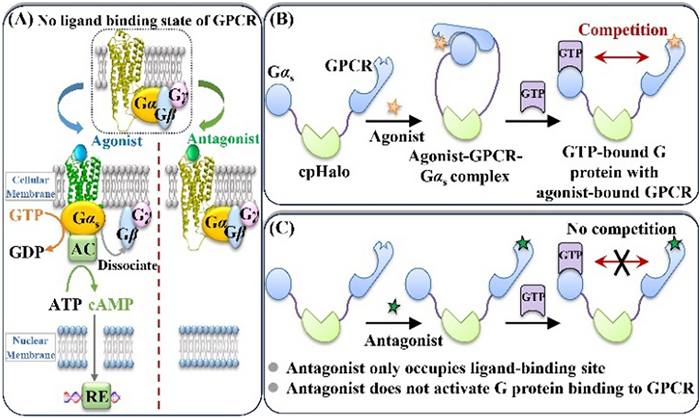
Figure 1.
Pharmacological process of GPCR upon binding to different ligands (A) and strategy for engineering G protein-GPCR miniature from cpHaloTag (B, C).
A chromatographic method for pursuing potential GPCR ligands with the capacity to characterize their intrinsic activities of regulating downstream signaling pathway
Ting Li , Xinxin Zheng , Lejing Qu , Yuanyuan Ou , Sai Qiao , Xue Zhao , Yajun Zhang , Xinfeng Zhao , Qian Li

G protein-coupled receptors (GPCRs) have historically been the dominant targets for drugs approved by the Food and Drug Administration (FDA), and continue to be attractive for ongoing drug discovery [1]. Despite successfully developing over 40% of current drugs, they exhibit a declining probability of producing a final drug from a new ligand in the last decades [2]. More than 90% of the lead compounds undertaking Phase Ⅰ trials have been withdrawn by the FDA [3]. This necessitates chemists and pharmacologists rethinking the techniques that generate a desirable lead compound, in particular, affinity-based assays with consideration of efficacy during the screening.
High throughput screening (HTS) in combination with the fluorescent-based methods allow the isolation of functional GPCRs for measuring the binding of ligands through a competition and displacement format [4,5]. Owing to the merits of conduction in plates, relatively low volumes, and optical readouts, these methods have provided alternatives for GPCR ligand screening [6]. Aside from the use of soluble forms, the availability of functional purified GPCR has opened a new avenue for pursuing GPCR ligands by the utilization of immobilized GPCRs [7] like surface plasmon resonance (SPR) [8] and high-performance receptor chromatography (RC) [9,10]. However, they may result in hits that are not potent enough for further in vivo or in vitro examination ascribed to: (1) Nonspecific bindings induced by stability and orientations of the immobilized receptor, especially in SPR methods as it compromises the throughput; (2) the lack of readout that can reflect the regulation of ligand-receptor binding on effector coupling or second messenger generation [11,12]; (3) unable to simultaneously evaluate intrinsic activities of the potential GPCR ligands during the screening. Previous work using RC to distinguish agonist and antagonist is mainly achieved by stabilizing the conformation of immobilized GPCR via specific aptamer or agonist/antagonist [13–15]. Resulting in the retention of only one type of ligands (agonist or antagonist only) on the RC column without intrinsic activity evaluation. Each method works through a unique aptamer or proper agonist/antagonist that cannot be applied universally. As such, a general strategy for simultaneously screening and evaluation the intrinsic activity of GPCR ligand would be highly valuable.
To this end, we have designed a protein miniature to develop the intrinsic activity-responsive method for GPCR ligand discovery. The pharmacological role of GPCRs is directly associated with the activation of heterotrimeric Gαβγ-proteins (Fig. 1A) [16]. (1) Once an agonist binds to GPCR, the conformation of the receptor will change and lead to the formation of agonist-GPCR-G protein complex, as well as the dissociation of Gα with Gβ-Gγ dimer. This process is associated with the downstream signaling pathway and pharmacological effects of the agonist. (2) Antagonist only occupies ligand-binding site on GPCR and the GPCR-G protein complex will not form. Actually, the dissociation of Gα with Gβ-Gγ dimer (i.e., the activation of Gα) relies on the binding of GTP with Gα, indicating that GTP is a key molecule in Gα activation. Upon these signaling proofs, we designed a miniature using circular permutation HaloTag (cpHalo) [17] as a scaffold to link GPCR and its downstream G protein. As shown in Fig. 1B, in the presence of agonist, the agonist-GPCR-Gα complex will form. Upon addition of GTP, it will bind to Gα competitively with the agonist-GPCR complex. The affinity and dissociation rate constant of GTP binding to Gα is associated with the intrinsic activity of the agonist. Therefore, agonists with different intrinsic activity could regulate the GTP affinity and kinetics to Gα. When an antagonist joined to GPCR binding (Fig. 1C), no GPCR-G protein complex forms and no competition occurs between GTP and antagonist-GPCR complex with G protein, thus the GTP affinity to Gα will not change.
We designed the receptor/G-protein complex taking β2-adrenoceptor (β2AR) as a probe, since it couples to Gαs while having a relatively clear crystal structure with good stability. We utilized cpHalo to fuse them as a whole protein (Fig. S1 in Supporting information) since (1) the linker is really flexible, which allows the two protein units to be environmentally sensitive for their partner bindings; (2) upon ligand binding, the receptor unit should be activated through conformation changes, while the Gαs unit has highly tunable photophysical and chemical properties associated with the activated receptor; (3) the self-labeling property of cpHalo enables direct immobilization of the fusion protein by specific reaction between the tag and haloalkane-modified supporter (Fig. 2A).
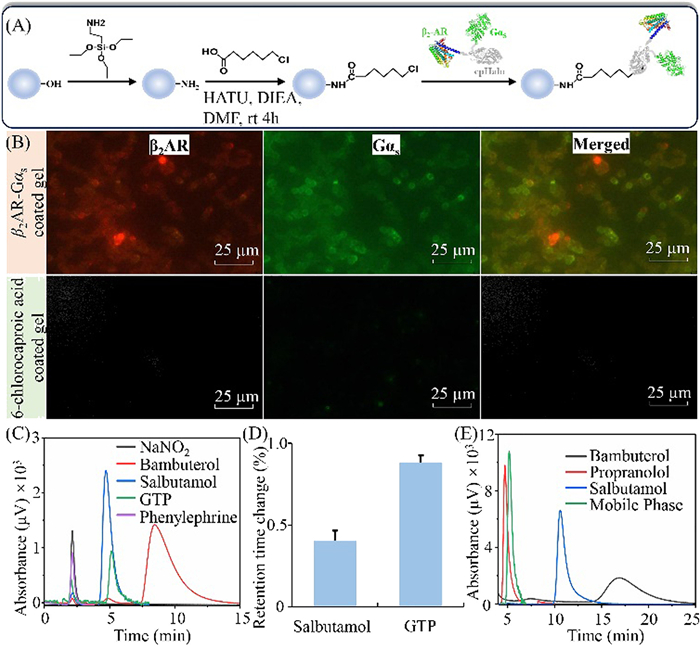
According to the assumption, we reconstituted and expressed Gαs-cpHalo-β2AR in E. coli systems and the details are in Fig. S2 (Supporting information). Even though the fusion protein mainly expressed as an inclusion body, it maintained binding specificity to the antibodies of the two functional units. As such, we dissolved the precipitation in phosphorate buffer (20 mmol/L, pH 7.2) to capture fusion Gαs-cpHalo-β2AR onto silica gel. This was realized by the bioorthogonal reaction between the cpHaloTag and haloalkanes modified on silica gels. To prove the reaction specificity, we determined the free 6-chlorocaproic acid in the mixture pro- and post-reaction with Gαs-cpHalo-β2AR. As shown in Fig. S3A (Supporting information), we found a continuous loss of the 6-chlorocaproic acid when the reaction time was shorter than 40 min. Afterward, it appeared to be constant even though the reacting time extended to 2.0 h. By this data, we reasoned Gαs-cpHalo-β2AR maintained the activity to specifically react with the enzyme's substrates. It is feasible to use it for immobilization of the fusion protein onto silica gels.
We introduced the chlorohexyl group on aminopropyl silica gel for immobilization of Gαs-cpHalo-β2AR. The efficiency was calculated by determining the protein pro- and post-reaction. As illustrated in Fig. S3B (Supporting information), obvious protein loss was observed when the reaction proceeded 2, 5, 10, 20, 30, and 40 min. The highest loss appeared to occur after 40 min with a reaction efficiency of 26%. Given an approximate efficiency, the speed was slightly slower than the data in the solution. This result is reasonable since the flexibility of the substrate was decreased after immobilization. Relying on the intensity loss, the immobilized Gαs-cpHalo-β2AR was measured by western blot. We examined the specificity of this reaction by a competitive displacement method by including 6-chlorocaproic acid in the reaction between the modified gels and the cell lysates. As anticipated, we observed a recovery of the intensity loss compared with the reaction without the inclusion of the substrate (Fig. S4 in Supporting information). This is worthwhile to be stated since it confirmed the specificity of the reaction between Gαs-cpHalo-β2AR and the chlorohexyl group on the gel surface. These, in summary, proved that Gαs-cpHalo-β2AR specifically reacted with the cpHalo's substrate even though the liquid-solid interface and complex matrices were involved.
As illustrated in Fig. 2B, we observed intensive red and green spherical particles when treated the Gαs-cpHalo-β2AR silica gels with the corresponding fluorescent antibodies of β2AR and Gαs. By merging the two fluorescence channels, we observed fully overlapped images. However, almost no fluorescent was found in the control gels, demonstrating that the fusion was successfully immobilized on the gels with the function of antibody recognition. Surface characterization of the immobilized Gαs-cpHalo-β2AR by X-ray photoelectron spectroscopy revealed that the nitrogen content increased 1.5-folds when the control gels were functionalized with Gαs-cpHalo-β2AR (Fig. S5 in Supporting information). The fusion protein is rich in nitrogen element, evidently, the increased nitrogen was introduced by the successfully immobilization of Gαs-cpHalo-β2AR.
Ligand binding specificity of the immobilized Gαs-cpHalo-β2AR was examined by analyzing chromatographic behaviors of the specific ligands of β2AR (salbutamol, bambuterol), GTP (a compound specifically binding to Gαs), sodium nitrite (a compound without interaction with immobilized protein), and phenylephrine (a specific ligand of α1AR). In Fig. 2C, sodium nitrite and phenylephrine presented void time of 2.17 ± 0.12 min for the chromatographic system. Unlike them, salbutamol, bambuterol, and GTP exhibited good retentions on the immobilized Gαs-cpHalo-β2AR column with retention times of 4.8, 8.3, and 5.2 min. These results, to sum up, demonstrated that immobilized β2AR-cpHalo-Gαs maintained the binding activity of the receptor and the G-protein subunits. It allowed the recognition of β2AR ligands and Gαs partners in a specific way.
We examined the stability of the immobilized Gαs-cpHalo-β2AR column by analyzing the retention times of salbutamol and GTP within three weeks with three samplings for each day. As illustrated in Fig. 2D, the relative standard deviations of the retention times of salbutamol and GTP were within 0.4% and 0.8% for fifteen days. This demonstrated that immobilized β2AR-cpHalo-Gαs remained stable in terms of the ligand binding capacity in fourteen days.
Upon ligand binding, GPCRs change their conformations to couple with G proteins [18]. Among the G proteins, Gαs is only responsive to the agonist-bound receptor, while lacking the capacity to interact with the antagonist-binding receptor [19]. According such a typical cascade, agonists and antagonists should exhibit distinct effects on the retentions of GTP on the immobilized Gαs-cpHalo-β2AR column. As illustrated in Fig. 2E, GTP presented a retention time of 5.18 min with an inclusion of 10 µmol/L propranolol (an antagonist of β2AR) in the mobile phase. This appeared to be identical with the retention when phosphorate buffer was applied as the mobile phase, thus providing proof of little effects on the GTP binding by the antagonist. Conversely, the inclusion of either bambuterol or salbutamol gave rise to longer retention times of GTP on the column. Bambuterol exhibited a stronger effect on GTP retention than salbutamol when their concentrations remained identical. As such, we reasoned that immobilized Gαs-cpHalo-β2AR had the capacity to distinguish the receptor agonists from the antagonists by their retentions.
Fig. 3 illustrates the effects of ligand concentrations on GTP retentions when immobilized Gαs-cpHalo-β2AR was utilized as a stationary phase. β2AR agonists, salbutamol, terbutaline, and bambuterol right shifted GTP retention times in a concentration-dependent manner, while the antagonist (propranolol) lacked such an effect (Figs. 3A-D). As a long-acting agonist, bambuterol gave rise to a change of 5.35 min in the retention time when its concentrations increased to 80 µmol/L. Such change was clearly reduced when the short-agonists (salbutamol and terbutaline) were applied. In a second examination, we did not observe such effects when the ligands were co-injected into the system using the mobile phase without the inclusion of them (Figs. 3E-H). These results revealed that the loss of GTP retention times was caused by the formation of ligand-β2AR-Gαs ternary complex rather than the changes in mobile phase compositions.
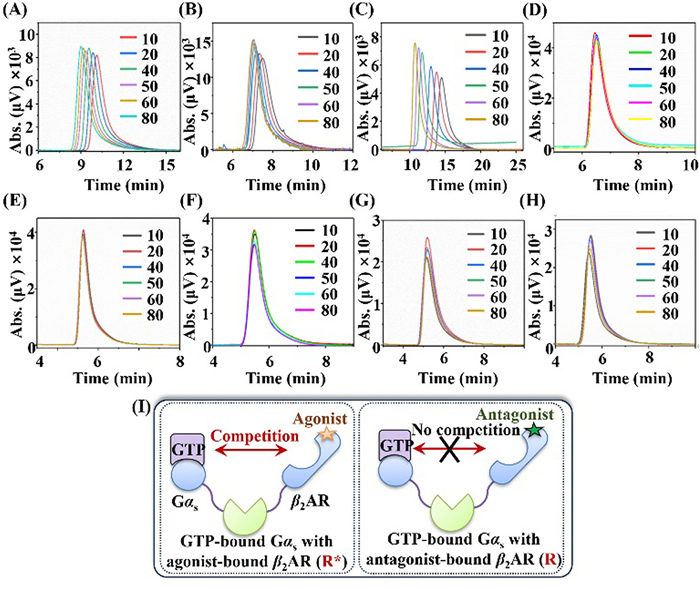
The ternary complex theory is a typical model for describing the GPCR activation procedure (Fig. 3I) [20]. This model assumes a dynamic equilibrium between the inactive state (R) and the active state (R*) of the receptor [21]. Agonists prove to stabilize R* through binding to the receptor. Partial agonists are less effective, whereas true antagonists block agonist binding without influence on the equilibrium between R and R* [22]. In this work, the retention times of GTP remained stable when diverse concentrations of propranolol were applied as the mobile phase. This demonstrated that the binding of propranolol to β2AR occurred at the R state. Unlike propranolol, all three agonists induced clear loss of GTP retention time through the binding to R*. In comparison with bambuterol, salbutamol and terbutaline presented reduced loss of the retention time thereby being less effective in stabilizing R*. Taking together, we reasoned that the loss of GTP retention time was possible to provide an index for assessing the efficacy of β2AR ligands by chromatographic method.
To demonstrate the inherent mechanism of the ligand enhancement for GTP retention times on the immobilized Gαs-cpHalo-β2AR, we performed nonlinear chromatography to examine the changes in GTP binding kinetics induced by the four ligands. As illustrated in Fig. S6 (Supporting information), we observed gradually decreased retention times when diverse concentrations of GTP were injected into the chromatographic system with the utilization of blank phosphorate buffer as the mobile phase. The peak profiles of GTP were tailed and broadened, indicating that nonlinear chromatography is suitable for revealing the interactions between the GTP and the immobilized Gαs-cpHalo-β2AR. Such interactions are supposed to be driven by affinity forces. With inclusion in the mobile phase, propranolol and the receptor-specific antagonist (ICI 118551) exhibited little effect on these retention patterns. On the contrary, all three agonists resulted in clear changes in GTP retentions at desired ligand concentrations. These results, again, confirmed the specificity of immobilized Gαs-cpHalo-β2AR to distinguish agonists from antagonists in terms of chromatographic methods. We found that the addition of GTP (0, 5, 10, 20, and 40 µmol/L) in mobile phase caused the distortion of agonists (i.e., retention time and half-peak-width changes), but had no effect on antagonists (Fig. S7 and Table S1 in Supporting information). This also confirmed the feedback regulation of the downstream GTP-Gαs binding to agonist rather than antagonist, providing another proof for distinguishing the agonist/antagonist of β2AR.
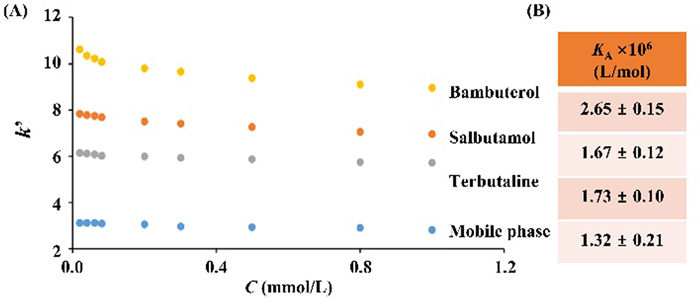
Upon the representative chromatograms in Fig. 4A, we calculated the retention factors (k′) of GTP with 5.0 µmol/L bambuterol, salbutamol, or terbutaline in the mobile phase. Compared with the use of the blank mobile phase, the changes in k′ values of GTP were substantially enhanced. As anticipated, bambuterol exhibited the greatest effect, while terbutaline appeared to be the weakest ligand. These results indicated the impact of ligands on GTP retentions may cause by the changes in binding thermodynamics or kinetics. As such, we calculated the association constants (KA) by injection amount-dependent method and rate constants of the GTP by nonlinear chromatography. We applied diverse concentrations of GTP on immobilized Gαs-cpHalo-β2AR and calculated KA values. As illustrated in Fig. 4B, the KA values of GTP were (1.67 ± 0.12) × 106 L/mol, (1.73 ± 0.10) × 106 L/mol, and (2.65 ± 0.15) × 106 L/mol when the three agonists were separately included in the mobile phase. Such results were clearly higher than the data using the blank mobile phase, thus demonstrating a much higher affinity of GTP to Gαs-cpHalo-β2AR.
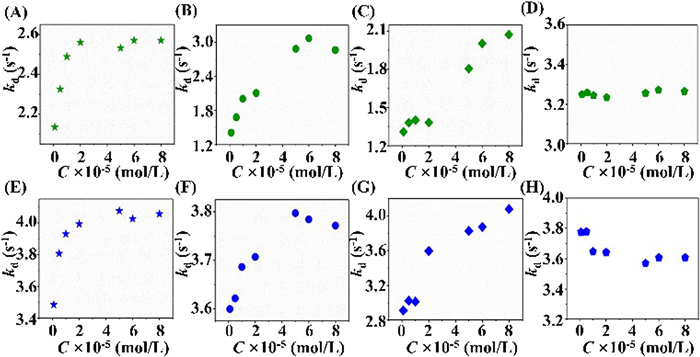
It was notable that all three agonists resulted in a maximum impact on kd values of GTP with a relatively stable value (Fig. 5). This was reasonable since their capacities to activate Gαs differed from each other. The numbers of active Gαs on the column were inconsistent when different ligands were applied even though their concentrations remained identical. Namely, diverse numbers of R* are involved in the stabilization of Gαs. Among the three ligands, bambuterol appeared to be weakest in terms of GTP kd′s regulation, while salbutamol presented the strongest effects. These, to a certain extent, were in good line with the intrinsic activity of the three drugs reported in the literature: salbutamol > terbutaline > bambuterol [23–26]. As such, we concluded that the plate values of kd provided an alternative for assessing ligand efficacy by chromatographic method. Given the constant size of the column, retention times of GTP were potent to rapidly determine GPCR ligand intrinsic activity.
In summary, we have developed a universal chromatographic-based method that uses GTP as an indicator to determine the intrinsic activity of GPCR ligands. Similar to a signal transduction, an agonist binds to GPCR, changes the conformation, leads to the formation of agonist-GPCR-G protein complex, and trigger activation of a physically coupled, functional unit. An antagonist does not have such effects. Therefore, we introduce the downstream signaling G protein as a functional unit to construct the bio-intelligent miniature. (1) It is easy to prepare such a miniature-based stationary phase since the immobilization method belongs to bioorthogonal reaction, which avoids protein purification and sophisticated synthesizing steps. (2) Non-GPCR ligand, agonist and antagonist can be clearly distinguished. (3) Intrinsic activity of different agonists can be evaluated. (4) Given an extension to the other classes of GPCRs, such a method has the possibility of improving the attributes of drug discovery efforts owing to the providence of a second information on signaling procedure. Although only Gαs was presented, this work is possible to afford a blueprint for developing new chromatographic methods that enable the discovery of potential GPCR ligands from diverse libraries like extracts of natural plants with minimizing false-positive results.
All authors disclosed no relevant relationships.
Ting Li: Conceptualization, Investigation, Methodology, Writing – original draft. Xinxin Zheng: Investigation. Lejing Qu: Investigation. Yuanyuan Ou: Data curation. Sai Qiao: Data curation. Xue Zhao: Data curation. Yajun Zhang: Conceptualization, Funding acquisition. Xinfeng Zhao: Conceptualization, Funding acquisition, Supervision. Qian Li: Conceptualization, Funding acquisition, Supervision.
We thank the National Natural Science Foundation of China (Nos. 22374116, 22074118, 82174088), Natural Science Basic Research Program of Shaanxi (2024JC-TBZC-21), and Shaanxi Administration of Traditional Chinese Medicine (No. 2022-SLRH-YQ-007).
Supplementary material associated with this article can be found, in the online version, at doi:
H. Jiang, D. Galtes, J. Wang, H.A. Rockman, Am. J. Physiol. Cell. Physiol. 323 (2022) C731–C748. doi: 10.1152/ajpcell.00210.2022
J.D. McCorvy, B.L. Roth, Pharmacol. Ther. 150 (2015) 129–142. doi: 10.1016/j.pharmthera.2015.01.009
A.S. Hauser, M.M. Attwood, M. Rask-Andersen, H.B. Schiöth, D.E. Gloriam, Nat. Rev. Drug. Discov. 16 (2017) 829–842. doi: 10.1038/nrd.2017.178
X. Zheng, S. Dong, J. Zheng, et al., Biotechnol. Adv. 32 (2014) 564–574. doi: 10.1016/j.biotechadv.2014.02.003
L. Zhang, D. Duan, Y. Liu, et al., J. Am. Chem. Soc. 136 (2014) 226–233. doi: 10.1021/ja408792k
K. Zheng, T.P. Jensen, D.A. Rusakov, Nat. Protoc. 13 (2018) 581–597. doi: 10.1038/nprot.2017.154
X. Wang, J. Wang, B. Ge, Protein. Pept. Lett. 20 (2013) 1272–1279. doi: 10.2174/09298665113209990043
A. Olaru, C. Bala, N. Jaffrezic-Renault, H.Y. Aboul-Enein, Crit. Rev. Anal. Chem. 45 (2015) 97–105. doi: 10.1080/10408347.2014.881250
Q. Liang, H. Zuo, T. Yang, et al., Eur. J. Med. Chem. 233 (2022) 114212. doi: 10.1016/j.ejmech.2022.114212
J. Wang, Q.Q. Yao, M.Z. Wang, Chin. Chem. Lett. 21 (2010) 860–863. doi: 10.1016/j.cclet.2010.01.024
J. Du, J. Guo, D. Kang, et al., Chin. Chem. Lett. 31 (2020) 1695–1708. doi: 10.1016/j.cclet.2020.03.028
J. Fu, W. Qin, L.Q. Cao, Z.S. Chen, H.L. Cao, Drug. Discov. Today. 28 (2023) 103576. doi: 10.1016/j.drudis.2023.103576
J. Gao, X. Yuan, X. Zheng, et al., Biomater. Sci. 9 (2021) 7934–7943. doi: 10.1039/D1BM01222C
J. Liu, T. Li, G. Wang, et al., iScience 25 (2022) 105361. doi: 10.1016/j.isci.2022.105361
R. Tian, J. Yin, Q. Yao, et al., Anal. Chem. 94 (2022) 9048–9057. doi: 10.1021/acs.analchem.2c01210
W. Zhang, X.-Y. Zhou, Q.-Y. Yu, et al., Chin. Chem. Lett. 27 (2016) 185–189. doi: 10.1016/j.cclet.2015.12.002
C. Deo, A.S. Abdelfattah, H.K. Bhargava, et al., Nat. Chem. Biol. 17 (2021) 718–723. doi: 10.1038/s41589-021-00775-w
Q. Zhou, D. Yang, M. Wu, et al., eLife 8 (2019) e50279. doi: 10.7554/eLife.50279
A. Kaiser, I. Coin, Molecules 25 (2020) 4724. doi: 10.3390/molecules25204724
S.G. Rasmussen, B.T. DeVree, Y. Zou, et al., Nature 477 (2011) 549–555. doi: 10.1038/nature10361
W.I. Weis, B.K. Kobilka, Annu. Rev. Biochem. 87 (2018) 897–919. doi: 10.1146/annurev-biochem-060614-033910
A. Vizurraga, R. Adhikari, J. Yeung, M. Yu, G.G. Tall, J. Biol. Chem. 295 (2020) 14065–14083. doi: 10.1074/jbc.REV120.007423
Y.L. Chou, C.C. Wu, H.W. Wang, Eur. Arch. Otorhinolaryngol. 267 (2010) 1305–1311. doi: 10.1007/s00405-009-1173-7
M. Johnson, Am. J. Respir. Crit. Care. Med. 158 (1998) S146–S153. doi: 10.1164/ajrccm.158.supplement_2.13tac110
B.L. Kallstrom, J. Sjoberg, B. Waldeck, Br. J. Pharmacol. 113 (1994) 687–692. doi: 10.1111/j.1476-5381.1994.tb17047.x
S.M. Abd Elhaleem, F. Elsebaei, S. Shalan, F. Belal, J. Fluoresc. 33 (2023) 1717–1725. doi: 10.1007/s10895-023-03182-7

Figure 1 Pharmacological process of GPCR upon binding to different ligands (A) and strategy for engineering G protein-GPCR miniature from cpHaloTag (B, C).

Figure 2 Immobilization and characterization of Gαs-cpHalo-β2AR. (A) Schematic drawing for capturing the fusion protein onto macroporous silica gels. (B) Immuno-fluorescent analysis of the functionalized gels. (C) The chromatograms of different ligands on the Gαs-cpHalo-β2AR column. (D) Retention time changes of salbutamol and GTP over fifteen days. (E) Chromatograms of GTP in the presence of different β2AR ligands in the mobile phase. GTP concentration: 1.0 mmol/L.

Figure 3 Chromatograms of GTP on the Gαs-cpHalo-β2AR column in the presence of various concentrations of β2AR ligands in the mobile phase (A-D) or mixed with the injected GTP (E-H). The ligands were salbutamol (A, E), terbutaline (B, F), bambuterol (C, G), and propranolol (D, H). The injected concentration of GTP was 0.1 mmol/L. (I) Typical model for the GPCR activation procedure. The unit of all the ligand concentration in the mobile phase is µmol/L.

Figure 4 The changes of GTP retention factors (k′, A) and association constants (KA, B) on the Gαs-cpHalo-β2AR column in the presence of different concentrations of salbutamol, terbutaline, and bambuterol.

Figure 5 The changes of dissociation constants (kd) of GTP on the Gαs-cpHalo-β2AR column in the presence of various concentrations of β2AR ligands in the mobile phase (A-D) or mixed with the injected GTP (E-H). The ligands were salbutamol (A, E), terbutaline (B, F), bambuterol (C, G), and propranolol (D, H).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: