Citation:
Shaohua Zhang, Xiaojuan Dai, Wei Hao, Liyao Liu, Yingqiao Ma, Ye Zou, Jia Zhu, Chong-an Di. A first-principles study of the Nernst effect in doped polymer[J]. Chinese Chemical Letters,
2024, 35(12): 109837.
doi:
10.1016/j.cclet.2024.109837
A first-principles study of the Nernst effect in doped polymer
English
A first-principles study of the Nernst effect in doped polymer
Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
b.
School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
c.
Laboratory of Theoretical and Computational Nanoscience, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Chinese Academy of Sciences, Beijing 100190, China
Received Date:
19 February 2024 Accepted Date:
26 March 2024 Revised Date:
21 March 2024 Available Online:
15 December 2024
Abstract:
Since the discovery of the Nernst effect in 19th century, it has been an important transverse thermoelectric charge transport phenomenon in solid states. Conjugated polymers have recently attracted great attention as promising optoelectronic materials. However, the Nernst effect is yet to be explored for conducting polymers. Here, we report the first theoretical investigations of the Nernst effect in doped conducting polymers by first-principles calculations under the frame work of Fermi-liquid theory. Specifically, the Nernst coefficients of PBTTT are found to be ranging from 0.0029 to 0.039 µV K−1 T−1. They are monotonically decreased with the doping level due to both much enhanced Fermi energy and the decreased charge mobility at high doping level. Our theoretical findings not only enhance our fundamental understanding of the doping mechanism that controls the charge transport properties of conducting polymers, but more importantly, they also offer initial predictions of the transverse thermoelectric conversion capability of conducting polymers. These predictions are crucial for the development of future flexible thermoelectric applications based on the Nernst effect.
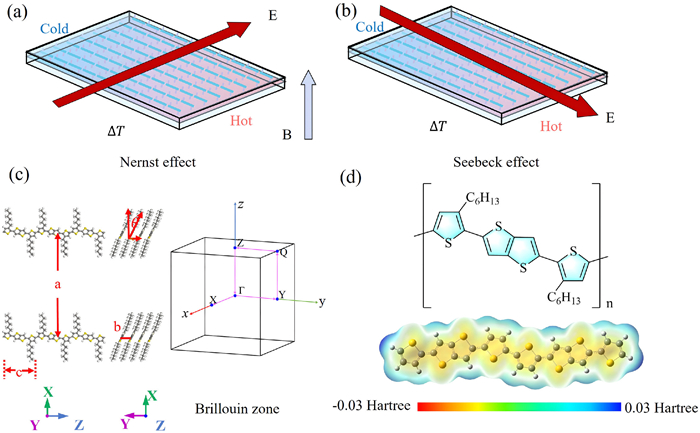
The Nernst effect is a fundamental magneto-thermoelectric phenomenon. It generates a transverse electric field Ey, when a longitudinal temperature gradient ∇xT is applied in the presence of a perpendicular magnetic field Bz. The Nernst coefficient ν is defined as ν = Ey/(Bz∇xT). As the transverse counterpart of the longitudinal Seebeck effect, the Nernst effect has also garnered significant attention [1–16]. This is not only due to its merits as the thermoelectric (TE) Hall effect in probing fundamental quasiparticle behavior [1–8], but also because of its unique geometry in producing efficient TE conversion on curved heat source (Figs. 1a and b) [9–14], which are particularly suitable for building flexible devices to power the Internet of Things and wearable electronics. As a result, conjugated polymers with intrinsic flexibility, large-area solution-processability, and most importantly the demonstrated promising TE conversion capability [17,18], could be potentially the perfect match for Nernst devices. However, even with this dreaming picture, the Nernst effect is yet to be explored in conjugated polymers possibly due to common experimental challenges in the magneto-transport measurements of polymers [19]. This motivates us to theoretically investigate the Nernst effect in conjugated polymers, for both exploring their transverse organic thermoelectric (OTE) potential and gaining more insights into their elusive charge transport nature.
Figure 1
Figure 1.
Comparison of the Nernst effect and Seebeck effect. (a) The Nernst effect produces a transverse electric field under a perpendicular magnetic field and a longitudinal temperature gradient. (b) The Seebeck effect generates a voltage in response to a temperature gradient, inducing an electric current flow. (c) Sketch of the structure and Brillouin zone of PBTTT-C6. (d) The electrostatic potential surfaces and structural formula of PBTTT using density functional theory (DFT).
Theoretically, within the boundary of the Landau’s Fermi-liquid (FL) picture, the Nernst coefficient of solid states is set by the ratio of charge mobility μ to Fermi energy EF [15,16]. This scaling relationship has demonstrated to be remarkably successful over several orders of magnitudes in a variety of different material systems including organic crystals [15,16]. Therefore, by assuming the polymers also follow this linear relationship, we are able to tentatively perform the initial theoretical investigation of their Nernst effect by first-principle investigations of the polymer electronic structure as well as charge transport properties μ and EF. However, even with this feasible route, it remains challenging to achieve the Nernst coefficients. Because, for this study, polymer models are required to be doped to approach the metallic state to achieve meaningful EF values, while the microscopic doping mechanisms such as the accurate doping sites are elusive for most polymers [20]. Moreover, the introduction of charged dopants significantly complicates the first-principle calculations, as it disrupts structural symmetry and requires incorporation of electric fields, charges, and spins.
Herein, we focus on the poly[2,5-bis(3-alkylthiophen-2-yl)thieno(3,2-b)thiophene] (PBTTT) system, not only due to its great interests as representing high performance conducting polymers, but also due to its proved metallic transport in high doping level regime [21,22], good crystalline orderings which enables reasonable first-principle band structure calculations [23], and relatively high charge mobility, which could potentially produce a large Nernst coefficient [24]. In this study, we report the first-principle investigations of the Nernst effect as well as charge transport properties in PBTTT-C6 with different ferric doping level. We first identify the optimized doping site of a common p-type dopant ion Fe3+ by calculating the charge density distribution as well as the binding energy of different possible doping site configurations based on the previously established PBTTT molecular structure. After finding the lowest energy doping site, three crystal models for PBTTT with different Fe3+ doping levels are constructed for subsequent electronic structure calculations. As doped PBTTT is considered to be semi-metallic with coherent transport nature, the band structure model is applied to calculate its charge transport properties including the charge mobility and effective mass [19]. Eventually, based on these calculated physical properties, we predict the Nernst coefficients of doped PBTTT by assuming it follows the Fermi-liquid picture, which are 0.039, 0.018, and 0.0029 µV K−1 T−1 for PBTTT with a Fe3+ doping level of 33%, 50%, and 100%, respectively. Our results reveal the Nernst coefficients decrease with the doping level monotonically, while the mobility initially increase with doping level and then decrease in the high doping level regime.
We first construct the PBTTT-C6 crystal orthorhombic unit cell based on the previous theoretical as well as experimental investigations [25,26], with lattice vectors b = 3.7 Å, c = 13.5 Å. The distance between neighboring sheets a is intentionally set to be sufficiently large to minimize interactions between interdigitated alkyl chains of adjacent sheets. First-principles calculations were performed using density functional theory (DFT) with the Vienna Ab initio Simulation Package (VASP) code to further optimize the PBTTT structure [27], [28]. The generalized gradient approximation (GGA) in Perdew-Burke-Ernzerhof (PBE) form was employed [29]. The calculations utilized a plane wave basis along with the frozen-core projector augmented wave (PAW) potential [30]. A plane wave cutoff energy of 500 eV was set for cell relaxation. To include the long-range van der Waals (vdW) interactions, a Grimme-type dispersion energy (DFT-D) was incorporated into the Kohn-Sham DFT energy calculations. The Brillouin zone sampling was conducted using a 1 × 6 × 1 k grid and the Gamma technique [31]. The convergence criteria for energy optimization were set at 10−4 eV, while for force convergence, it was set at 0.02 eV/Å per atom. The optimized PBTTT-C6 crystal structure and Brillouin zone are shown in Fig. 1c, the lattice parameters are as follows: b equals 3.93 Å, c equals 13.94 Å, with an adjusted angle between the backbone and lattice set at 23°.
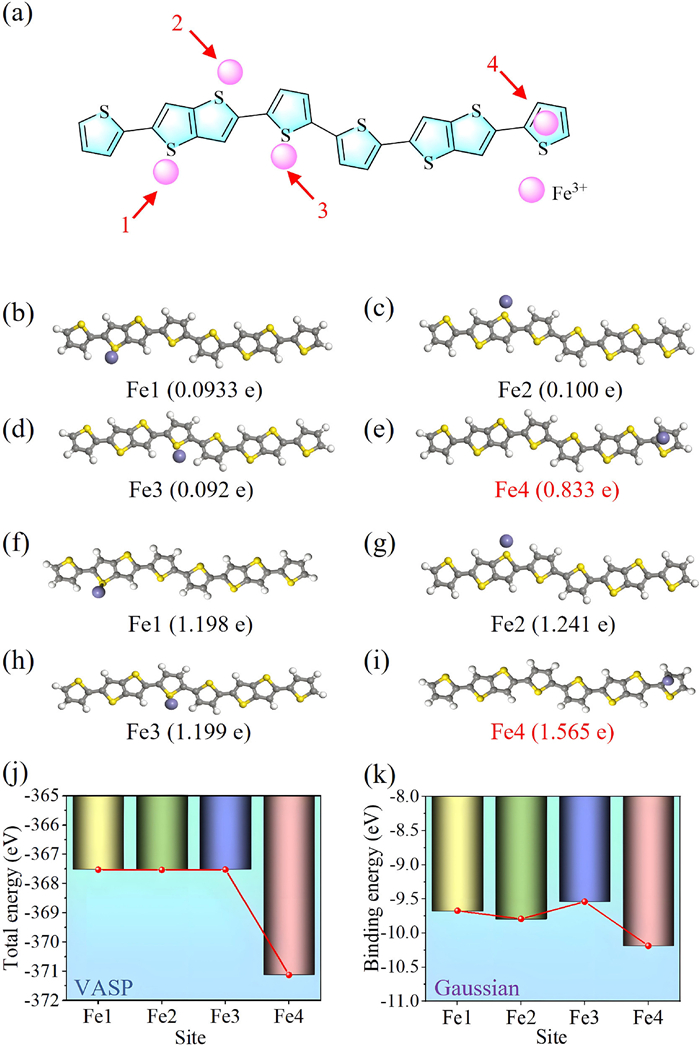
To determine the potential doping site of Fe3+, we calculated the molecular electrostatic potential distribution of PBTTT, as PBTTT is commonly p-doped and the Fe3+ dopants should locate around lowest potential site. This calculation was carried out for PBTTT oligomers without alkyl side chains for simplification, using the Gaussian 09 A.01 version with the B3LYP functional and the 6–31G(d, p) basis set in a nitromethane implicit solvent environment [32]. The resulting electrostatic potential distribution along the PBTTT skeleton is shown in Fig. 1d, in which the yellow regions represent the lowest potential site where the p-type dopants should locate for optimal doping efficiency. Specifically, four possible optimal doping sites of Fe3+ are quantitatively identified based on variations in the magnitude of the electrostatic potential surfaces, include the proximity of the electronegative sulfur elements as well as within the π electron cloud on the thiophene ring, as indicated by Fe1, Fe2, Fe3, and Fe4 (Fig. 2a). To search for the most strongly bonded site, the total energy of the four possible doping site configurations were calculated carefully, as shown in Figs. 2b–e. Three units of positive charge were introduced and the DFT+U method was employed for doped system calculations [33]. To verify the result of optimal doping site, in addition to the VASP, Gaussian is applied to calculate the binding energy as well as the charge transfer insights of the four possible doping site configurations, with a total charge of +3 and a spin multiplicity of 2 for the trivalent iron (Figs. 2f–i). As shown in Fig. 2j, the configuration Fe4, which is on the thiophene ring, is found to be the most energetically favorable doping site. The calculated binding energies were plotted in Fig. 2k, in which the Fe4 site is still found to be the lowest energy doping site with the largest charge transfer quantity, thus confirming the above calculations results.
Figure 2
Figure 2.
(a) Four doping sites determined based on the magnitude of electrostatic potential surfaces. (b–e) Four possible sites (such as near the electronegative sulfur atom, on the thiophene ring formed by the π electron cloud) were chosen to calculate, optimized structures doped at Fe1, Fe2, Fe3 and Fe4 sites and their charge transfer by VASP. (f–i) Optimized structures doped at Fe1, Fe2, Fe3, Fe4 sites and their charge transfer by Gaussian. (j) The total energy of four doping sites selected based on symmetry by VASP. (k) The binding energy of four doping sites selected based on symmetry by Gaussian.
After we found that the Fe4 on the single thiophene ring is the optimal doping site for PBTTT-C6, three different PBTTT unit cells of 1 Fe3+ per monomer, dimer, and trimer with a doping concentration of 100%, 50%, and 33%, respectively, were constructed for subsequent band structure calculations to explore the doping level dependent charge transport properties. The resulting band structure and density of states (DOS) plots of pristine PBTTT-C6 as well as 33%, 50%, and 100% doped PBTTT-C6 were presented in Figs. 3a–d, respectively. For pristine PBTTT, its band structure and DOS plots clearly reveals obvious semiconducting nature with an indirect band-gap of 0.7378 eV (Fig. 3a), where the valence band’s maximum located at Γ while the conduction band’s minimum at Q point in the Brillouin zone. As the doping level increases, the band-gap is gradually reduced and the Fermi level is shifted downward toward the valence band, indicating clear p-doping behavior (Figs. 3b–d). Meanwhile, the continuous polaron bands induced by Fe3+ appear around and cross the Fermi level, which reveals the metallic characteristics of doped PBTTT and consistent with previous experimental studies that indicate coherent transport as well as metallic temperature dependence of the conductivity in highly doped PBTTT [22].
Figure 3
Figure 3.
The crystal structure, band structure and density of states of PBTTT-C6 and doped systems. (a) PBTTT-C6. (b) Trimer doped iron ferric which doping concentration is 33%. (c) Dimer doped ferric ions which doping concentration is 50%. (d) Monomer doped ferric ions which doping concentration is 100%.
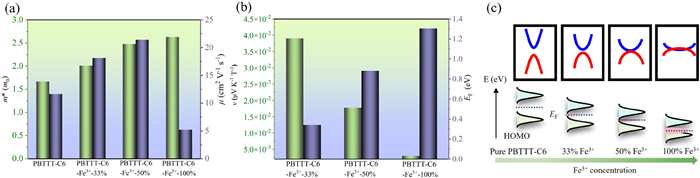
The charge transport properties including charge effective mass and interchain mobility can be quantitatively calculated based on the band structure of doped PBTTT. The charge effective mass of PBTTT with different doping level can be calculated by m* = ħ2/(∂2E/∂k2), where k is parallel to its spatial vector; E is the energy of the band; the ħ is the reduced Planck constant. The resulting effective mass are plotted in Fig. 4a, which are 1.67 electron mass m0, 2.01 m0, 2.48 m0 and 2.63 m0, corresponding to the doping level 0, 33%, 50%, 100%. The revealed charge effective mass monotonically increases with the doping level. This is because, as the PBTTT become metallic induced by the increased doping level, its energy bands will become relatively flat as similar to most metals. Subsequently, since the charge transport in doped PBTTT is considered to be two-dimensional [19], the two-dimensional deformation potential theoretical treatment is applied to achieve the charge mobility based on the effective mass m* (Eq. 1).
(1)
Figure 4
Figure 4.
(a) Variation of effective mass and mobility with Fe3+ doping concentration. (b) Variation of Nernst coefficient and fermi energy with Fe3+ doping concentration. (c) A conceptual representation explains the mechanism: Changes in fermi energy and Nernst coefficient with doping concentration.
where T is the temperature, and kb is Boltzmann constant; B is elastic constant; εac is the deformation potential constant [34,35]. The elastic constant can be obtained by fitting the parabolic relationship between the total lattice energy and the deformation variable, while the deformation potential constant is calculated by fitting the linear relationship between the band edge energy and the lattice deformation variable. The resulting calculated PBTTT charge mobility reveals an initially positive doping level dependence which is 11.66 cm2 V−1 s−1, 18.15 cm2 V−1 s−1, 21.42 cm2 V−1 s−1, corresponding to the 0, 33%, and 50% doping level, respectively, and then significantly decreases to 5.23 cm2 V−1 s−1 when the doping level is enhanced to 100% (Fig. 4a).
The doped PBTTT Fermi energy is further calculated within the Fermi gas electron filling picture, in which the Fermi energy is positively correlated with the number of DOS at the Fermi level ρ(EF) by Eq. 2:
(2)
As shown in Fig. 4b, due to the enhanced number of DOS at the Fermi level induced by the increased doping level, the Fermi energy monotonically increases with doping, which is consistent with typical doping effect.
With the calculated charge mobility as well as Fermi energy, the Nernst coefficients ν of doped PBTTT can be obtained by assuming it follows the Fermi-liquid picture (Eq. 3) [36–39]:
(3)
The resulting room-temperature (300 K) Nernst coefficients are calculated to be 0.039 µV K−1 T−1, 0.018 µV K−1 T−1, 0.0029 µV K−1 T−1, for 33%, 50%, and 100% doped PBTTT respectively.
The revealed non-monotonic doping level dependence of the charge mobility is unusual regarding the common belief of enhanced charge mobility with doping level in conducting polymers [40]. This abnormal behavior is due to the intricate interplay between various factors. A conceptual representation elucidates the mechanism underlying the changes in density of states in Fig. 4c. In the initial stage of doping up to 50% doping level, the schematic illustrates an increase in electron density near the Fermi energy level due to doping, resulting in a reduced bandgap which facilitates electron transitions and charge transport, thus enhancing the charge mobility. Meanwhile, as the doping concentration approaches 100%, the bandgap stops reducing and the PBTTT enters the metallic regime, while the effective mass is continuously increased due to flat bands, which eventually leads to a decrease in charge mobility. Regarding the Nernst effect, similar to the Seebeck effect, the Nernst coefficients also monotonically decrease with the doping level, which is mostly due to the enhanced Fermi energy.
In summary, we present the initial first-principle study of the Nernst effect in doped conducting polymers. Our comprehensive analysis enables the identification of molecular doping sites, deepen our understanding of the microscopic doping mechanism and molecular dynamics. Moreover, the doping level dependent band structure and charge transport properties calculations provide essential insights into the evolution of charge transport in conducting polymers upon doping. Our findings are pivotal for the strategic design and performance optimization of organic electronic devices. Although the Nernst coefficients calculated under the Fermi-liquid picture are found to be much lower than common Seebeck coefficients of conducting polymers, they might still promise surprising Nernst effect. Because current first-principle calculations cannot effectively consider the intrinsic structural, energetic, as well as dynamic disorder in polymer systems, which disorder effects will become significantly more notable and complicated in realistic doped Nernst systems when charged dopants are incorporated. Moreover, in fact, the charge transport in conducting polymers could be more exotic beyond the Fermi-liquid regime due to its quasi-one-dimensional interchain charge transport nature, even though there are not much solid experimental evidence to support this idea. Therefore, the experimental measurements of the Nernst effect in polymers remain to be the key path to evidently reveal their transverse TE conversion capability, and we anticipate an exciting experimental breakthrough in this field in a near future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Shaohua Zhang: Data curation, Formal analysis. Xiaojuan Dai: Formal analysis, Software. Wei Hao: Formal analysis. Liyao Liu: Writing – original draft, Writing – review & editing. Yingqiao Ma: Conceptualization, Writing – original draft. Ye Zou: Funding acquisition, Software. Jia Zhu: Formal analysis, Writing – original draft. Chong-an Di: Conceptualization, Formal analysis, Writing – original draft, Writing – review & editing.
Acknowledgments
The authors acknowledge financial support from the National Natural Science Foundation of China (Nos. 22125504, 22175186, 62205347, 22305253, 62075224, 22021002 and 21805285), the Natural Science Foundation of Beijing (No. Z220025) and the K.C. Wong Education Foundation (No. GJTD-2020-02).
[1]
O. Cyr-Choiniere, R. Daou, F. Laliberté, et al., Nature 458 (2009) 743–745. doi: 10.1038/nature07931
[2]
Z. Zhu, H. Yang, B. Fauque, et al., Nat. Phys. 6 (2010) 26–29. doi: 10.1038/nphys1437
[3]
T. Liang, Q. Gibson, J. Xiong, et al., Nat. Commun. 4 (2013) 2696. doi: 10.1038/ncomms3696
[4]
T. Yamashita, Y. Shimoyama, Y. Haga, et al., Nat. Phys. 11 (2015) 17–20. doi: 10.1038/nphys3170
T.J. Aubry, J.C. Axtell, V.M. Basile, et al., Adv. Mater. 31 (2019) 1805647. doi: 10.1002/adma.201805647
Figure 1
Comparison of the Nernst effect and Seebeck effect. (a) The Nernst effect produces a transverse electric field under a perpendicular magnetic field and a longitudinal temperature gradient. (b) The Seebeck effect generates a voltage in response to a temperature gradient, inducing an electric current flow. (c) Sketch of the structure and Brillouin zone of PBTTT-C6. (d) The electrostatic potential surfaces and structural formula of PBTTT using density functional theory (DFT).
Figure 2
(a) Four doping sites determined based on the magnitude of electrostatic potential surfaces. (b–e) Four possible sites (such as near the electronegative sulfur atom, on the thiophene ring formed by the π electron cloud) were chosen to calculate, optimized structures doped at Fe1, Fe2, Fe3 and Fe4 sites and their charge transfer by VASP. (f–i) Optimized structures doped at Fe1, Fe2, Fe3, Fe4 sites and their charge transfer by Gaussian. (j) The total energy of four doping sites selected based on symmetry by VASP. (k) The binding energy of four doping sites selected based on symmetry by Gaussian.
Figure 3
The crystal structure, band structure and density of states of PBTTT-C6 and doped systems. (a) PBTTT-C6. (b) Trimer doped iron ferric which doping concentration is 33%. (c) Dimer doped ferric ions which doping concentration is 50%. (d) Monomer doped ferric ions which doping concentration is 100%.
Figure 4
(a) Variation of effective mass and mobility with Fe3+ doping concentration. (b) Variation of Nernst coefficient and fermi energy with Fe3+ doping concentration. (c) A conceptual representation explains the mechanism: Changes in fermi energy and Nernst coefficient with doping concentration.

 DownLoad:
DownLoad:


 下载:
下载:





 下载:
下载:
