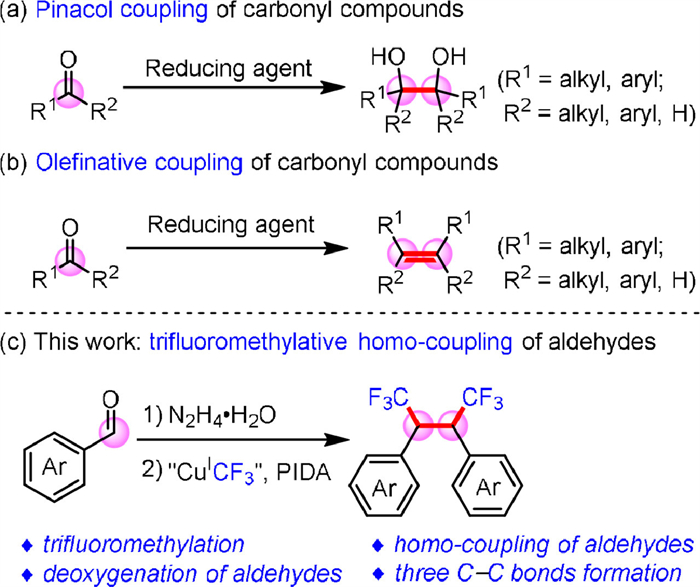
Scheme 1.
Homo-coupling of carbonyl compounds.

In light of the continual depletion of fossil energy reserves, the matter of the energy crisis has attracted growing interest within the field of chemistry [1,2]. Biomass resources, a pivotal facet of renewable resources, present an ideal substitute for fossil fuels [3]. Unlike fossil resources, biomass resources possess a notably higher oxygen content, primarily in the form of carbonyl or hydroxyl groups [4–7]. Consequently, the efficient harnessing of the abundant carbonyl compounds within biomass resources has become increasingly imperative.
Carbonyl compounds are extensively employed in organic synthesis due to their abundance and chemical versatility. Over the course of extensive research, homo-coupling of carbonyl compounds has emerged as an efficient means of forging C–C bonds. The classic Pinacol coupling reaction (Scheme 1a) [8,9] is a well-established process in organic chemistry. Traditionally, stoichiometric quantities of metal reagents are utilized as reducing agents to convert carbonyl compounds into ketone radicals through a single electron transfer (SET) process. Subsequently, intermolecular radical coupling reactions yield pinacol products. With the advent of photochemical reactions catalyzed by metal photosensitizers, [Ir] photoredox catalysis for generating ketone radicals from carbonyl compounds through the SET process was reported by Rueping group in 2019 [10]. At the same time, our group reported Pinacol coupling of ketone compounds relying solely on N2H4 as a hydrogen atom transfer (HAT) reducing agent [11,12]. Furthermore, our group reported a Ni-catalyzed homo-coupling reaction of carbonyl compounds via hydrazones, successfully synthesizing bibenzyls [6]. In 2022, Yamaguchi reported the generation of diarylphosphinates from diarylketones and diphenylphosphine through the phospha-Brook rearrangement, and benzyls were also successfully generated using a palladium catalyst [13]. Another significant method for homo-coupling of carbonyl compounds is olefinative coupling (Scheme 1b) [14–18]. The McMurry reaction enables the reductive homo-coupling of aldehydes and ketones to form alkenes under the influence of low-valent titanium. In 2017, we successfully implemented the hydrazone as organo-metallic equivalent (HOME chemistry) [19–24] strategy to achieve olefinative coupling of carbonyl compounds catalyzed by ruthenium(Ⅱ) [25]. Compared to the classic McMurry reaction, this process avoided the use of strong reducing agents under mild conditions and exhibited excellent functional group compatibility. In 2019, Konig et al. reported a photo-redox catalytic reaction using B2(pin)2 as the terminal reducing agent, successfully achieving olefinative coupling of carbonyl compounds [26]. In 2022, Nagib et al. developed a novel olefinative coupling model that converted aldehydes into carbenes via zinc carbenoids, thereby accomplishing olefinic coupling of aldehydes through effective catalysis using cobalt salts [27]. Very recently, Nagib group reported cross-olefinative coupling reactions between different carbonyl compounds, producing Z- or E-olefins with Fe or Cr catalysis, respectively [28].
While there has been notable progress in the homo-coupling of carbonyl compounds in recent years, significant limitations continue to hinder their practical application in synthesis. Currently, carbonyl compounds tend to primarily yield either pinacols, as a result of the high bond energy of C–O bonds making deoxygenation difficult, or olefins through the elimination of hydroxyl groups in homo-coupling reactions. This limitation restricts our ability to fully harness the synthetic potential inherent in C=O bonds [29]. Consequently, substantial challenges persist in achieving deoxy-functionalization homo-coupling of carbonyl compounds.
Trifluoromethyl groups are known for their strong electron-withdrawing and the strongest lipophilicity properties. Their introduction into organic molecules can greatly alter electron distribution and enhance lipophilicity [30,31]. Meanwhile, owing to the high stability of the C–F bond, the introduction of trifluoromethyl significantly enhances the metabolic stability of organic molecules in organisms [31]. Therefore, trifluoromethylation reactions are a hot topic in organic chemistry, attracting substantial interest from researchers.
Building on our earlier success in homo-coupling carbonyl compounds, we present a new advancement: the trifluoromethylative homo-coupling of carbonyl compounds [32] via hydrazones (Scheme 1c). Significantly, this protocol not only introduces the CF3 group conveniently into natural and abundant aldehydes but also accomplishes the deoxygenative homo-coupling of aromatic aldehydes. This process leads to the creation of three C–C bonds in a single step, establishing a novel pathway for homo-coupling reactions of carbonyl compounds.
At the onset of our investigation, p-methylbenzaldehyde hydrazone (2a) was used as the model substrate, and “CuⅠCF3” (prepared in situ from CuI, TMSCF3 and CsF in 1.0 mL DMF solvent) [33] as trifluoromethyl source, to optimize the reaction conditions. When K2S2O8 was used as an oxidant, trifluoromethylative homo-coupling product 3a was detected in 5% 19F NMR yield (Table 1, entry 1). Considering the importance of oxidants for this transformation, various types of oxidants were tested (Table 1, entries 2–7). (Diacetoxyiodo)benzene (PIDA) gave the best yield (52%) of the homo-coupling product 3a among these oxidants (entry 7). Adjusting the amount of PIDA or reaction temperature did not significantly improve the yield of 3a (entries 8–11). Exploring various bases did not improve the yield of the target product 3a (entries 12–18). When 4 Å molecular sieve (MS 4 Å) was added to the reaction system, the yield of the target product 3a was increased to 55% (entry 19). Then, we investigated the influence of the amounts of 4 Å molecular sieve and CuI (entries 20–22), neither of which increased the yield of target product 3a. Extending the reaction duration or conducting the reaction in an argon atmosphere did not enhance the production of 3a either (entries 23 and 24). Finally, we obtained the optimal conditions for trifluoromethylative coupling of aldehydes via hydrazones (entry 19).
With the optimized reaction conditions in hand, the substrate scope for the aromatic aldehydes was explored (Scheme 2). Alkyl substituted benzaldehydes (R = Me, n-Pr, t-Bu) underwent the reaction smoothly and provided the target compounds in moderate yields (3a–3c). Even sterically hindered substrate, such as pentamethylbenzaldehyde (1d), yielded the desired product 3d in 51% yield, indicating that the steric effect did not significantly impact the reaction. Strong electron-donating groups, such as methoxy and thiomethyl on the arene ring, led to trifluoromethylative homo-coupling products in moderate to good yields (3e-3h). It is worth noting that this transformation is also applicable with heterocyclic structures (3i–3k). Weakly electron-withdrawing halogen substituents (X = F, Cl, Br and I) proved compatible (3l–3o); in particular, polyhalogen substituted benzaldehydes also successfully produced the corresponding products in medium yields (3p, 3q). For aromatic aldehyde hydrazones with strong electron-withdrawing substituents, we observed the partial generation of hydrotrifluoromethylation products [23], resulting in lower yields of trifluoromethylative homo-coupling products (3r, 3s). Polycyclic hydrazones, like 2-naphthalene formaldehyde 1t, also successfully achieved this transformation in 41% yield (3t). Moreover, when a hetero-aromatic aldehyde was subjected to the standard conditions, 53% yield was obtained with benzofuran-5-carbaldehyde 1u. The reactivities of 2-furancarbaldehyde 1v, 2-thiophenecarbaldehyde 1w, and 3-pyridinecarbaldehyde 1x, which were aldehydes containing heterocyclic aromatic rings, were less than satisfactory. The following reasons may affect the overall yield of products: (1) The benzyl radical underwent a radical substitution reaction with an aromatic ring to form by-products; (2) Deoxygenative hydrotrifluoromethylation products [23] can be detected as by-product.
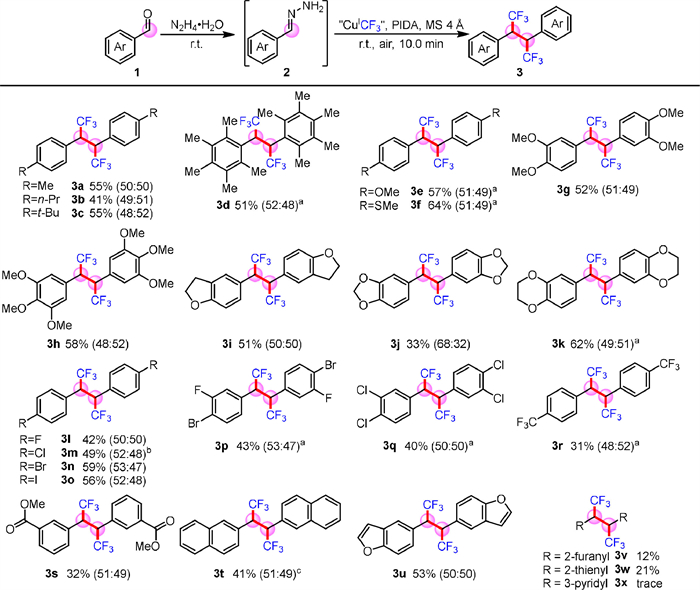
Furthermore, the demethylation of trifluoromethylative homo-coupling product 3e has unique applications in medicine and materials. It can significantly inhibit tumor growth in hormone- dependent mammary carcinomas [34], as well as has important applications in waterproof materials and 19F MRI (Scheme 3) [35].
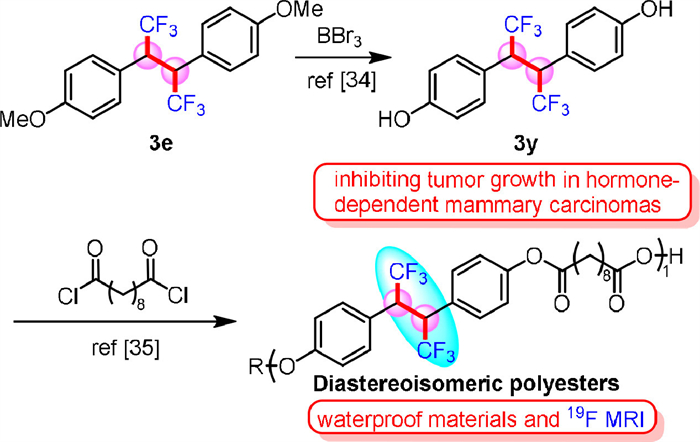
To elucidate the mechanism of this reaction, controlled experiments were carried out (Scheme 4). Firstly, N-tosylhydrazone 4 was used instead of hydrazone 2a under standard conditions, and no target product 3a was obtained, indicating that the reaction did not involve diazo compounds or metal carbene intermediates (Scheme 4a). Considering that PIDA might oxidize “CuⅠCF3” to generate “CuⅢ(CF3)3″ species, which might be responsible for this reaction, we used NBu4CuⅢ(CF3)4 as the CF3 source (Scheme 4b) and no target product 3a was detected. The addition of CBrCl3 (0.5 equiv.) as a radical inhibitor to the reaction system led to complete inhibition of target product generation, indicating that the reaction process might involve radical intermediates (Scheme 4c). When TEMPO was added as a radical capture agent to the reaction system, the trifluoromethylative homo-coupling product 3a was inhibited, and the formation of compound 5 showed that the trifluoromethyl substituted benzyl radical might be an important intermediate (Scheme 4d).
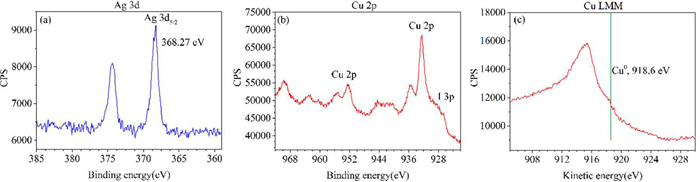
Furthermore, the oxidation state of Cu was examined subsequent to the reaction (please see Section ΙΙ in Supporting information for details), with the Ag 3d5/2 signal at 368.27 eV serving as the reference for charge calibration (Fig. 1a). X-ray photoelectron spectroscopy (XPS) analysis of copper showed that the CuⅡ signal was clearly detected under general condition (Fig. 1b). Auger electron spectroscopy (AES) analysis of copper showed that there was no obvious peak at the kinetic energy position of Cu0 at 918.6 eV (Fig. 1c), which revealed that the reaction system did not contain Cu0 (Fig. 1).
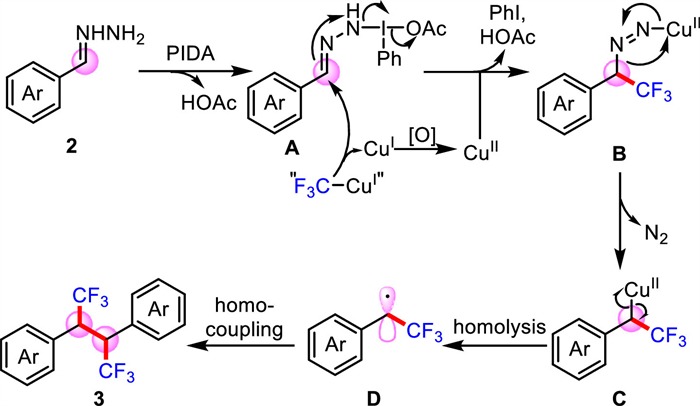
Based on the above experimental results, a possible reaction mechanism is proposed. As shown in Scheme 5, PIDA is attacked by terminal N atoms of hydrazone 2 to form intermediate A and loose acetic acid. Then, under the attack of the trifluoromethyl anion from “CuⅠCF3”, intermediate A releases iodobenzene and HOAc to form CuⅡ coordinated intermediate B. Subsequently, the intermediate B releases nitrogen gas to generate organic copper(ΙΙ) species C, which forms trifluoromethyl substituted benzyl radical D due to the homolysis of the C–CuⅡ bond [36,37]. Finally, the benzyl radical D is homo-coupled to generate trifluoromethylative homo-coupling product 3.
In conclusion, we have successfully pioneered the trifluoromethylative homo-coupling of aldehydes, which resulted in the formation of three C–C bonds in a single step. Mechanistic studies showed that the trifluoromethyl substituted benzyl radical was an important intermediate. This protocol introduces an innovative pathway for the deoxy-functionalization homo-coupling of aldehydes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xinlong Han: Methodology, Writing – original draft. Huiying Zeng: Conceptualization, Project administration, Writing – review & editing. Chao-Jun Li: Conceptualization, Writing – review & editing.
We thank the National Natural Science Foundation of China (NSFC, No. 21971093), the Fundamental Research Funds for the Central Universities (No. lzujbky-2021-sp53), the International Joint Research centre for Green Catalysis and Synthesis (No. 2016B01017), The Science and Technology Major Program of Gansu Province of China (No. 22ZD6FA006) and the 111 project for support of our research. We also thank the Canada Research Chair (Tier I) foundation, the E.B. Eddy Endowment Fund, the CFI, NSERC, and FQRNT to C.-J. Li.
Supplementary material associated with this article can be found, in the online version, at doi:
M. He, Y. Sun, B. Han, Angew. Chem. Int. Ed. 52 (2013) 9620–9633. doi: 10.1002/anie.201209384
C.J. Li, Chem 1 (2016) 423–437. doi: 10.1016/j.chempr.2016.08.007
A. Corma, S. Iborra, A. Velty, Chem. Rev. 107 (2007) 2411–2502. doi: 10.1021/cr050989d
J.R. Dethlefsen, P. Fristrup, ChemSusChem 8 (2015) 767–775. doi: 10.1002/cssc.201402987
L.J. Donnelly, S.P. Thomas, J.B. Love, Chem. Asian J. 14 (2019) 3782–3790. doi: 10.1002/asia.201901274
D. Cao, Z. Chen, L. Lv, et al., iScience 23 (2020) 101419. doi: 10.1016/j.isci.2020.101419
G.W.K. Moore, S.E.L. Howell, M. Brady, X. Xu, K. McNeil, Nat. Commun. 12 (2021) 1. doi: 10.1038/s41467-020-20314-w
T. Wirth, Angew. Chem. Int. Ed. 35 (1996) 61–63. doi: 10.1002/anie.199600611
K. Suzuki, M. Tamiya, Pinacol coupling reactions, in: P. Knochel (Ed.), Comprehensive Organic Synthesis, 2nd Ed, Elsevier, Amsterdam, 2014, pp. 580–620.
M. Nakajima, E. Fava, S. Loescher, Z. Jiang, M. Rueping, Angew. Chem. Int. Ed. 54 (2015) 8828–8832. doi: 10.1002/anie.201501556
Z. Qiu, H.D.M. Pham, J. Li, et al., Chem. Sci. 10 (2019) 10937–10943. doi: 10.1039/c9sc03737c
M. Liu, T. Tan, R.T. Rashid, et al., Chem. Sci. 11 (2020) 7864–7870. doi: 10.1039/d0sc02718a
M.B. Kurosawa, K. Kato, K. Muto, J. Yamaguchi, Chem. Sci. 13 (2022) 10743–10751. doi: 10.1039/d2sc03720c
J.E. McMurry, M.P. Fleming, J. Am. Chem. Soc. 96 (1974) 4708–4709. doi: 10.1021/ja00821a076
J.E. McMurry, M.P. Fleming, J. Org. Chem. 41 (1976) 896–897. doi: 10.1021/jo00867a038
B.E. Kahn, R.D. Rieke, Chem. Rev. 88 (1988) 733–745. doi: 10.1021/cr00087a002
J.E. McMurry, Chem. Rev. 89 (1989) 1513–1524. doi: 10.1021/cr00097a007
T. Takeda, A. Tsubouchi, The McMurry coupling and related reactions, 2013, doi:
C.J. Li, Pure Appl. Chem. 95 (2023) 465–474. doi: 10.1515/pac-2022-1003
C.J. Li, J. Huang, X.J. Dai, et al., Synlett 30 (2019) 1508–1524. doi: 10.1055/s-0037-1611853
P. Pan, Y. Lang, D. Cao, H. Zeng, C.J. Li, CCS Chem. 4 (2022) 3254–3263. doi: 10.31635/ccschem.022.202101685
P. Pan, S. Liu, Y. Lan, H. Zeng, C.J. Li, Chem. Sci. 13 (2022) 7165–7171. doi: 10.1039/d2sc01909d
H. Zeng, Z. Luo, X. Han, C.J. Li, Org. Lett. 21 (2019) 5948–5951. doi: 10.1021/acs.orglett.9b02072
L. Jia, C.J. Li, H. Zeng, Chin. Chem. Lett. 33 (2022) 1519–1523. doi: 10.1016/j.cclet.2021.08.125
W. Wei, X.J. Dai, H. Wang, et al., Chem. Sci. 8 (2017) 8193–8197. doi: 10.1039/C7SC04207H
S. Wang, N. Lokesh, J. Hioe, R.M. Gschwind, B. König, Chem. Sci. 10 (2019) 4580–4587. doi: 10.1039/c9sc00711c
L. Zhang, B.M. DeMuynck, A.N. Paneque, J.E. Rutherford, D.A. Nagib, Science 377 (2022) 649–654. doi: 10.1126/science.abo6443
L. Zhang, D.A. Nagib, Nat. Chem. 16 (2024) 107–113. doi: 10.1038/s41557-023-01333-8
J. Li, C.Y. Huang, C.J. Li, Angew. Chem. Int. Ed. 61 (2022) e202112770. doi: 10.1002/anie.202112770
R. Filler, Y. Kobayashi, Biomedicinal Aspects of Fluorine Chemistry, Elsevier, Amsterdam, 1982.
H.J. Böhm, D. Banner, S. Bendels, et al., ChemBioChem 5 (2004) 637–643. doi: 10.1002/cbic.200301023
B. Gao, Y. Zhao, C. Ni, J. Hu, Org. Lett. 16 (2014) 102–105. doi: 10.1021/ol403083e
M. Hu, C. Ni, J. Hu, J. Am. Chem. Soc. 134 (2012) 15257–15260. doi: 10.1021/ja307058c
R.W. Hartmann, A. Heindl, M.R. Schneider, H. Schoenenberger, J. Med. Chem. 29 (1986) 322–328. doi: 10.1021/jm00153a004
J. Lv, S. Chen, Z. Xu, S. Zhang, Y. Li, Y. Zhao, Macromolecules 54 (2021) 3716–3724. doi: 10.1021/acs.macromol.0c02762
T.W. Liwosz, S.R. Chemler, J. Am. Chem. Soc. 134 (2012) 2020–2023. doi: 10.1021/ja211272v
Y. Miller, L. Miao, A.S. Hosseini, S.R. Chemler, J. Am. Chem. Soc. 134 (2012) 12149–12156. doi: 10.1021/ja3034075

Scheme 2 The scope of hydrazone substrates. General conditions: 2 (0.2 mmol), “CuⅠCF3” (2.0 equiv., 1.0 mL DMF as solvent), PIDA (1.5 equiv.) and 4 Å molecular sieves (50.0 mg) were stirred for 10.0 min at r.t. in 20.0 mL tube under air. Unless otherwise indicated, the 19F NMR yields were shown, diastereoselectivity (syn/anti) were determined by 19F NMR given in parentheses. aUnder Ar. bIsolated yield. c1.0 mL DMSO as solvent.

Figure 1 X-ray photoelectron spectroscopy (XPS) and Auger electron spectroscopy (AES) of the Cu-containing mixture. (a-c) The remaining solids in the trifluoromethyl homo-coupling reaction system, respectively.

Scheme 5 Possible mechanism of trifluoromethylative coupling of carbonyl compounds via hydrazones.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:


 下载:
下载: