7–9 steps) were required to obtain diverse axially chiral β-naphthols, which indicated that it is highly appreciable to develop more efficient synthetic methodology for such type skeletons. Lately, a landmark achievement was realized by Sparr and coworkers [30] through a bioinspired non-canonical polyketide cyclizations, which enabled rapid access to β-binaphthols (Scheme 1c).
Alkynes, readily available precursors and known as valuable building blocks for organic synthesis, can undergo numerous useful transformations [36–38]. Despite many achievements, the catalytic asymmetric cyclization of alkynes to construct axially chiral β-naphthols have rarely been reported. To the best of our knowledge, only Alcarazo [31] and co-workers reported an enantioselective synthesis of the β-naphthol units through a Au-catalyzed hydroarylation of alkynones (Scheme 1c). In this regard, the discovery and identification of novel types of non-2-binaphthol atropisomer as ligands/catalysts might strongly depend on the development of new catalytic reaction model that however cannot draw from α-binaphthol synthesis. As our continuous interest in alkyne-based catalytic asymmetric tandem reactions [39,40], in conjunction with the inspiring contributions by other groups [30,31,41–43], we envisioned that a tandem hydrolysis of alkynes and intramolecular aldol condensation of ortho-alkynylarylketone might enable a one-pot synthesis of β-naphthol effectively. However, realizing this tandem reaction, especially with high regio- and stereoselectivity, is quite challenging. Firstly, there are two possible reaction pathways for the cyclization (5-exo or 6-endo), thus to control the regioselectivity is a difficult issue need to be address [44]. Second, due to the low reactivity of the resulting diaryl ketone intermediate, it is inevitably required to identify higher active catalyst or harsher conditions, which is usually detrimental to asymmetrical control. To overcome these challenges, electron-withdrawing ester groups were designed as substituents to stabilize the enol intermediates, which is also benefit to catalyst coordination. Herein, we describe the realization of such a synergistic Brønsted/Lewis acid-catalyzed tandem reaction of ortho-alkynylarylketone, which allows efficient and practical synthesis of valuable aryl-β-naphthol with high regio- and enantioselectivity.
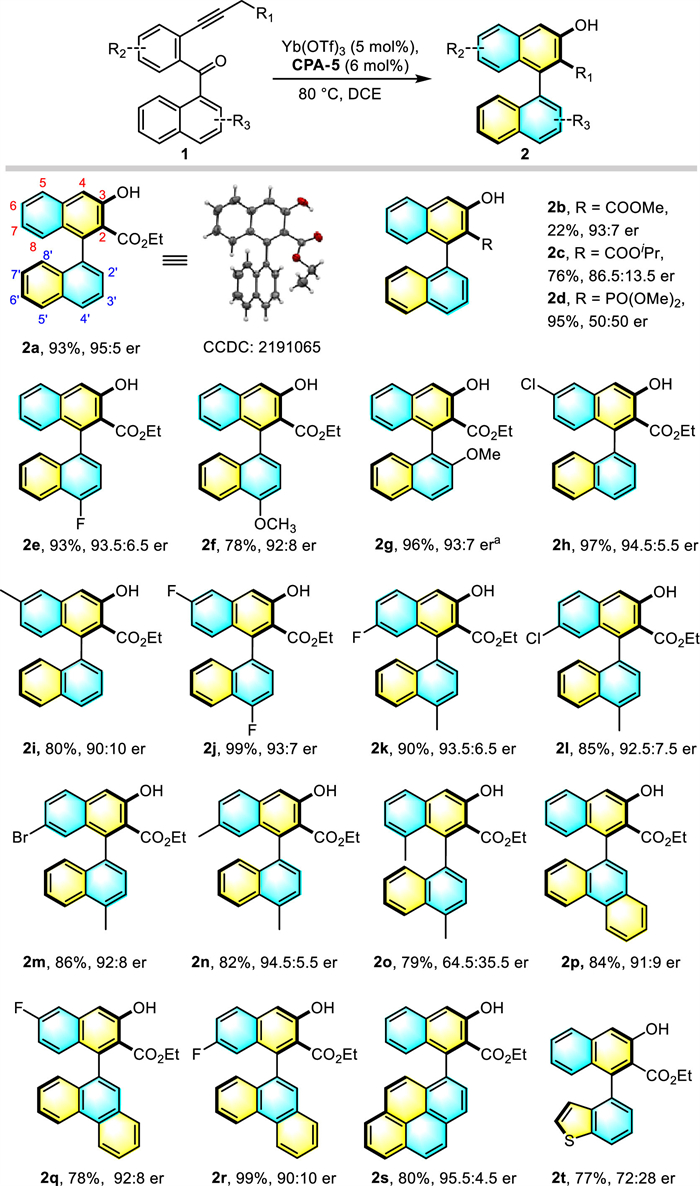
Due to the oxophilic properties of rare earth metals, a series of rare earth metals were intuitively selected as our candidate cocatalysts, to promote a tandem hydrolysis/cyclization of the engineered enynone (1a). We evaluated Sc(OTf)3 with different chiral phosphric acids (CPA), which are now prevailing in asymmetric catalysis. As expected, the combination of Sc(OTf)3 (5 mol%) and CPA-1 (6 mol%) promoted the reaction at 80 ℃ in 1,2-dichloroethane (DCE), affording the desired 2a in 71% yield, but the atropisomeric ratio was only 56:44 (Table 1, entry 1). Various other chiral CPAs were then evaluated (entries 2–6), the results revealed that the CPA-5 having 3,3′-di-(2,4,6-tri-isopropyl)phenyl substituents gave the highest enantioselectivity (80:20 er, entry 5). The CPA-6, bearing a more sterically hindered substituent, showed almost no enantioselectivity (entry 6). Different Lewis acids were then studied with the privileged CPA-5 (entries 7–11). Delightfully, the Y(OTf)3, La(OTf)3, Yb(OTf)3 afforded good enantioselectivities (up to 95:5 er, entry 11), despite Y(OTf)3 showed relatively low yields. Further evaluation of the reaction parameters such as solvent and temperature by using Yb(OTf)3 and CPA-5 as co-catalyst partner were conducted. However, when the reaction was carried out at 60 ℃, the enantioselectivity decreased slightly (entry 12). Interestingly, further decreasing the temperature to 40 ℃ not only resulted in retard the reaction rate but also decreased stereoselectivity (91:9 er, entry 13), which goes against the experience wherein lower temperature typically is beneficial to the enantioselectivity. The use of solvent C6H5Cl did not markedly alter the yield or enantioselectivity, whereas no reaction was observed with the coordinative tetrahydrofuran (entries 14 and 15).
With the established optimal conditions in hand (Table 1, entry 11), we next explored the scope of this reaction for the atroposelective construction of axially chiral β-naphthols (Scheme 2). The absolute configuration of 2a was deduced to be (S) by X-ray single-crystal diffraction analysis (CCDC: 2191065) and other products were assigned by analogy. First, examination the steric hindrance of the different electron withdrawing groups at the end of alkyne showed that the introduction of a smaller methyl ester or a bulkier isopropyl ester substituent resulted in reduced both yields and enantioselectivities (2b, 2c). Interestingly, the enantiomeric ratio of the product (2d) decreased sharply (50:50) but still with excellent yield, when the substrate with a phosphonate terminal was applied. Then the electronic effects on the naphthalene ring were evaluated by the variation of substituents. Firstly, different substituents such as fluorine and methoxy groups at the C-4′ position of naphthalene ring of substrates were tested, delivering the products 2e, 2f with good yields (up to 93%) and excellent enantioselectivities (up to 93.5:6.5). However, when 2′‑methoxy substituted substrate was subjected to standard conditions, the enantioselectivity was dismal, which could be restored (2g, 96%, 93:7 er) by using octahydrosubstituted CPA* as cocatalyst under reduced reaction temperature (60 ℃). Next, substrates with a chlorine (2h) and methyl (2i) at the phenyl rings were also investigated, and excellent yields and enantioselectivities were observed. Excellent result was also achieved with substrate (2j) bearing a fluorine group at both the naphthalene and the phenyl ring. The 4′-methyl-substituted substrates with either electron-donating or electron-withdrawing groups at the phenyl of alkynyl substituted benzene were also tolerated and gave products 2k-2n in excellent yields and enantioselectivities. Yet the atroposelectivity of 8-methyl substituted substrate (2o) was reduced dramatically, indicating the great sterical impact of the C-8 substituents. To our delight, phenanthrene (2p-2r) and pyrene (2s) substituted axially β-naphthol were well tolerated (78%−99%, 91:9–95.5:4.5 er). Benzo-heterocyclic compound (2t) was also suitable for this transformation in good yield but moderate enantiomer ratio (77%, 72:28 er), implying more steric hinderance difference between the left and right part of heteroarene is beneficial to the enantioselectivity.
