Organic stoichiometric cocrystals with a subtle balance of charge-transfer degree and molecular stacking towards high-efficiency NIR photothermal conversion
Citation:
Jieqiong Xu, Wenbin Chen, Shengkai Li, Qian Chen, Tao Wang, Yadong Shi, Shengyong Deng, Mingde Li, Peifa Wei, Zhuo Chen. Organic stoichiometric cocrystals with a subtle balance of charge-transfer degree and molecular stacking towards high-efficiency NIR photothermal conversion[J]. Chinese Chemical Letters,
2024, 35(10): 109808.
doi:
10.1016/j.cclet.2024.109808
Organic stoichiometric cocrystals with a subtle balance of charge-transfer degree and molecular stacking towards high-efficiency NIR photothermal conversion
English
Organic stoichiometric cocrystals with a subtle balance of charge-transfer degree and molecular stacking towards high-efficiency NIR photothermal conversion
Institutes of Physical Science and Information Technology, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui Graphene Engineering Laboratory, Anhui University, Hefei 230601, China
b.
Molecular Science and Biomedicine Laboratory (MBL), State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, Aptamer Engineering Center of Hunan Province, Hunan University, Changsha 410082, China
c.
Department of Chemistry and Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Shantou 515063, China
* Corresponding authors. E-mail addresses: lskhnu@126.com (S. Li)
zhuochen@hnu.edu.cn (Z. Chen). 1 These authors contributed equally to this work.
Received Date:
16 January 2024 Accepted Date:
20 March 2024 Revised Date:
15 March 2024 Available Online:
15 October 2024
Abstract:
Charge-transfer (CT) stoichiometric cocrystals are promising choice of organic materials for unveiling the structure-property relationship. However, due to the contradiction between large CT degree required for strong NIR absorption and flexible molecular stacking, construction of stoichiomorphism-based cocystals with near-infrared (NIR) photothermal property remains challenging. Herein, the first example of stoichiomorphism-based photothermal cocrystals were accomplished through the adaptive assembly of 3,3′,5,5′-tetramethylbenzidine (TMB) donor and 1,2,4,5-tetracyanobenzene (TCNB) acceptor. The selective cocrystallization could be controlled by varying the donor-acceptor stoichiometries via a surfactant-assisted method, resulting in two cocrystals with 1:1 (T1C1) and 1:2 (T2C1) stoichiometries. The absorbance intensity of T1C1 at 808 nm was nearly twice that of T2C1, while the photothermal conversion efficiency (PCE) of the former was 60.3% ± 0.6%, approximately 80% of that for the latter (75.5% ± 2.6%), which might be caused by the different intermolecular interactions in distinct molecular stacking patterns. Notably, both excellent PCEs of stoichiometric cocrystals were attributed to the nonradiative transition process, including internal conversion and charge dissociation processes, as elucidated by femtosecond transient absorption spectroscopy measurements. Furthermore, T1C1 was used as an NIR heater for preparing agarose-based photothermal hydrogel, showing great potential for light-controlled in-situ gelation. This strategy of balancing the CT degree and molecular packing orientation not only uncovered the relationship between stoichiometric stacking and photothermal property, but also provided an opportunity to develop advanced organic optoelectronic materials.
Organic charge transfer (CT) cocrystal, is a class of single crystalline material, consisted of electron donor (D) and electron acceptor (A) through intermolecular CT interactions at a certain stoichiometric ratio [1,2]. Due to the superiorities of structural flexibility, reliable synergies, low cost, and solution processability, CT cocrystal engineering has emerged as a universal and effective strategy for construction of organic functional materials with desired optoelectronic features, such as high conductivity [3], ambipolar charge transport [4], room-temperature phosphorescence [5], nonlinear optics [6]. Molecular orbital theory holds that CT transition is a process in which partial electrons transfer from the highest occupied molecular orbital (HOMO) of the donor to the lowest unoccupied orbital (LUMO) of the acceptor [7,8]. Therefore, the intermolecular CT nature can effectively narrow the energy gap, resulting in obvious absorption redshift of cocrystals and promoting the non-radiative decay [9–20]. A plentiful of co-assemblies of donors and acceptors have been attempted to pursue photothermal materials with broad near-infrared (NIR) absorption and glorious photothermal performance, and the resultant cocrystals show great application perspectives in the fields of photothermal imaging [9,11,12,14,20], photothermal therapy [15,17–19], and seawater desalinations [12,14,16]. Although CT cocrystals-based photothermal materials have got swift and violent progress recently [21], the inadequate understanding of photothermal mechanism makes them still in the stage of gradual discovery.
It is well-know that the long-range order, free-defects and precise crystal architectures of organic cocrystals provide the opportunities in revealing the structure-property and CT-property relationships [22–25]. In particular, the discovery of cocrystals polymorph phenomenon, most of which are stoichiometric cocrystals, composed of the same building blocks with different D/A ratios and packing arrangements, allows for meticulous investigation of the relationship between molecular stacking fashions and fascinating physicochemical properties [26–33]. For instance, perylene and 7,7,8,8-tetracyanoquinodimethane (TCNQ) can be assembled into three types of PxTy cocrystals with 1:1, 2:1 and 3:1 stoichiometries and display different electron or holes transport capabilities [34–36]. Stoddart et al. construct two-photon cocrystals through the selective cocrystallization of naphthalenediimide-based triangle and coronene at D/A ratios of 1:1 and 1:2, respectively [37]. The former exhibits deep-red and NIR emission, while the latter only emits deep-red fluorescence. Moreover, the differences caused by different stoichiometric stacking are reflected in the mechanical response behaviors [38] and photomechanical motions [39]. In these reported stoichiometric cocrystals, their flexible and varied molecular stacking fashions depend on the smaller degrees of charge transfer (DCT) between donors and acceptors. While the large DCT required for effective NIR absorption of photothermal cocrystals might limit the constituent units to a specific molecular packing to some extent. Therefore, the effect of stoichiometric stacking in CT cocrystals on photothermal conversion has hardly been studied because it is difficult to achieve the coexistence of strong NIR absorption and stoichiomorphism in one system.
Herein, two stoichiometric photothermal supramolecular materials based on the cocrystallization of 3,3′,5,5′-tetramethylbenzidine (TMB) and 1,2,4,5-tetracyanobenzene (TCNB) are prepared via a green and simple surfactant-assisted strategy. Owing to the well-matched structures and electron clouds, the donor TMB and the acceptor TCNB can form two cocrystals with 1:1 and 2:1 stoichiometries, which are denoted by T1C1 and T2C1, respectively. The definite donor-acceptor stoichiometric ratio is essential to control the selective cocrystallization. Analyses of the single-crystal structures reveal that T1C1 exhibits a more efficient donor-acceptor overlap with a mixed-stack mode compared with that of T2C1 with typical 2:1 stacking, which contributes to a stronger CT interaction in T1C1. As a result, T1C1 has narrower energy gap and an absorption cutoff edge of 1100 nm, while T2C1 can only absorb light before 957 nm. Although there is a significant difference of nearly twice the light absorption intensity at 808 nm, the photothermal heating capacity of T2C1 is comparable to that of T1C1. This is mainly due to that the photothermal conversion efficiency (PCE) of T2C1 (75.5% ± 2.6%) is higher than that of T1C1 (60.3% ± 0.6%). Femtosecond transient absorption spectroscopy demonstrates that both excellent PCEs are attributed to the non-radiation decay process, including internal conversion and charge dissociation processes. Furthermore, we in-situ construct a composite agarose hydrogel by using T1C1 as an NIR photothermal heater, showing great potential for remote light-controlled in-situ gelation of functional hydrogels.
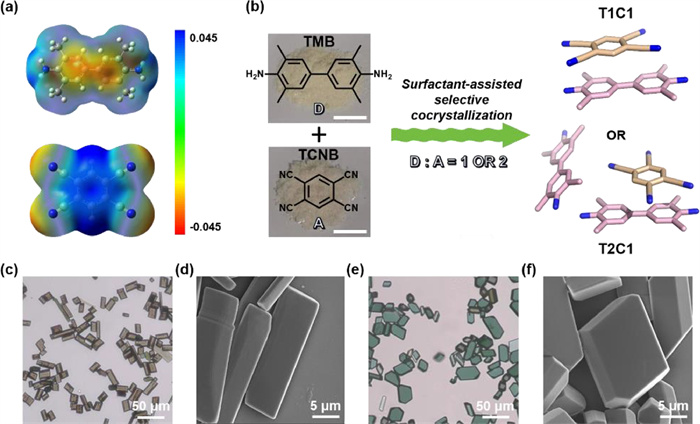
TCNB molecules are known as one of the most popular acceptors, which can accept electrons from most of the donor molecules to form large amounts of organic functional cocrystals [23,40–42]. Taking into account the balance between the DCT and the flexibility of packing modes, TMB with suitable electron-donating capability was selected to match TCNB for constructing CT cocrystals [43]. Molecular electrostatic potential (ESP) analyses showed that the aromatic nuclei of TMB was highly electron-rich, while TCNB had deep π pores, ensuring strong π-π interactions between TMB and TCNB (Fig. 1a). A green surfactant-assisted coprecipitation method was adopted to prepare TMB-TCNB cocrystals in aqueous solutions. The surfactant C18-PEG played a key role in promoting cocrystallization, due to its solubilization effects for hydrophobic TMB and TCNB molecules in water (Fig. 1b) [44]. The proposed mechanism for the formation of stoichiometric TMB-TCNB cocrystals was shown in Fig. S1 (Supporting information). When TMB and TCNB molecules were added to the dilute surfactant solutions, two kinds of hydrophobic molecules could be quickly encapsulated by surfactant molecules C18-PEG and dispersed in water [45]. Under different TMB-TCNB molar ratios, molecules contacted with each other and formed two stoichiometric cocrystals through intermolecular CT interactions. More importantly, the donor-acceptor stoichiometries played a decisive role in the cocrystal morphologies in Fig. S2 (Supporting information). With a TMB-TCNB molar ratio of 1:1, brownish T1C1 cocrystals with uniform cuboid shape were obtained. Alternatively, when TMB-TCNB molar ratio was 2:1, a quite another greenish T2C1 cocrystals with polygon shape occurred as shown in Figs. 1c-f and Fig. S3 (Supporting information). Compared to the colorless original components, the dark color of two cocrystals clearly indicated the presence of intermolecular CT interactions between TMB and TCNB molecules in Fig. S4 (Supporting information).
Figure 1
Figure 1.
(a) Electrostatic potential (ESP) analyses of TMB (top) and TCNB (bottle). (b) Surfactant-assisted selective cocrystallization of TMB and TCNB toward T1C1 cocrystal or T2C1 cocrystal, together with photographs of TMB and TCNB powders. Optical microscopy images of (c) T1C1 cocrystals and (e) T2C1 cocrystals. SEM images of (d) T1C1 cocrystals and (f) T2C1 cocrystals.
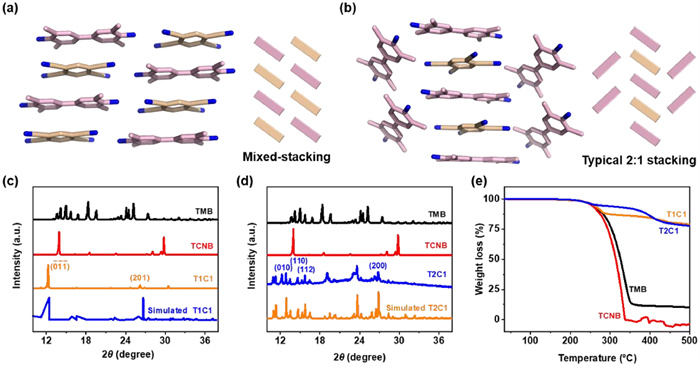
Single-crystal X-ray diffraction (XRD) analysis of T1C1, in which the molar ratio between TMB and TCNB was 1:1, revealing that it belonged to the triclinic P-1 space group in Table S1 (Supporting information). As shown in Fig. 2, TMB and TCNB packed alternately along their face-to-face stacking direction (-D-A-D-A-) based on CT, π···π and hydrogen bonding interactions. The average plane-to-plane distance between TMB and TCNB was 3.30 Å, and the TMB and TCNB molecules were almost parallel with a dihedral angle of 3.13° (Figs. S5a and b in Supporting information), suggesting the presence of π···π interactions. Moreover, three kinds of hydrogen bonds including N—H···N (2.32 Å) and C—H···N (2.70, 2.74 Å) between TCNB and adjacent molecules facilitated the crystal growth along the [100] direction (Fig. S5c in Supporting information). In contrast, T2C1, with a 2:1 TMB-TCNB stoichiometry, crystallized in a triclinic P-1 space group (Table S1). With the change in D–A ratio, the extra donor formed another column besides the mixed-stacking mode (-D-A-D-A-D-A-D-A and -D-D-D-D-, Fig. 2b). The average intermolecular distance between TMB and TCNB was 3.34 Å in the mixed-stacking column with a dihedral angel of 3.93° (Figs. S6a and b in Supporting information), indicative of the existence of CT and π···π interactions. Besides the C—H···N (2.76 Å) hydrogen bond between adjacent TCNB molecules, there were two kinds of N—H···N (2.70 Å) and C—H···N (2.78 Å) between TCNB and TMB in the individual donor column in T2C1 (Fig. S6c in Supporting information). In addition, C—H···π (3.26 Å) interaction between adjacent TMB molecules in the TMB column also promoted this typical 2:1 stacking pattern.
Figure 2
Figure 2.
Crystalline packing structures of (a) T1C1 cocrystal and (b) T2C1 cocrystal and schematics of their stacking patterns. PXRD patterns of experimental and simulated (c) T1C1 cocrystal and (d) T2C1 cocrystal. (e) Thermogravimetric curves of TMB, TCNB, T1C1 and T2C1 cocrystals.
The Hirshfeld surfaces and two-dimensional (2D) fingerprint were utilized to further analyze diverse intermolecular interactions in stoichiomorphic cocrystals (Figs. S7 and S8 in Supporting information) [46]. Diverse areas on the surfaces represented that the intermolecular contact distances were shorter (red), equal (white) to and longer (blue) than their van der Waals distances, respectively. In the 2D fingerprint plots, the proportion of intermolecular C···C interactions were 17.9% and 14.4% of the total Hirshfeld surfaces in T1C1 and T2C1, respectively (Figs. S7d and S8d), which further confirmed the existence of intermolecular π···π stacking. Importantly, the intermolecular N···H bonding in all forms was a main factor in the crystal packing, which made up 50.2% to 51.8% of the Hirshfeld surface in T1C1 and T2C1 cocrystals (Figs. S7e and S8e), respectively. These contacts were mainly due to the interaction of N atoms with protons from neighboring molecules.
Powder XRD measurements displayed different diffraction peak patterns for stoichiomorphic cocrystals (Figs. 2c and d). T1C1 and T2C1 cocrystals had several new sharp peaks compared to the pattern of TMB and TCNB crystals, which were highly consistent with the simulated pattern due to the high phase purities of two cocrystals. Moreover, thermogravimetric (TG) analysis in Fig. 2e indicated that both stoichiometric cocrystals had higher decomposition temperatures than TMB and TCNB crystals, suggesting cocrystallization was in favor of improving the thermal stability of original components.
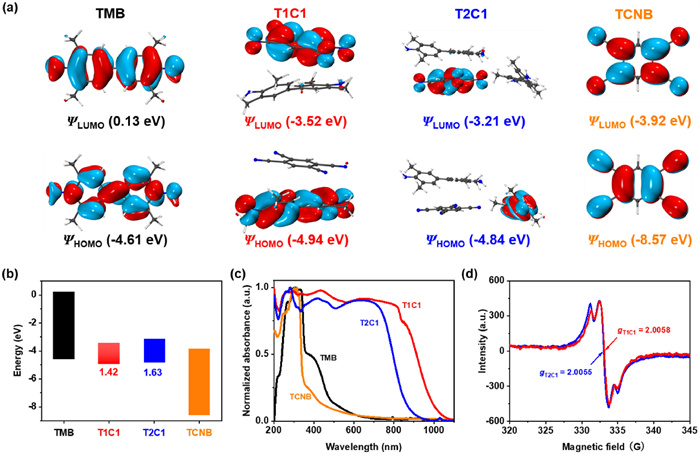
The frontier molecular orbital diagrams (Fig. 3a) showed that the HOMOs of T1C1 and T2C1 were concentrated on the electron-donating TMB molecule with energies of −4.94 and −4.84 eV, respectively. These values were close to that of TMB (−4.61 eV). The LUMOs of T1C1 and T2C1 were localized on the electron-accepting TCNB molecule with energies of −3.52 and −3.21 eV, respectively, resembling that of TCNB (−3.92 eV). These observations demonstrated that the HOMOs of stoichiometric cocrystals were principally derived from the HOMO of the donor TMB, while the LUMOs of cocrystals were close to that of the acceptor TCNB. However, different stoichiometric stacking could lead to subtle differences in the energy levels of cocrystals. Therefore, the calculated HOMO-LUMO energy gaps of T1C1 and T2C1 cocrystals were 1.42 and 1.63 eV, respectively (Fig. 3b). Notably, the calculated CT degree of T1C1 and T2C1 cocrystals were 0.075e and 0.071e (Fig. S9 in Supporting information), which was due to the electronic coupling between the HOMO of TMB and the LUMO of TCNB [9].
Figure 3
Figure 3.
The calculated (a) molecular orbital diagrams and (b) energy diagrams of TMB, TCNB, T1C1 and T2C1 cocrystals. (c) Solid-state absorption spectra of TMB, TCNB, T1C1 and T2C1 cocrystals. (d) ESR spectra of T1C1 and T2C1 cocrystals.
On the other hand, solid-state UV–vis absorption spectra were also recorded to elucidate the CT nature in stoichiometric cocrystals. As shown in Fig. 3c, TMB crystals exhibited strong absorption in the region of 200−600 nm, and TCNB crystals absorbed primarily at 200−500 nm. Distinctive absorption spectra were observed after selective cocrystallization. Compared with the individual TMB and TCNB crystals, T2C1 showed a significantly red-shifted broad absorption ranging from 200 nm to 957 nm, owing to the effective intermolecular CT interaction in cocrystals. By contrast, the absorption band of T1C1 extended up to 1100 nm, which was 143 nm red-shifted compared to that of T2C1. This difference might arise from a more efficient D–A overlap in the solid-state superstructure of T1C1 than in that of T2C1. As shown in Fig. S10 (Supporting information), the deep color of the two stoichiometric cocrystals powders also further indicated strong CT interactions between TMB and TCNB.
Electron spin resonance (ESR) tests showed two strong signals of T1C1 and T2C1 with g factors of 2.0058 and 2.0055, respectively, indicating the existence of unpaired electron, which was consistent with the CT interaction in ground state (Fig. 3d). Both Fourier transform infrared (FTIR) spectra and Raman spectra of stoichiometric cocrystals were the sum of main peaks for TMB and TCNB crystals (Figs. S11 and S12 in Supporting information), but subtle shift changes might be caused by distinct chemical environments or electron cloud densities. For instance, the characteristic IR peak of C≡N stretching vibrations at 2249 cm−1 in TCNB were red-shifted to 2244 and 2246 cm−1 in T1C1 and T2C1 cocrystals, respectively. Moreover, the characteristic Raman peak of C≡N stretching vibrations at 2245 cm−1 in TCNB were red-shifted by 19 and 7 cm−1 in T1C1 and T2C1 cocrystals, respectively. These results not only powerfully verified the significant CT interaction between TMB and TCNB, but also suggested the T1C1 cocrystals owned a larger DCT than T2C1 cocrystals, in agreement with the theoretical calculation results.
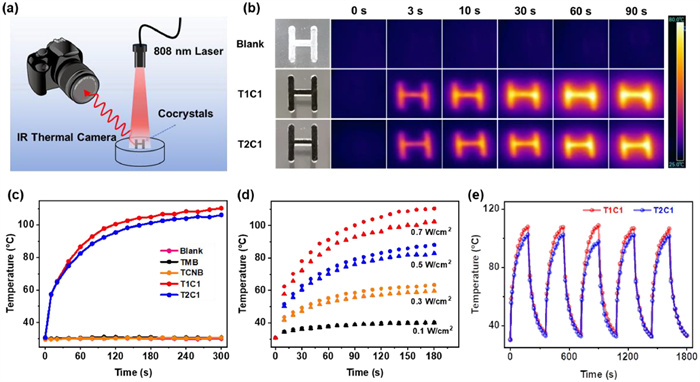
Benefiting from the sufficient NIR light absorption and outstanding thermal stability, both stoichiometric cocrystals demonstrated remarkable photothermal conversion performance. Under irradiation with an 808 nm laser, the thermal images and rapid temperature changes of the cocrystal powders were recorded by an IR thermal camera. As shown in Fig. 4a, the photothermal imaging capability of cocrystals was evaluated by irradiating the capital letter H, filled with cocrystals. At the beginning of 808 nm laser irradiation (0.5 W/cm2), both cocrystals pattern could be quickly captured, indicative of the high-quality imaging effects. With increasing irradiation time, the thermal image of letter H containing cocrystals became more and more bright (Fig. 4b), showing great potential to be excellent NIR photothermal imaging reagents.
Figure 4
Figure 4.
(a) Diagram of the photothermal imaging measurement. (b) Thermal images of two cocrystal-patterned letter H under 808 nm laser irradiation (0.5 W/cm2). (c) Photothermal heating curves of TMB, TCNB, T1C1 and T2C1 cocrystals on quartz glass under 808 nm laser irradiation (0.7 W/cm2). (d) Temperature changes of T1C1 (circular dots) and T2C1 (triangular dots) at different NIR laser densities. (e) Photothermal cycling curve of T1C1 and T2C1 cocrystals under 808 nm laser irradiation (0.7 W/cm2).
Then, two cocrystal powders and original components were put on a quartz glass substrate and irradiated by an 808 nm laser to quantitatively compare their NIR photothermal conversion capabilities. As shown in Fig. 4c, the surface temperatures of T1C1 and T2C1 cocrystals could rise up to 110.3 ℃ and 106.2 ℃ when the power density was 0.7 W/cm2, respectively, while their individual components had almost no temperature response. It was worth noting that although the absorption intensity of T1C1 at 808 nm (1.258) was nearly twice the absorption value of 0.622 at 808 nm for T2C1 (Fig. S13 in Supporting information), the photothermal heating capacity of T2C1 was not much weaker than that of T1C1. This contrast phenomenon might be closely related to the difference in the actual photothermal conversion efficiency (PCE) of two cocrystals.
From the cooling curves, the PCE of T1C1 cocrystal was determined to reach 60.3% ± 0.6% at 0.5 W/cm2 density power (Fig. S14 and Table S2 in Supporting information). In contrast, T2C1 cocrystal reached 75.5% ± 2.6% PCE under the same irradiation conditions (Fig. S15 and Table S2 in Supporting information), which was comparable than other reported similar photothermal materials (Table S3 in Supporting information). Both stoichiometric cocrystals were non-emission due to the strong CT interactions (Fig. S16 in Supporting information), which was also a key reason for the high values of PCE. The surface temperatures of cocrystals sharply increased under laser irradiation with different power densities, and both temperature changes were positively correlated with the laser power (Fig. 4d and Fig. S17 in Supporting information). Five on/off cycles of 808 nm laser irradiation confirmed the superior photo-stabilities of T1C1 and T2C1 cocrystals (Fig. 4e). In addition, the consistent powder XRD patterns of the two cocrystals before and after 2 h of laser irradiation also indicated their excellent photothermal stability (Fig. S18 in Supporting information).
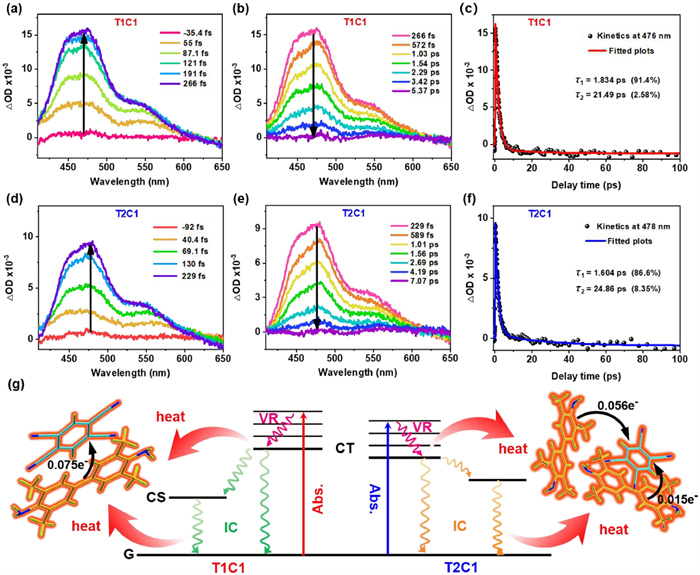
Ulteriorly, femtosecond transient absorption (fs-TA) spectra were applied to elucidate the excited state dynamics and photothermal conversion properties of stoichiomorphic cocrystals [11]. Two excited state absorption (ESA) signals emerged instantly after being photoexcited via an 800 nm laser in both cocrystals. Specifically, in T1C1 cocrystal, the absorption intensity reached a maximum at 266 fs (Fig. 5a), which could be due to the excited CT state. Then, the ESA signals diminished rapidly in the time range from 266 fs to 5.37 ps, as given in Fig. 5b, which was ascribed to the immediate deactivation of the excited CT state via the nonradiative transitions of the internal conversion (IC) process from 1CTn* to 1CT1* or vibrational relaxation (VR). To investigate the kinetics of the excited state of the cocrystals, a two-exponential fitting was utilized. The corresponding decay lifetimes τ1 and τ2 were obtained by fitting the fs-TA result at 476 nm. In Fig. 5c, the first lifetime τ1 of 1.8 ps (amplitude = 91.4%) represented the superfast VR and IC transition from the excited state 1CTn* to the ground state (CT0). The second lifetime τ2 of 21.5 ps (amplitude = 2.6%) might jointly contributed by the structural relaxation of the hot CT0 state and ground state bleaching (GSB).
Figure 5
Figure 5.
Fs-TA spectra of the T1C1 cocrystal in the range of (a) −35.4 fs to 266 fs and (b) 266 fs to 5.37 ps. (c) Kinetic fitting results of the T1C1 cocrystal at 476 nm. Fs-TA spectra of the T2C1 cocrystal in the range of (d) −92 fs to 229 fs and (e) 229 fs to 7.07 ps. (f) Kinetic fitting results of the T2C1 cocrystal at 478 nm. (g) Jablonski diagram of stoichiomorphic cocrystals.
Similarly, the ESA intensity of T2C1 cocrystal reached a maximum at 229 fs (Figs. 5d and e), and then began to decay with two lifetimes. As shown in Fig. 5f, the first lifetime τ1 of the IC transition from the excited state 1CTn* to the ground state (CT0) of T2C1 was 1.6 ps, and the second lifetime τ2 represented the structural relaxation and GSB was 24.9 ps. Two major decay processes accounted for 86.6% and 8.4% of the total decay process of T2C1 cocrystal, respectively. All of these decay processes contributed to the thermal generation in this stoichiometric cocrystals system. Based on the above analyses of their excited state dynamics, the possible Jablonski diagram of the stoichiometric cocrystals was proposed in Fig. 5g to elaborate the photophysical processes of the photothermal conversion. When cocrystals were excited, the electronic transitions taken place from their ground CT state to the excited states. Then the excited electrons undergone nonradiative decay of IC, VR and charge dissociation and returned to the ground states, which were in favor of high-efficiency photothermal conversion [47]. Notably, the ultra-strong CT interactions played a dominant role in determining these rapid excited state relaxation dynamics.
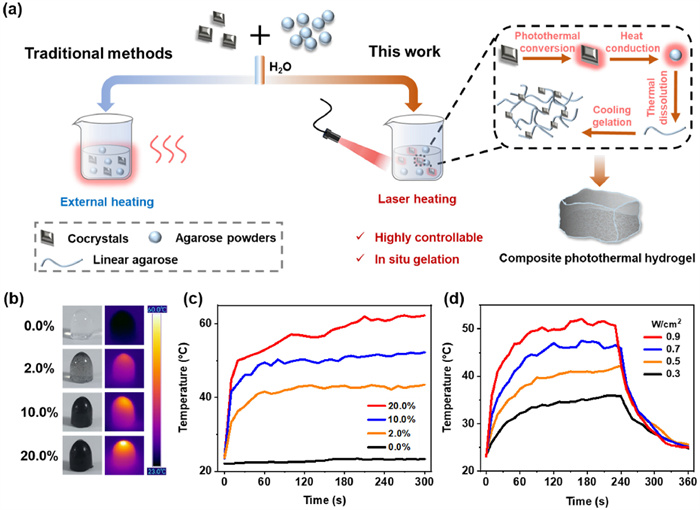
Having demonstrated the superb photothermal properties and photothermal mechanisms of stoichiomorphic cocrystals, we sought to exploit one of the two cocrystals T1C1, which can promisingly be selected in generally converting light into heat for in-situ gelation. Due to the excellent biocompatibility, agarose hydrogels with polyhydroxyl groups are often doped with some photothermal reagent molecules for functional medical dressings and photothermal therapy [48]. However, agarose usually requires a higher temperature to dissolve in water by unfolding its linear chains, and then which forming a hydrogel through strong hydrogen bonding interactions between the hydroxyl groups on the chains. In traditional methods, photothermal agarose hydrogels are often prepared by external heating ways, such as microwave heating and water-bath heating, which makes it a challenge to achieve in-situ non-invasive gelation in the future biomedical field.
In this work, we proposed a novel in-situ gelation method to prepare photothermal agarose hydrogels by utilizing the excellent photothermal properties and stability of CT cocrystals (Fig. 6a). Firstly, the T1C1 cocrystal powders was mixed with the agarose powders and added to the water, gently stirred to disperse it evenly, and then exposed to an 808 nm laser. Under the NIR light irradiation, T1C1 cocrystals in the mixed solution absorbed a large amount of light and efficiently converted it into thermal energy. The heat generated by the cocrystals was then transferred to the surrounding agarose molecules through the conduction of heat, which facilitated the unfolding of the agarose linear chain. Finally, a composite photothermal agarose hydrogel was obtained after cooling. T1C1 cocrystal powders of different masses were doped to prepare three composite agarose hydrogels with 2.0 wt%, 10.0 wt% and 20.0 wt% cocrystal concentrations, as shown in Fig. 6b. In the process of photothermal gelation, the higher the concentration of doped cocrystals was conducive to increasing the temperature of the whole system, and the required irradiation time was thus shorter. Meanwhile, a blank agarose hydrogel without cocrystal was prepared by microwave heating as a control. In order to evaluate the photothermal property of composite hydrogels, an 808 nm laser was selected to irradiate these hydrogels with 1 W/cm2. There was almost no obvious temperature change in the blank agarose hydrogel, and the surface temperature of the agarose hydrogel increased with the increase of doped cocrystal proportion (Figs. 6b and c). Noticeably, for 2.0 wt% of the composite agarose hydrogel, most of the cocrystal sank in the bottom owing to the long irradiation time. Moreover, the heating capacity of composite agarose hydrogels was also closely related to the power of the irradiated laser. Taking 20.0 wt% composite agarose hydrogel as an example, the surface temperature was positively correlated with the laser power density (Fig. 6d). These results fully demonstrated that T1C1 cocrystal hold promising applications as infrared heater for remotely controlled in-situ gelation.
Figure 6
Figure 6.
(a) The schematic diagram of preparation of photothermal agarose composite hydrogel by in-situ photothermal gelation using T1C1 cocrystals. (b) Brightfield photographs and corresponding thermal images and (c) heating curves of four agarose composite hydrogels with different cocrystal concentrations under 808 nm laser irradiation. (d) Temperature changes of 20.0 wt% agarose composite hydrogel at different NIR laser densities.
In summary, we construct a novel photothermal cocrystal stoichiomorphism system by a subtle balance between DCT and molecular stacking. Two stoichiometric CT cocrystals have been selectively obtained by controlling the donor-acceptor stoichiometries via a green and convenient surfactant-assisted method. By altering the donor-acceptor ratio from 1:1 (T1C1) to 2:1 (T2C1), the intermolecular CT interactions become weaker and the energy bandgap becomes wider in T2C1. T2C1 exhibits a high PCE of 75.5% ± 2.6%, while T1C1 owns a 60.3% ± 0.6% PCE under 808 nm laser irradiation. Both high PCEs are attributed to the nonradiative transition process, including internal conversion and charge dissociation processes as demonstrated by fs-TA measurements. Finally, by using T1C1 as an infrared heater, we develop an in-situ photothermal gelation strategy for preparing a composite agarose hydrogel. This work provides an example of how to gain better insights, through experiments supported by theory, into the relationship between the stoichiometric stacking of materials and their photothermal properties.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared toinfluence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 22001006, 22375002, 22273057, 22225401), the Anhui Provincial Natural Science Foundation (No. 2308085Y10) and the Open Project of Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Anhui University, Ministry of Education. The National Key Research and Development Program of China (Nos. 2022YFC2403500, 2020YFA0210800). The Universities Joint Laboratory of Guangdong, Hong Kong and Macao (No. 130/07422011), the Natural Science Foundation of Guangdong Province (No. 2022A1515011661), the China Postdoctoral Science Foundation (Nos. 2023M730760, 2023M740010).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109808.
[1]
L. Sun, Y. Wang, F. Yang, et al., Adv. Mater. 31 (2019) 1902328. doi: 10.1002/adma.201902328
[2]
L. Sun, W. Zhu, X. Zhang, et al., J. Am. Chem. Soc. 143 (2021) 19243–19256. doi: 10.1021/jacs.1c07678
Figure 1
(a) Electrostatic potential (ESP) analyses of TMB (top) and TCNB (bottle). (b) Surfactant-assisted selective cocrystallization of TMB and TCNB toward T1C1 cocrystal or T2C1 cocrystal, together with photographs of TMB and TCNB powders. Optical microscopy images of (c) T1C1 cocrystals and (e) T2C1 cocrystals. SEM images of (d) T1C1 cocrystals and (f) T2C1 cocrystals.
Figure 2
Crystalline packing structures of (a) T1C1 cocrystal and (b) T2C1 cocrystal and schematics of their stacking patterns. PXRD patterns of experimental and simulated (c) T1C1 cocrystal and (d) T2C1 cocrystal. (e) Thermogravimetric curves of TMB, TCNB, T1C1 and T2C1 cocrystals.
Figure 3
The calculated (a) molecular orbital diagrams and (b) energy diagrams of TMB, TCNB, T1C1 and T2C1 cocrystals. (c) Solid-state absorption spectra of TMB, TCNB, T1C1 and T2C1 cocrystals. (d) ESR spectra of T1C1 and T2C1 cocrystals.
Figure 4
(a) Diagram of the photothermal imaging measurement. (b) Thermal images of two cocrystal-patterned letter H under 808 nm laser irradiation (0.5 W/cm2). (c) Photothermal heating curves of TMB, TCNB, T1C1 and T2C1 cocrystals on quartz glass under 808 nm laser irradiation (0.7 W/cm2). (d) Temperature changes of T1C1 (circular dots) and T2C1 (triangular dots) at different NIR laser densities. (e) Photothermal cycling curve of T1C1 and T2C1 cocrystals under 808 nm laser irradiation (0.7 W/cm2).
Figure 5
Fs-TA spectra of the T1C1 cocrystal in the range of (a) −35.4 fs to 266 fs and (b) 266 fs to 5.37 ps. (c) Kinetic fitting results of the T1C1 cocrystal at 476 nm. Fs-TA spectra of the T2C1 cocrystal in the range of (d) −92 fs to 229 fs and (e) 229 fs to 7.07 ps. (f) Kinetic fitting results of the T2C1 cocrystal at 478 nm. (g) Jablonski diagram of stoichiomorphic cocrystals.
Figure 6
(a) The schematic diagram of preparation of photothermal agarose composite hydrogel by in-situ photothermal gelation using T1C1 cocrystals. (b) Brightfield photographs and corresponding thermal images and (c) heating curves of four agarose composite hydrogels with different cocrystal concentrations under 808 nm laser irradiation. (d) Temperature changes of 20.0 wt% agarose composite hydrogel at different NIR laser densities.

 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载:
