College of Chemistry and Chemical Engineering, and Key Laboratory of Photonic and Electronic Bandgap Materials, Ministry of Education, Harbin Normal University, Harbin 150025, China
b.
Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, China
c.
Institute for Advanced Study, Chengdu University, Chengdu 610106, China
d.
College of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Ji'nan 250014, China
Received Date:
15 January 2024 Accepted Date:
20 March 2024 Revised Date:
20 March 2024 Available Online:
15 August 2024
Abstract:
Electrocatalytic synthesis of urea through CN bond formation, converting carbon dioxide (CO2) and nitrate (NO3–), presents a promising, less energy-intensive alternative to industrial urea production process. In this communication, we report the application of Mo2C nanosheets-decorated carbon sheets (Mo2C/C) as a highly efficient electrocatalyst for facilitating CN coupling in ambient urea electrosynthesis. In CO2-saturated 0.2 mol/L Na2SO4 solution containing 0.05 mol/L NO3–, the Mo2C/C catalyst achieves an impressive urea yield of 579.13 µg h–1 mg–1 with high Faradaic efficiency of 44.80% at –0.5 V versus the reversible hydrogen electrode. Further theoretical calculations reveal that the multiple Mo active sites enhance the formation of *CO and *NH2 intermediates and facilitate their CN coupling. This research propels the use of Mo2C-based electrodes in electrocatalysis and accentuates the capabilities of binary metal-based catalysts in CN coupling reactions.
The extensive utilization of urea (nitrogen fertilizers, medical products, pesticides) has significantly propelled the growth of the urea industry [1,2]. Presently, industrial synthesis of urea involves two key thermochemical stages: (i) The conversion of nitrogen (N2) and hydrogen (H2) to ammonia (NH3) at temperatures ranging from 450 ℃ to 600 ℃ and pressures between 100 bar and 250 bar; and (ii) the subsequent reaction of NH3 with carbon dioxide (CO2) to form urea at temperatures of 180–230 ℃ and pressures of 150–250 bar [3,4]. Despite being a prevalent technique for decades, the future of this technology faces challenges due to its stringent reaction conditions and the substantial requirement for liquid ammonia NH3 [5]. In contrast, the emerging method of urea electrosynthesis, which involves coupling CO2 with a nitrogen source under ambient conditions, offers an innovative pathway [6–11]. This pathway not only significantly reduces energy consumption but also plays a crucial role in addressing the energy crisis and mitigating global warming caused by excessive CO2 emissions.
In the realm of urea electrosynthesis, the use of N2 as a primary source for urea production has recently captured considerable attention [7,12-16]. The limited solubility of N2 in water at ambient conditions and the high energy required to break its N≡N bond (941 kJ/mol), nevertheless, typically results in suboptimal performance [17–22]. Conversely, nitrate (NO3–), being water-soluble, is a more favorable nitrogen source for its relatively lower dissociation energy of the N=O bond (204 kJ/mol), which facilitates enhanced reaction kinetics for urea electrosynthesis [6,10,23-28]. In addition, the electrochemical reduction of NO3– is considered as an environmentally friendly method, particularly for its ability to remove the abundant yet harmful NO3– in groundwater [29,30]. At the mechanistic level, introducing NO3– as a nitrogen source involves the electrocatalytic reduction of CO2 to *CO and NO3– to *NH2 on the catalyst's active surface, which followed by a non-electrocatalytic coupling that forms C—N bond for urea production [31]. Nonetheless, significant advancements in this field are currently hindered by the challenge of identifying efficient catalysts that can effectively facilitate the C—N bonding process.
Current research primarily center on indium (In)- [25,32-34], ruthenium (Ru)- [35,36], and copper (Cu)-based catalysts [9,26,27,37-40] for the electrosynthesis of urea. However, their on-scale applications are hindered by the high costs of In and Ru, and the susceptibility of Cu to corrosion during NO3– reduction [41–44]. In natural processes, the reduction of NO3– is accomplished via enzymatic cascades involving NO3– reductase with a molybdenum (Mo) cofactor, and Mo sites are also known to catalyze CO2 reduction [45–47]. Recent study has also demonstrated the effectiveness of molybdenum oxide nanoclusters in urea electrolysis through the conversion of CO2 and NO3– [48]. In comparison, 2D molybdenum carbide (Mo2C) is emerging as a highly appealing material due to its higher conductivity and fully exposed Mo active sites [49–51]. Mo2C has been reported to show activity in both NO3– and CO2 reduction reaction [51–53]. It is thus anticipated that Mo2C can significantly enhance the C—N coupling process required for efficient urea electrolysis.
Herein, we propose for the efficient urea electrosynthesis via the co-reduction of NO3– and CO2 on Mo2C nanosheets-decorated carbon sheets (Mo2C/C). The as-prepared Mo2C/C affords a high urea yield rate of 579.13 µg h–1 mg–1 and a high Faradaic efficiency (FE) of 44.80% at –0.5 V versus reversible hydrogen electrode (RHE). Furthermore, it demonstrates outstanding electrochemical stability, consistently performing well over a duration of 12 h and through 8 successive cycles. Additionally, we employed density functional theory (DFT) calculations to delve deeper into the underlying mechanisms of the urea electrosynthesis process.
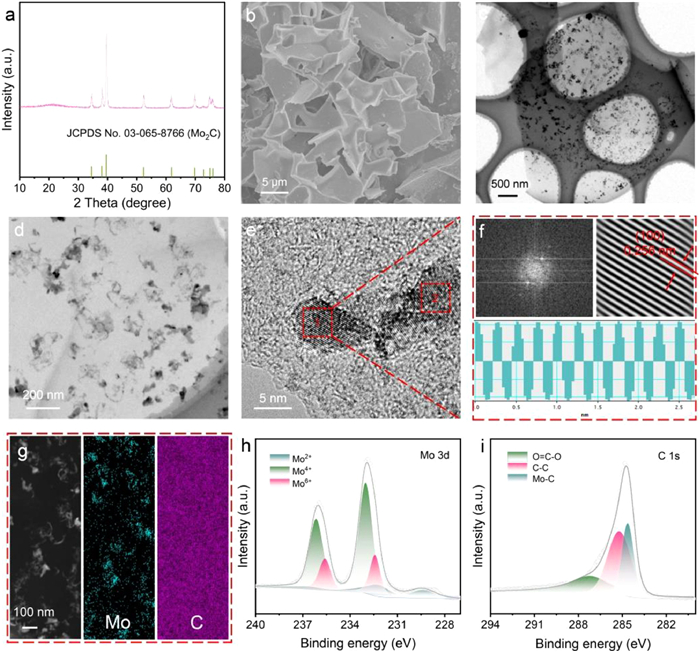
The X-ray diffraction (XRD) pattern of Mo2C/C, as shown in Fig. 1a, exhibits distinct diffraction peaks at approximately 34.53°, 38.05°, 35.59°, 52.31°, 61.87°, 69.77°, 72.83°, 74.99° and 75.99°. These peaks closely align with those of standard Mo2C (JCPDS No. 03–065–8766) [52,54]. Additionally, a broadened diffraction peak at around 21.6° is attributed to carbon [55], originating from the pyrolysis of glucose. Scanning electron microscopy (SEM) image (Fig. 1b) reveals interconnected, relatively smooth carbon sheets. Transmission electron microscopy (TEM) images (Figs. 1c and d) depict densely packed nanosheets on the surfaces of carbon sheet. In Fig. 1e, the high-resolution TEM (HRTEM) image of Mo2C/C is presented. The selected area electron diffraction (SAED) patterns and line scan of the HRTEM image, indicated by red boxes 1 and 2 in Fig. 1f and Fig. S1 (Supporting information) from Fig. 1e, confirm the good crystallinity of Mo2C, featuring a lattice spacing of 0.256 nm, which corresponds to the (100) plane of Mo2C. Elemental distribution analysis using high-angle annular. High-angle annular dark field scanning TEM (HAADF-STEM) and corresponding elemental imaging in Fig. 1g reveal a homogeneous distribution of Mo and C atoms across the structure, indicating that the sheet-like nanoparticles are composed of Mo2C uniformly distributed within the carbon sheet. Subsequent X-ray photoelectron spectroscopy (XPS) analysis of Mo2C/C provide insights into the surface electronic states. The Mo 3d spectra (Fig. 1h) show peaks at 228.9 and 231.98 eV, corresponding to the binding energies of Mo2+, indicative of Mo2C. Other doublets observed at 229.5/232.7 and 232.6/235.8 eV can be attributed to oxidized molybdenum phases Mo4+ and Mo6+, respectively, likely due to surface oxidation [56,57]. The C 1s spectra (Fig. 1i) consist of three subpeaks at 284.8, 285.6, and 288.8 eV, which are assigned to Mo-C, C—C, and O=C—O bonds, respectively [55,58,59].
Figure 1
Figure 1.
(a) XRD pattern of Mo2C/C. (b) SEM image of Mo2C/C. (c) Low and (d) high magnification TEM images of Mo2C/C. (e) High-resolution TEM image of Mo2C/C. (f) SAED pattern and line scan and of the HRTEM image indicated by the red box 1 in (e). (g) HAADF-STEM and its corresponding mapping images of Mo2C/C. XPS spectra of Mo2C/C in the (h) Mo 3d, (i) C 1s regions.
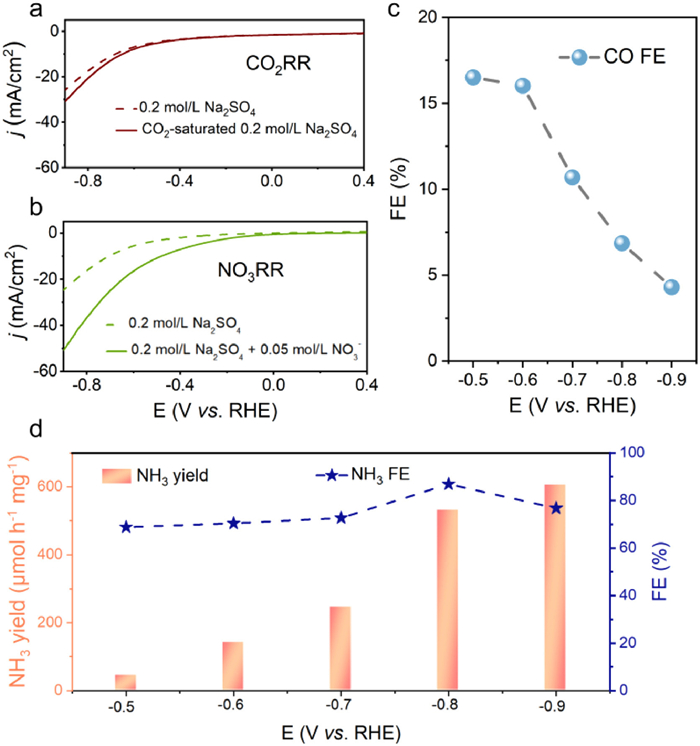
The intrinsic CO2 reduction reaction (CO2RR) and NO3– reduction reaction (NO3RR) performance of Mo2C/C were initially evaluated in an H-type electrochemical cell. The linear sweep voltammetry (LSV) curve in CO2-saturated 0.2 mol/L Na2SO4 (Fig. 2a) exhibited a higher current density compared to that in pure 0.2 mol/L Na2SO4, indicating significant CO2RR activity. Similarly, an obviously increase in current density was observed in 0.2 mol/L Na2SO4 containing 0.05 mol/L NO3–, demonstrating effective NO3RR (Fig. 2b). Based on these LSV curves, the potential window from –0.5 V to –0.9 V was selected for subsequent CO2RR and NO3RR experiments, respectively. Mo2C/C displayed commendable CO2RR capacity for CO generation (Fig. 2c, Figs. S2 and S3 in Supporting information). The production of NH3 was quantified using the indophenol blue method, with the corresponding calibration curve presented in Fig. S4 (Supporting information). Mo2C/C also showed notable NO3RR efficiency in generating NH3 (Fig. 2d, Figs. S5 and S6 in Supporting information). In essence, the promising electrocatalytic performance of Mo2C/C in both CO2RR and NO3RR underscores its potential for efficient electrocatalytic urea synthesis.
Figure 2
Figure 2.
(a) LSV curves of Mo2C/C in 0.2 mol/L Na2SO4 with and without saturated CO2, respectively. (b) LSV curves of Mo2C/C in 0.2 mol/L Na2SO4 with and without 0.05 mol/L NO3−, respectively. (c) FEs of CO in CO2RR process at various potentials on Mo2C/C. (d) NH3 yields and FEs on Mo2C/C for NO3RR in 0.2 mol/L Na2SO4 + 0.05 mol/L NO3− with various applied potentials.
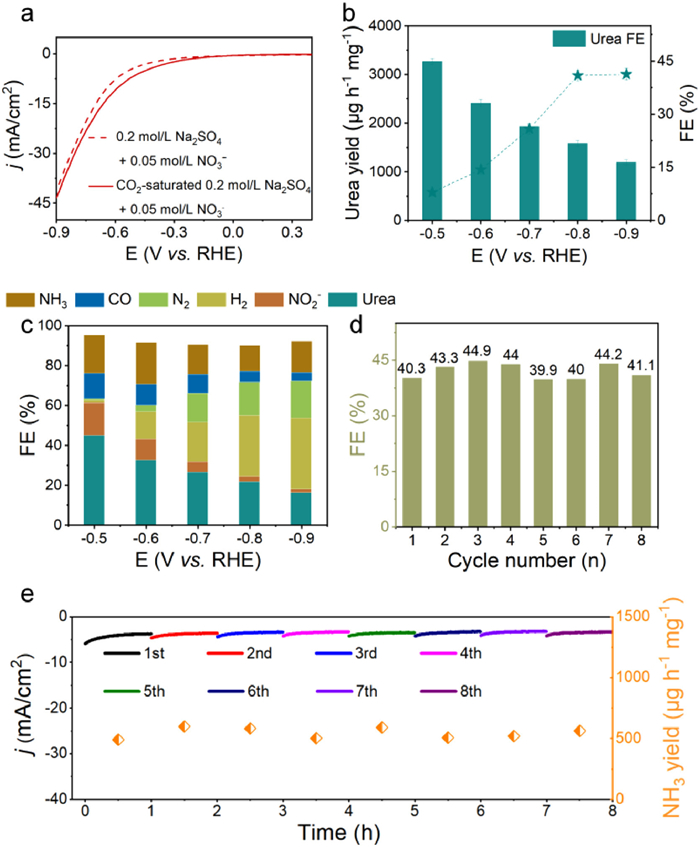
In the process of urea electrosynthesis, CO2 was continuously bubbled into a solution of 0.2 mol/L Na2SO4 containing 0.05 mol/L NO3–. Initially, the potential performance was assessed using LSV curves (Fig. 3a). The presence of CO2 elicited a current response, suggesting the potential occurrence of electrocatalytic C–N bonding crucial for urea synthesis. Subsequent chronoamperometry experiments were conducted to further confirm urea production (Fig. S7 in Supporting information). Urea detection was performed using the urease decomposition method, with corresponding UV–vis absorption spectra shown in Fig. S8 (Supporting information). And calculated urea yields and FEs at various potentials are presented in Fig. 3b. The UV–vis spectrum indicates a prominent absorbance at 655 nm at –0.9 V, corresponding to a urea yield of 3004.70 µg h–1 mg–1. A high FE of 44.80% was recorded at –0.5 V, surpassing previously most of the reported urea electrosynthesis catalysts (Table S1 in Supporting information). Comprehensive evaluation of urea electrosynthesis also involved quantitative detection of byproducts. Gas products (N2, H2, and CO) were monitored using online gas chromatography. The concentration of produced NO2– was determined using the Griess method (Figs. S9 and S10 in Supporting information). As depicted in Fig. 3c, combined with Fig. 3b, at potentials below –0.5 V, the excess release of NH3 and H2 from intensified hydrogen evolution blocks the adsorption sites of CO2 and NO3–. This impacts the urea electrosynthesis performance, leading to a diversified distribution of products, with NO2–, N2, and CO also being detected. Enhanced hydrogen evolution significantly hinders the formation of C–N bonds by coupling *CO with *NH2. The recyclability and stability of the electrocatalyst are crucial for industrial applications. Therefore, an 8-cycle electrosynthesis of urea was conducted, with results shown in Fig. S11 (Supporting information), Figs. 3d and e. The consistent current response, urea FEs, and yields indicate the durable stability of Mo2C/C. Furthermore, a 12-h long-term electrolysis test at –0.5 V in CO2-saturated 0.2 mol/L Na2SO4 containing 0.05 mol/L NO3– showed a steady current density, underlining the catalyst's potential for practical applications. The stability of Mo2C/C during NO3RR is further confirmed by a steady current density over 12 h of prolonged electrolysis (Fig. S12 in Supporting information). Additionally, post-electrolysis analysis shows nearly identical LSV curves (Fig. S13 in Supporting information), well-preserved morphology (Fig. S14 in Supporting information), and consistent XRD pattern (Fig. S15 in Supporting information), collectively demonstrating the high durability and stability of Mo2C/C in this process.
Figure 3
Figure 3.
(a) LSV curves of Mo2C/C in Ar- and CO2-saturated 0.2 mol/L Na2SO4 + 0.05 mol/L NO3−. (b) Urea yields and FEs of Mo2C/C at different potentials. (c) FEs of NH3, CO, N2, H2, NO2− and urea on Mo2C/C at different potentials during the electrosynthesis of urea. (d) Urea FEs at –0.5 V during consecutive recycling test. (e) Urea yields and chronoamperometry curves during consecutive recycling test.
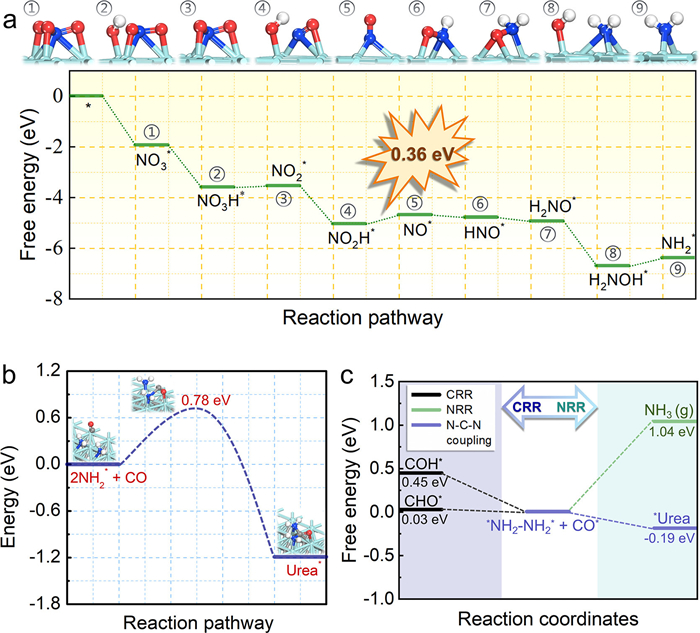
To further reveal the underlying mechanism of urea electrosynthesis on 2D Mo2C nanosheets from NO3– and CO2, DFT computations were carried out. The atomic structure of Mo2C is depicted in Fig. S16 (Supporting information). These calculations explored the entire free energy landscape of urea formation. The NO3– reduction follows a multi-step pathway: NO3– → NO3* → NO2* + OH* → NO2* → NO* + OH* → NO* → HNO*→ H2NO* → NH2*+ OH* → NH2* [31]. Here, the formation of NO* represents the potential rate-limiting step, exhibiting a ΔG value of 0.36 eV (Fig. 4a). The presence of multiple Mo active sites significantly enhances the dissociation and hydrogenation of NO3*, NO2*, and H2NO* intermediates. The CO2 reduction process follows the pathway CO2 → COOH* → CO*, easily proceeding to *CO (Fig. S17 in Supporting information). Two mechanisms, Eley–Raideal and Langmuir-Hinshelwood, were then considered for the N—C-N coupling process. As depicted in Fig. 4b, the energy barrier for inserting CO between two independently adsorbed NH2* intermediates, leading to urea formation, is only 0.78 eV. This step is also thermodynamically favorable, with a ΔG value of –0.19 eV, indicating that CO and NH2* coupling is energetically advantageous both kinetically and thermodynamically. To assess Mo2C nanosheet's selectivity for urea production, potential competitive reactions, such as NO3– reduction to NH3, CO* hydrogenation, and the hydrogen evolution reaction (HER), were examined. The results revealed higher ΔG values for NH2* reduction to NH3 and CO* hydrogenation (1.04 eV and 0.03 eV, respectively) compared to N—C-N coupling (–0.19 eV) (Fig. 4c), suggesting the thermodynamic unfeasibility of these reactions. For HER, its selectivity was evaluated by comparing the free energy change of H* adsorption with NO3– adsorption on Mo active sites. As shown in Fig. S18 (Supporting information), ΔGH* is –0.70 eV, more positive than ΔGNO, thus indicating a preference for NO3– reduction over HER. Consequently, the side reactions of NO3– reduction, CO* hydrogenation, and HER are substantially less favored compared to urea production, affirming the high selectivity of Mo2C towards urea formation. Ab initio molecular dynamic (AIMD) simulations confirm that at 500 K, Mo2C maintains its structure during 10 ps of annealing, with minimal fluctuations in total energy and temperature (Fig. S19 in Supporting information), further demonstrating Mo2C's robust stability in urea formation.
Figure 4
Figure 4.
(a) Free energy diagram and optimized geometrical structures of the involved reaction intermediates for NO3– reduction on Mo2C. (b) The minimum energy pathway from (NH2* + NH2*+ CO) to Urea* and the corresponding energy barrier. (c) The reaction energies in co-adsorbed *NH2–NH2* and CO further reaction.
In summary, we have successfully developed Mo2C/C as a high-active catalyst for urea synthesis through the electrocatalytic coupling of NO3– and CO2. The synthesized Mo2C/C demonstrates exceptional catalytic efficiency, achieving a high urea production rate of 579.13 µg h–1 mg–1 and a FE of 44.80% at −0.5 V. Additionally, it exhibits remarkable electrochemical, maintaining its performance throughout 12 h of continuous electrolysis. Theoretical insights into the mechanism indicate that the formation of *NH2 and the retention of *CO on the Mo2C's multiple active sites is crucial for facilitating C—N bond formation during urea electrosynthesis. This research not only showcases the potential of Mo2C/C as an electrocatalyst for the ambient conversion of NO3– and CO2 but also paves the way for future experimental and theoretical studies in developing 2D electrocatalysts with multiple active sites for sustainable production of organonitrogen compounds through C—N coupling.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figure 1
(a) XRD pattern of Mo2C/C. (b) SEM image of Mo2C/C. (c) Low and (d) high magnification TEM images of Mo2C/C. (e) High-resolution TEM image of Mo2C/C. (f) SAED pattern and line scan and of the HRTEM image indicated by the red box 1 in (e). (g) HAADF-STEM and its corresponding mapping images of Mo2C/C. XPS spectra of Mo2C/C in the (h) Mo 3d, (i) C 1s regions.
Figure 2
(a) LSV curves of Mo2C/C in 0.2 mol/L Na2SO4 with and without saturated CO2, respectively. (b) LSV curves of Mo2C/C in 0.2 mol/L Na2SO4 with and without 0.05 mol/L NO3−, respectively. (c) FEs of CO in CO2RR process at various potentials on Mo2C/C. (d) NH3 yields and FEs on Mo2C/C for NO3RR in 0.2 mol/L Na2SO4 + 0.05 mol/L NO3− with various applied potentials.
Figure 3
(a) LSV curves of Mo2C/C in Ar- and CO2-saturated 0.2 mol/L Na2SO4 + 0.05 mol/L NO3−. (b) Urea yields and FEs of Mo2C/C at different potentials. (c) FEs of NH3, CO, N2, H2, NO2− and urea on Mo2C/C at different potentials during the electrosynthesis of urea. (d) Urea FEs at –0.5 V during consecutive recycling test. (e) Urea yields and chronoamperometry curves during consecutive recycling test.
Figure 4
(a) Free energy diagram and optimized geometrical structures of the involved reaction intermediates for NO3– reduction on Mo2C. (b) The minimum energy pathway from (NH2* + NH2*+ CO) to Urea* and the corresponding energy barrier. (c) The reaction energies in co-adsorbed *NH2–NH2* and CO further reaction.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