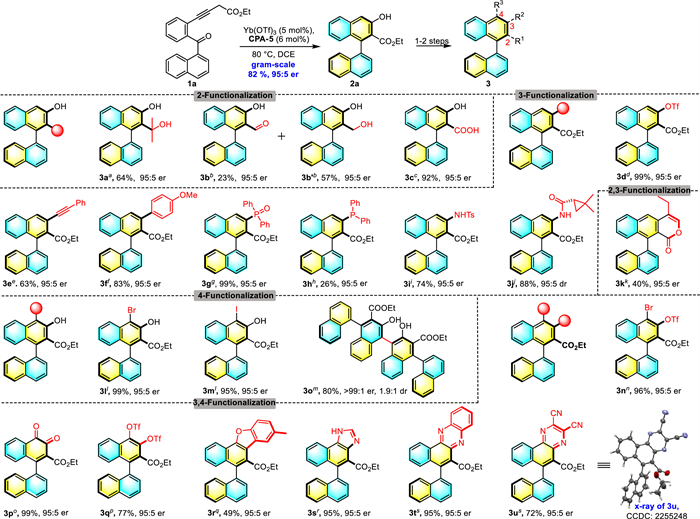
A gram-scale synthesis was carried out (2.5 mmol) and afforded the desired product (S)−2a in 82% yield and 95:5 er. As mentioned, the axially chiral aryl-β-naphthol 2a could be a promising framework, thus diverse transformations were then conducted to expand the synthetic utility (Scheme 3). As a versatile functionality, the ester unit allows to readily further transformations. For example, 2a could undergo nucleophilic addition with Grignard reagent or DIBAL-H to give corresponding alcohol (3a) or atropisomeric aldehyde (3b), which recently emerged as a new type biomimic catalyst [30,34]. More importantly, axially chiral carboxylic acid, which exhibits unique properties in enantioselective catalysis [45–49], could be obtained (3c) via simple hydrolysis with almost no erosion of the enantiopurity. This methodology might provide a new avenue for the synthesis of axially chiral carboxylic acid. In order to take full advantage of the phenolic hydroxyl group, the naphthol 2a was protected with Tf2O to give the triflate 3d in quantitative yield, which provided good opportunities for downstream coupling reactions, allowing quick access to a variety of structurally diversified atroposelective binaphthyls. For instance, Sonogashira coupling reaction with phenylacetylene proceeded smoothly to give substituted product 3e. Meanwhile, triflate 3d also allowed Suzuki coupling on the 3-position of naphthol moiety to produce the corresponding derivative 3f in 83% yield and excellent enantiopurity. Likewise, a palladium-catalyzed coupling with phosphite gave the aryl phosphonates 3g in good yield, then followed by reduction to furnish the corresponding chiral phosphine 3h, which could potentially be used as the organocatalyst or ligand in asymmetric transformations. In addition, triflate 3d could undergo Buchwald-Hartwig amination with chiral amide afforded product 3i in 88% yield and 95:5 er, or with enantiopure primary amine afforded product 3j. Interestingly, a lactone 3k was obtained in moderate yield when an alkyl aldehyde was used as coupling partner. The product 3l was formed in almost quantitative yield without loss of enantiomeric purity, upon treatment of 2a with NBS in dichloromethane at 0 ℃ for 1 h, which could be extended to other halogen sources, affording 4-iodonaphthol 3m in high yield and enantioselectivity as well. The resulting 3m was further transferred into a triflate bromide 3n, which holds great potential in orthogonal reactions. Furthermore, the recrystallized 2a (>99:1 er) could readily react with another molecule 2a under the promotion of oxidant to give a multiple axially chiral dimer (3o) in 80% yield and 1.9:1 dr. Oxidation of the enol group of 2a with IBX led to the formation of o-quinone 3p in quantitative yield, which was further transformed into the desired 3q in 77% yield upon selective reduction and triflation. The synthetic utility of the newly synthesized 1,2-naphthoquinone 3p was demonstrated by its divergent transformations to complex heterocyclic compounds. As shown in Scheme 3, a polyannulated axial chirality dibenzofuran 3r was obtained in moderate yield by treatment of quinone 3p with 4-methylcyclohexanone under the catalysis of FeCl3. The atroposelective imidazole 3s was also synthesized by condensation of 3p with ammonium acetate and formaldehyde. Similarly, the reactions of quinone 3p with benzene 1,2-diamine in the presence of hydrochloric acid and sodium sulfate proceeded smoothly to yield the corresponding products 3t in 95% yield and 95:5 er, and diaminomaleonitrile gave benzophenazine derivatives 3u in 85% yield. It is noteworthy that no stereochemical integrity loss was observed for all these reactions.
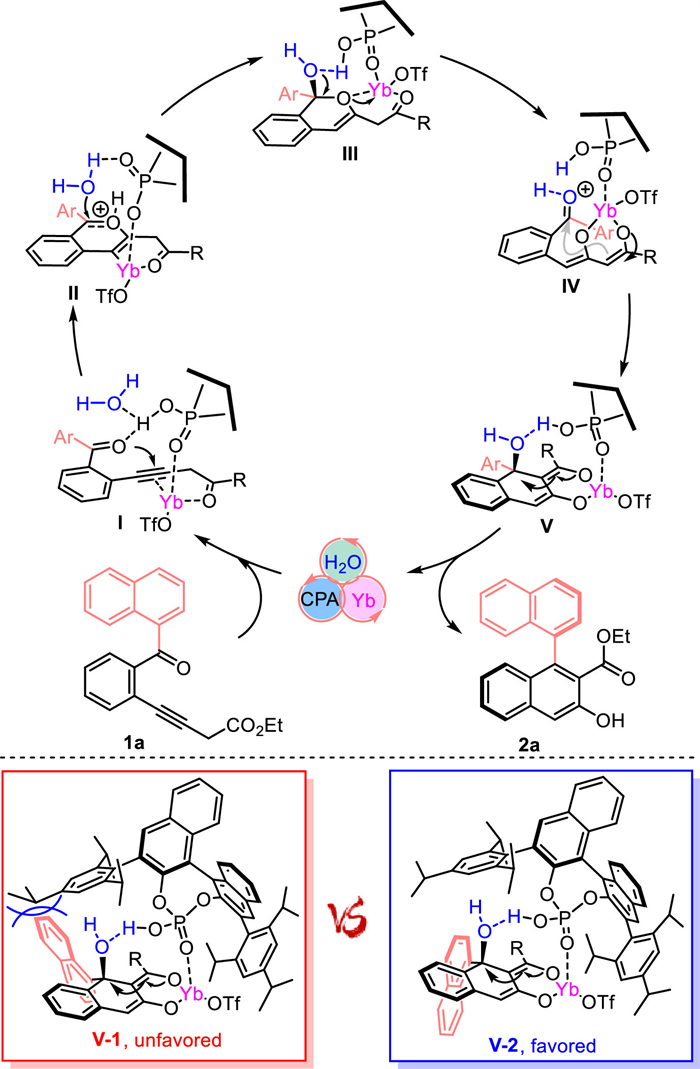
To probe the reaction mechanism, control experiments were carried out. When the reaction was ran only in the presence of chiral CPA-5, an excellent yield and moderate enantimeric ratio was observed (Scheme 4a, entry 1). When the Yb(OTf)3 was added as a single catalyst, the reaction could proceed more rapidly with almost equal yield (91% vs. 89%) obtained in a quite short time (16 h vs. 36 h, entry 2). These results imply that great background reaction need to be suppressed in order to achieve excellent enantioselectivity. Interestingly, when combination of CPA-5 with Yb(OTf)3 (entry 3), a superior enantioselectivity was observed and the reaction time was further shorted, demonstrating a synergistic or cooperative effect exerted important effect in the catalytic processes. Then, the diketone 1aa, serendipitously isolated from gram-scale reaction, was also subjected to the optimal reaction condition, which only afforded the corresponding 2a in 73% yield and compromised stereoselectivity (88:12 er vs. 95:5 er, entry 5). The result indicated that 1aa is likely to be the intermediate, but substrate 1a may be a more effective substrate in this system. We then checked reaction process and no obvious 1aa was accumulated during the reaction. Finally, a series of thermal racemization experiments with the prepared axially chiral β-naphthol 2a were carried out to gain insight into its conformational stability. The half-life of enantiopure 2a was about 4537 h at 110 ℃ in toluene and the rotation barrier ΔG‡ was identified to be 36.6 kcal/mol, thus rule out of the probable racemic effect of prolong reaction time (Scheme 4b).

Based on these results, a possible working mechanism with 1a as an example was then proposed. As shown in Scheme 5, the coordinative complex Ⅰ undergoes an intramolecular 6-endo-dig cyclization to give oxonium intermeidate Ⅱ, which is attacted by CPA tethered water (adventitiously introduced by solvent) to form hemiketal Ⅲ, then followed by intramolecular aldol cyclization from the Re face of the ketone and E1cB elimination, delivering the desired β-naphthol 2a via central-to-axial chirality transfer. In addition, a tentative enantioselectivity induction model is proposed, wherein the sterically hinderance between naphthyl motif and the peripheral isopropyl of the CPA restrict the rotation of the atroposelectivity determining C—C bond [50]. Obviously, there is less steric clashes of rotamer Ⅴ-2, which make it favor over the rotamer Ⅴ-1, thus ensured the observed (S)-atropisomer.
In conclusion, we have successfully realized the synergistic Lewis acid/chiral Brønsted acid-catalyzed atroposelective reaction, giving efficient access to a class of axially chiral aryl-β-naphthol in excellent yields and enantiomer ratio. This efficient and good functional groups tolerant approach allowed the rapid construction of versatile axially chiral compounds providing a platform for the synthesis of structurally diverse derivatives via easily late-stage chemical modification. Mechanistic study revealed that the synergistic effect is critical to suppress the background reaction, allowing the atroposelective determining central-to-axial chirality transfer in high stereochemical fidelity.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jiajun Lu: Investigation, Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft. Zhehui Liao: Investigation. Tongxiang Cao: Data curation, Formal analysis, Funding acquisition, Resources, Supervision, Validation, Writing – review & editing. Shifa Zhu: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing.
We appreciate financial support from the National Natural Science Foundation of China (Nos. 22071062, 22001077 and 22271096), Guangdong Science and Technology Department (Nos. 2021A1515012331, 2023A1515011001), the Fundamental Research Funds for the Central Universities (No. 2022ZYGXZR016) and South China University of Technology for start-up funds.
Supplementary material associated with this article can be found, in the online version, at doi:
J.K. Cheng, S.H. Xiang, S. Li, L. Ye, B. Tan, Chem. Rev. 121 (2021) 4805–4902. doi: 10.1021/acs.chemrev.0c01306
D. Liang, W. Xiao, S. Lakhdar, J. Chen, Green Synth. Catal. 3 (2022) 212–218. doi: 10.1016/j.gresc.2022.06.003
X.L. Min, X.L. Zhang, R. Shen, Q. Zhang, Y. He, Org. Chem. Front. 9 (2022) 2280–2292. doi: 10.1039/d1qo01699g
Y. Chen, S. Yekta, A.K. Yudin, Chem. Rev. 103 (2003) 3155–3212. doi: 10.1021/cr020025b
R.S. Griffith, F.B. Peck Jr., Antibiot. Annu. 3 (1955) 619–622.
Y.F. Hallock, K.P. Manfredi, J.W. Blunt, et al., J. Org. Chem. 59 (1994) 6349–6355. doi: 10.1021/jo00100a042
M. Nakajima, K. Kanayama, I. Miyoshi, S.I. Hashimoto, Tetrahedron Lett. 36 (1995) 9519–9520. doi: 10.1016/0040-4039(95)02063-2
H. Egami, K. Matsumoto, T. Oguma, T. Kunisu, T. Katsuki, J. Am. Chem. Soc. 132 (2010) 13633–13635. doi: 10.1021/ja105442m
S. Narute, R. Parnes, F.D. Toste, D. Pappo, J. Am. Chem. Soc. 138 (2016) 16553–16560. doi: 10.1021/jacs.6b11198
H. Kang, C. Torruellas, J. Liu, M.C. Kozlowski, Org. Lett. 20 (2018) 5554–5558. doi: 10.1021/acs.orglett.8b02183
J.M. Tian, A.F. Wang, J.S. Yang, et al., Angew. Chem. Int. Ed. 58 (2019) 11023–11027. doi: 10.1002/anie.201903435
Y.H. Chen, D.J. Cheng, J. Zhang, et al., J. Am. Chem. Soc. 137 (2015) 15062–15065. doi: 10.1021/jacs.5b10152
H. Gao, Q.L. Xu, C. Keene, et al., Angew. Chem. Int. Ed. 55 (2016) 566–571. doi: 10.1002/anie.201508419
J.Z. Wang, J. Zhou, C. Xu, et al., J. Am. Chem. Soc. 138 (2016) 5202–5205. doi: 10.1021/jacs.6b01458
Z.J. Jia, C. Merten, R. Gontla, et al., Angew. Chem. Int. Ed. 56 (2017) 2429–2434. doi: 10.1002/anie.201611981
Y.S. Jang, Ł. Woźniak, J. Pedroni, N. Cramer, Angew. Chem. Int. Ed. 57 (2018) 12901–12905. doi: 10.1002/anie.201807749
L.W. Qi, S. Li, S.H. Xiang, J. Wang, B. Tan, Nat. Catal. 2 (2019) 314–323. doi: 10.1038/s41929-019-0247-1
G. Coombs, M.H. Sak, S.J. Miller, Angew. Chem. Int. Ed. 59 (2020) 2875–2880. doi: 10.1002/anie.201913563
J.W. Zhang, L.W. Qi, S. Li, S.H. Xiang, B. Tan, Chin. J. Chem. 38 (2020) 1503–1514. doi: 10.1002/cjoc.202000358
G. Bringmann, D. Menche, Acc. Chem. Res. 34 (2001) 615–624. doi: 10.1021/ar000106z
K. Mori, T. Itakura, T. Akiyama, Angew. Chem. Int. Ed. 55 (2016) 11642–11646. doi: 10.1002/anie.201606063
C. Yu, H. Huang, X. Li, Y. Zhang, W. Wang, J. Am. Chem. Soc. 138 (2016) 6956–6959. doi: 10.1021/jacs.6b03609
G.A.I. Moustafa, Y. Oki, S. Akai, Angew. Chem. Int. Ed. 57 (2018) 10278–10282. doi: 10.1002/anie.201804161
L.a. Hu, Y. Zhang, G.Q. Chen, et al., Org. Lett. 21 (2019) 5575–5580. doi: 10.1021/acs.orglett.9b01907
B.A. Jones, T. Balan, J.D. Jolliffe, C.D. Campbell, M.D. Smith, Angew. Chem. Int. Ed. 58 (2019) 4596–4600. doi: 10.1002/anie.201814381
G. Yang, D. Guo, D. Meng, J. Wang, Nat. Commun. 10 (2019) 3062. doi: 10.1038/s41467-019-10878-7
M. Furusawa, K. Arita, T. Imahori, et al., Tetrahedron Lett. 54 (2013) 7107–7110. doi: 10.1016/j.tetlet.2013.10.080
Y. Liu, X. Wu, S. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 6491–6495. doi: 10.1002/anie.201801824
S. Jia, S. Li, Y. Liu, W. Qin, H. Yan, Angew. Chem. Int. Ed. 58 (2019) 18496–18501. doi: 10.1002/anie.201909214
R.M. Witzig, V.C. Fäseke, D. Häussinger, C. Sparr, Nat. Catal. 2 (2019) 925–930. doi: 10.1038/s41929-019-0345-0
J. Zhang, M. Simon, C. Golz, M. Alcarazo, Angew. Chem. Int. Ed. 59 (2020) 5647–5650. doi: 10.1002/anie.201915456
Y. Fukuyama, M. Toyota, Y. Asakawa, J. Chem. Soc., Chem. Commun. (1988) 1341–1342.
G.N. Zhang, L.Y. Zhong, S.W. Annie Bligh, et al., Phytochemistry 66 (2005) 1113–1120. doi: 10.1016/j.phytochem.2005.04.001
J. Chen, X. Gong, J. Li, et al., Science 360 (2018) 1438–1442. doi: 10.1126/science.aat4210
W. Wen, L. Chen, M.J. Luo, et al., J. Am. Chem. Soc. 140 (2018) 9774–9780. doi: 10.1021/jacs.8b06676
N.E. Schore, Chem. Rev. 88 (1988) 1081–1119. doi: 10.1021/cr00089a006
F. Alonso, I.P. Beletskaya, M. Yus, Chem. Rev. 104 (2004) 3079–3160. doi: 10.1021/cr0201068
J. Guo, Z. Cheng, J. Chen, X. Chen, Z. Lu, Acc. Chem. Res. 54 (2021) 2701–2716. doi: 10.1021/acs.accounts.1c00212
L. Chen, K. Chen, S. Zhu, Chem 4 (2018) 1208–1262. doi: 10.1016/j.chempr.2018.02.001
L. Chen, Z. Liu, S. Zhu, Org. Biomol. Chem. 16 (2018) 8884–8898. doi: 10.1039/c8ob02348d
Z.X. Zhang, T.Y. Zhai, L.W. Ye, Chem. Catal. 1 (2021) 1378–1412. doi: 10.1016/j.checat.2021.09.011
Y.D. Shao, M.M. Dong, Y.A. Wang, et al., Org. Lett. 21 (2019) 4831–4836. doi: 10.1021/acs.orglett.9b01731
P. Nimnual, K. Norseeda, B. Akkachairin, et al., Asian J. Org. Chem. 7 (2018) 932–945. doi: 10.1002/ajoc.201800025
B. Akkachairin, J. Tummatorn, N. Supantanapong, et al., J. Org. Chem. 82 (2017) 3727–3740. doi: 10.1021/acs.joc.7b00198
H. Egami, J. Asada, K. Sato, et al., J. Am. Chem. Soc. 137 (2015) 10132–10135. doi: 10.1021/jacs.5b06546
C. Min, D. Seidel, Chem. Soc. Rev. 46 (2017) 5889–5902. doi: 10.1039/C6CS00239K
X. Xu, L. Peng, X. Chang, C. Guo, J. Am. Chem. Soc. 143 (2021) 21048–21055. doi: 10.1021/jacs.1c11044
T. Zhou, P.F. Qian, J.Y. Li, et al., J. Am. Chem. Soc. 143 (2021) 6810–6816. doi: 10.1021/jacs.1c03111
J.Y. Li, P.P. Xie, T. Zhou, et al., ACS Catal. 12 (2022) 9083–9091. doi: 10.1021/acscatal.2c00962
V.S. Raut, M. Jean, N. Vanthuyne, et al., J. Am. Chem. Soc. 139 (2017) 2140–2143. doi: 10.1021/jacs.6b11079

Scheme 2 Substrate scope for chiral β-naphthol compounds. Reactions were performed on 0.1 mmol scale (0.1 mol/L solution). Isolated yield, er was determined by chiral-phase HPLC analysis. a Reactions were carried out with octahydrosubstituted CPA* (6 mol%) (see Supporting information for details).

Scheme 3 Preliminary application for chiral β-naphthol compounds. Reaction conditions: (1) Use 2a as the substrate: a CH3MgBr (5 equiv.), THF, 0 ℃, 3 h. b DIBAL-H (1.2 equiv.), DCM, −78 ℃, 1 h. c 10% NaOH, EtOH, reflux, 2 h. d Tf2O (2 equiv.), DCM, 0 ℃, 3 h. l NBS (or NIS) (1 equiv.), CH3CN, 0 ℃, 30 min. m TBHP (1 equiv.), FeCl3 (20 mol%), DCE:HFIP(1:1), 10 h. o IBX (1.2 mmol), DMSO, 6 h. (2) Use 3d as the substrate: e Pd(OAc)2 (10 mol%), PPh3 (0.4 equiv.), K3PO4 (1.2 equiv.), 4 Å MS, DMSO, 80 ℃, 24 h. f Pd(PPh4)3 (30 mol%), K3PO4 (1.5 equiv.), 4-methoxyphenylboronic acid (1.2 equiv.), 1,4-dioxane, 100 ℃, 24 h. g Ph2P(O)H (4 equiv.), Pd(OAc)2 (10 mol%), dppb (20 mol%), Et3N (6 equiv.), DMSO, 110 ℃, 12 h. h Et3N (7 equiv.), HSiCl3 (5 equiv.), toluene, 100 ℃, 18 h. i Pd2(dba)3 (5 mol%), t-BuXphos (6 mol%), 4-methylbenzenesulfonamide (1.2 equiv.), K3PO4 (1.2 equiv.), t-AmOH, 80 ℃, 15 h. j Pd(OAc)2 (10 mol%), Xantphos (15 mol%), Cs2CO3 (1.4 equiv.), enantiopure amide (2 equiv.), 4 Å MS, toluene, 100 ℃, 24 h.k Pd2(dba)3 (5 mol%), Xantphos (5 mol%), Cs2CO3 (2 equiv.), butyraldehyde (5 equiv.), 4 Å MS, 1,4-dioxane, 100 ℃, 16 h. n Hünig's base (2.2 equiv.), Tf2O (2.0 equiv.), DCM, 0 ℃, 16 h. (3) Use 3p as the substrate: p (1) Na2S2O4(1.5 equiv.), DCM, H2O, 0 ℃; (2) Tf2O (4 equiv.), pyridine (4.4 equiv.), −78 ℃, 2 h. q FeCl3 (3 equiv.), 4-methylcyclohexanone (5 equiv.), DCE, 12 h. r (CH2O)n (2 equiv.), ammonium acetate (10 equiv.), AcOH, reflux, 4 h. s o-phenylenediamine (or maleimide) (4 equiv.), Na2SO4 (1.8 equiv.), HCl, DCM, 5 min. Isolated yields. er was determined by chiral-phase HPLC analysis.

Scheme 4 Control experiments and racemization experiments. Reaction conditions: 1 (0.1 mmol), Yb(OTf)3 (5 mol%), CPA-5 (6 mol%) in DCE (1 mL) at 80 ℃ under N2 atmosphere. Isolated yields. The er was determined by HPLC analysis. Racemization study.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

 下载:
下载: