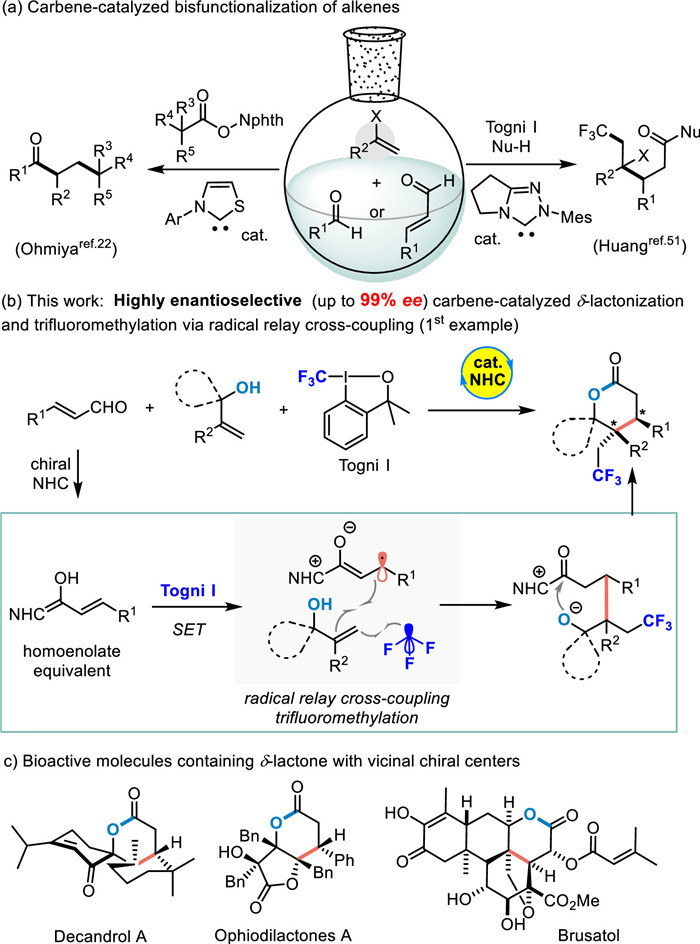
Scheme 1.
Background of NHC-catalyzed enantioselective radical coupling.

Over the past decades, N-heterocyclic carbenes (NHCs) have emerged as a powerful tool for synthetic reactions, medicinal skeletons and their biological functions [1-24]. Its catalytic abilities of HOMO or LUMO activation of aldehydes or carboxylic acids have been extensively studied [25-27]. Although the single-electron transfer capacity of carbene intermediates was predicted in the 1990s [28,29], there are extremely limited examples of NHC-catalyzed radical reactions [10,13]. The pioneering work of carbene-catalyzed radical reaction was reported by Studer and co-workers in 2008 [30]. Since then, this reaction mode has gradually become a hot spot in the field of NHC catalysis [31-34]. Intermediates generated from NHCs and carbonyl compounds can be converted via single-electron transfer to generate several new radical species, such as ketyl radical [35-45] and homoenolate radical [46-52]. The reactivity of these radical intermediates has been explored in recent years and used to construct a few important chemical structures [10,13,40,43-45]. Despite these achievements, only limited examples have been reported for NHC-catalyzed asymmetric radical reactions, such as enantioselective β-hydroxylation [46,48], alkylation [47], arylation [52] of enals and asymmetric synthesis of α-substituted ketones [45]. Later on, the cyclization via homoenolate radicals has also been achieved. Ye et al. accomplished the NHC catalyzed asymmetric [3 + 2] annulation of dioxindoles with enals [49,54]. To date, the enantioselective radical relay couplings involving the formation of multiple chemical bonds are considered to be one of the most effective methods for delivering complex molecules [53]. Since it does not require the use of stoichiometric oxidants or additives to achieve catalytic cycling, this strategy is considered a practical strategy with atomic economy. To our knowledge, NHC-catalyzed radical relay coupling has been explored to a certain degree. In 2019 [36], the Ohmiya group reported a distinguished SET process between Breslow intermediates and redox-active esters to generate ketyl radical and alkyl radical, thereby completing simple alkyl acylation of styrenes (Scheme 1a, left). Since then, a large number of ketyl radical precursors have been applied to such reactions, resulting in a variety of bifunctional olefins. Moreover, it’s associated highly enantioselective versions have rarely been reported. In 2019, the Li group achieved acylfluoroalkylation of olefins by ketone radical relay coupling, but its high enantioselectivity synthesis attempt was unsuccessful (20% ee) [37]. In 2021, the Huang group disclosed an elegant homoenolate radical relay cross-coupling reaction (Scheme 1a, right), accessing to a broad spectrum of β-substituted linear carboxylic acid derivatives, but also albeit in poor enantioselectivity control (27% ee) [51]. Recently, the relay coupling between ketyl radicals and prochiral carbon radicals was attempted by Zhao and coworkers, but the enantioselectivity control of this process remained unresolved (3% ee) [40]. It is no doubt that the NHC-catalyzed highly enantioselective radical relay cross-coupling still remains highly elusive.
Herein, we reported an NHC-catalyzed highly enantioselective radical relay cross-coupling reaction that constructs a spirocyclic δ-lactone structure with two adjacent chiral centers and also contains a CF3 fragment (Scheme 1b) [55-57]. It is worth noting that δ-lactones with vicinal chiral centers at β and γ position are a common skeleton in bioactive compounds (Scheme 1c), such as Decandrol A [58,59], Ophiodilactones A [60] and Brusatol [61].
We began our investigation with trans-cinnamaldehyde 1a, 1-(1-phenylvinyl)cyclohexan-1-ol 2a, and Togni I as model substrates. With the NHC pre-catalyst A, the desired product 4a was formed in 30% yield with 1:1 dr and moderated ee (entry 1). A briefly screen of chiral NHC pre-catalysts indicated that the aminoindanol-derived NHC-pre cat. D afforded better yield and enantioselectivity (entry 4). Replacing the N-phenyl unit of cat. C with electron-withdrawing trichlorophenyl or replacing the N-phenyl unit with a bulkier cyclohexyl group reduced enantioselectivity and did not significantly improve yield (entries 4 and 5). Using 4-tert-butylphenyl-derived aminoindanol F as pre-NHC cat., the ee value was increased slightly. To our delight, when introducing 3,5-di-tert-butylphenyl group to further increase the bulkiness of aminoindanol scaffold (pre-NHC cat. G), excellent enantioselectivity and moderate yield were achieved (entry 10). In addition, highly polar solvent DMF tent to provide better catalytic performance. Exchanged DBU with inorganic base did not offer satisfied results (entries 11 and 12). Finally, adjusting the ratio of substrates 1a and 2a afforded product 4a with 75% yield, 1:1 diastereoselectivity, and excellent ee (entry 13).
With the optimal conditions in hand, we investigated the scope by testing various aldehydes. As shown in Table 1, a broad range of aldehydes with diverse electronic properties was well tolerated. When a fluorine located at the 3-position of the phenyl ring (1b), a result of 79% yield, dr = 1:1, 98% and 90% ee, respectively, was observed (4b). When methoxy group replaced fluorine, a good yield and an excellent ee value were achieved (4c). Functional groups (e.g., methyl (4d), cyano (4e), chlorine (4f)) located at the 4-position were all well tolerated. When a fluorine located at the 2-position of the phenyl ring, a result of 68% yield, dr = 1:3, 98% and 95% ee, respectively, was observed (4g). Substrates bearing with (hetero)cycles, such as 2-naphthyl, 2-thienyl, 2-benzothiophenly, 2-dibenzofuranly, 2-furanyl, 3-pyridyl, 2-quinolinyl, 6-quinoline group, all afforded their corresponding chiral lactones (4h–4o) in moderate to high yields, 1:1 to 1:4 dr and with excellent ee values. Notably, the estrone and probenecid derived aldehydes had proved to be compatible with this system (4p and 4q). The absolute configurations of two diastereoisomers ((4S,5S)-4f (CCDC: 2303772) and ((4S,5R)-4f (CCDC: 2303773)) were determined by X-ray crystallography (Scheme 2), and other products were assigned by analogy.
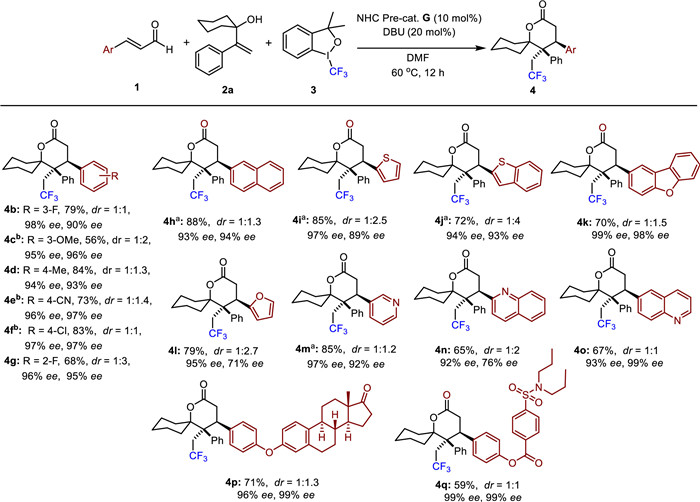
Next, we turned our attention to investigate the scope of alkenes (Scheme 3). meta-Substituted olefin afforded the product 5b in a 55% yield with a high ee value. The electronic properties of aryl substituents had little effect on chemical yields (5c-5e). Unfortunately, ortho-substituted olefins exhibit a lower conversion rate in the reaction, possibly because the radical intermediate formation is impeded, which greatly reduces the efficiency of the critical coupling step. Disubstituted styrene (5f) and naphthyl styrene (5g) were tolerable in our protocol. Heteroaryl olefin also gave the desired adduct in good yield and high ee value (5h). Then, we explored the impact of R group on substrate 2. When tetrahydro-2H-pyranyl and tetrahydro-2H-thiopyranyl were used, both good yields and high to excellent enantioselectivities were achieved (5i and 5j). Then, we varied the ring sizes. Decreased ee values of one obtained isomer were observed (5k and 5l, 80% ee and 70% ee, respectively). Additionally, only moderate ee’s for both isomers (5m) were obtained when cyclohexyl group was replaced by two hydrogens in olefin, indicating the key factor of steric hindrance of benzyl radicals for enantioselective control.
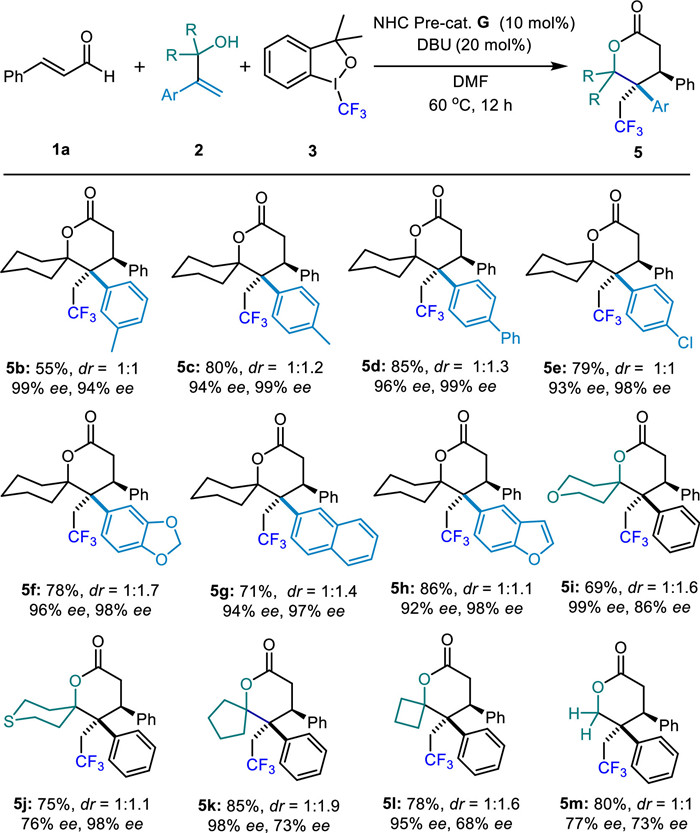
In addition, when we used 2-cyclohexylpropylpropyl-2-en-1-ol (2n) as substrate under standard conditions (Scheme 4a), we could still successfully obtain the corresponding lactone 5n (73% yield, dr = 1:1), but tolerate some losses in ee values (70% ee, 80% ee, respectively). Pleasingly, when we carried out the experiment by using 2o instead of 2a, the corresponding 7-membered lactone 5o was also achieved with an accepted result of 61% yield, dr = 1:2, 90% and 85% ee, respectively.
To further demonstrate the practicality and versatility of this method, we next replaced the trifluoromethyl group with pentafluoroethyl substituent in Togni reagent (6a), and the result showed that the reaction could still be well conducted under standard conditions (Scheme 5a). Methylation at α position of carbonyl group of isomer (4S,5S)-4a yielded the corresponding product 8 in good yield and excellent stereoselectivity (Scheme 5b). (4S,5S)-4e was successfully and equivalently converted to biphenyl compound 9 by Suzuki coupling reaction (Scheme 5c).

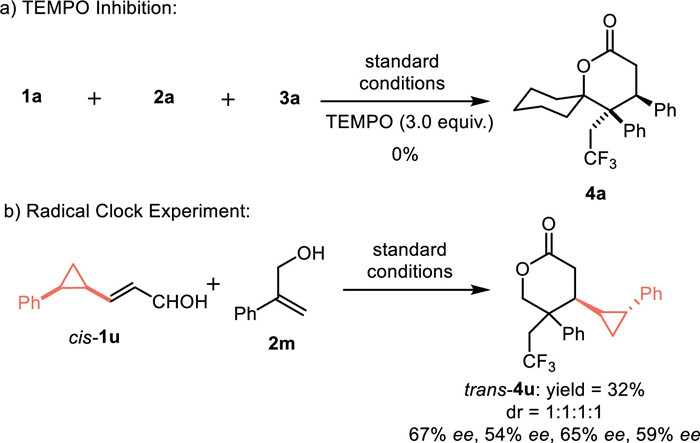
To verify the mechanism, several experiments were carried out. As shown in Scheme 6a, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) effectively inhibited the occurrence of the reaction, suggesting that the reaction may be a radical pathway. In addition, the radical clock experiment with cis-1u as substrate generated trans-lactone, which suggested the ring opening and closing processes of cyclopropane, and the results were consistent with these reported by the Huang group (Scheme 6b).
DFT calculations were performed to further deepen the understanding of the reaction mechanism, including enantioselective and diastereoselective control (For details, see Supporting information). Coupling process between Int3 (homoenolate radical) and Int4 (benzyl radical) is a critical step in determining enantioselectivity. Both Int3 and Int4 have Re and Si faces, so there are four coupling modes. The calculation shows that the energy barriers of TS3 (Int3 Si, Int4 Si, ΔG≠ = 5.6 kcal/mol lead to (S,S)-product) and TS4 (Int3 Si, Int4 Re, ΔG≠ = 5.7 kcal/mol lead to (S,R)-product) are very low and close to each other, while TS5 (Int3 Re, Int4 Re, ΔG≠ = 17.1 kcal/mol lead to (R,R)-product) and TS6 (Int3 Re, Int4 Si, ΔG≠ = 14.5 kcal/mol lead to (R,S)-product) have much higher barriers, indicating that the reaction will not proceed in these two ways.
How to explain the low diastereoselectivity observed in the experiment? The DFT calculation results (Fig. S1 in Supporting information) show that the difference in energy barrier between the Re and Si faces of Int4 is basically negligible, so Int3 can react with Int4 from two faces almost equally. This results in the energies of TS3 and TS4 being very close and produces 4a and 4a′ in an almost 1:1 ratio. DFT calculations are completely consistent with the observed low diastereoselectivity. All the calculations were done at the level of SMD [62]-(DMF)-(U)-M06-2X [63]-D3 [64]/def2-TZVPP//def2-SVP [65]. Lu’s Multiwfn [66] software is uesd for wavefunctin analysis. RDG plots [67] were drawn to display the noncovalent interactions in the transition state (Fig. S2 in Supporting information). Meanwhile, DIAS [68] analyses were also carried out to give insights on the deformation of fragments and interaction between them.
In summary, we have developed an N-heterocyclic carbene (NHC) catalyzed highly enantioselective cyclisation and trifluoromethylation of olefins with cinnamaldehydes via radical relay cross-coupling in the presence of Togni reagent, providing poly-substituted δ-lactones bearing with two adjacent stereogenic centers at β- and γ-positions in good to high yields, moderate diastereoselectivities, and high to excellent enantioselectivities. Further computational studies explain that the radical cross-coupling step is the key to determining the enantioselectivity. DFT calculations also reveal that the hydrogen-bonding interaction plays a vital role in the promotion of this chemistry. Further investigations on novel radical relay cross-couplings catalyzed by NHCs are ongoing in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Generous financial supports for this work are provided by the National Natural Science Foundation of China (Nos. 21871160, 21672121, 22071130), the Bayer Investigator fellow, the fellowship of Tsinghua-Peking centre for life sciences (CLS).
Supplementary material associated with this article can be found, in the online version, at doi:
D. Enders, O. Niemeier, A. Henseler, Chem. Rev. 107 (2007) 5606–5655. doi: 10.1021/cr068372z
N. Marion, S. Díez-González, S.P. Nolan, Angew. Chem. Int. Ed. 46 (2007) 2988–3000. doi: 10.1002/anie.200603380
R. Zhang, H. Yan, J. Zhang, et al., Nat. Prod. Res. 38 (2024) 2078–2081. doi: 10.1080/14786419.2023.2241973
S.J. Ryan, L. Candish, D.W. Lupton, Chem. Soc. Rev. 42 (2013) 4906–4917. doi: 10.1039/c3cs35522e
W.G. Chen, S.S. Zhang, S. Pan, et al., Drug Des. Dev. Ther. 16 (2022) 509–520. doi: 10.2147/dddt.s348836
D.M. Flanigan, F. Romanov-Michailidis, N.A. White, et al., Chem. Rev. 115 (2015) 9307–9387. doi: 10.1021/acs.chemrev.5b00060
Y. Li, H. Xu, H. Wang, et al., Biomed. Pharmacother. 165 (2023) 115218. doi: 10.1016/j.biopha.2023.115218
X.Y. Chen, Z.H. Gao, S. Ye, Acc. Chem. Res. 53 (2020) 690–702. doi: 10.1021/acs.accounts.9b00635
H. Zhou, H. Li, H. Wang, J. Tradit. Chin. Med. 43 (2023) 925–933.
T. Ishii, K. Nagao, H. Ohmiya, Chem. Sci. 11 (2020) 5630–5636. doi: 10.1039/d0sc01538e
J. Shang, L.Y. Liu, T. Dong, X. Wu, et al., J. Chemother. 34 (2022) 401–413. doi: 10.1080/1120009X.2021.2009991
A.P. Song, T.T. Ding, S.G. Zheng, et al., Toxicol. Appl. Pharm. 472 (2023) 116574. doi: 10.1016/j.taap.2023.116574
K. Liu, M. Schwenzer, A. Studer, ACS Catal. 12 (2022) 11984–11999. doi: 10.1021/acscatal.2c03996
Y.Y. Cao, Z. Wang, Z.H. Wang, et al., Int. Immunopharmacol. 90 (2021) 107218. doi: 10.1016/j.intimp.2020.107218
C. Zhang, J.F. Hooper, D.W. Lupton, ACS Catal. 7 (2017) 2583–2596. doi: 10.1021/acscatal.6b03663
Y.Y. Zhou, G.D. Wang, J. Han, et al., Int. Immunopharmacol. 111 (2022) 109137. doi: 10.1016/j.intimp.2022.109137
S.P. Sun, Y.Y. Du, Q. Sun, et al., Pharm. Biol. 58 (2020) 1286–1298. doi: 10.1080/13880209.2020.1859552
G. Wang, H. Zhang, J. Sun, et al., Int. Immunopharmacol. 96 (2021) 107744. doi: 10.1016/j.intimp.2021.107744
C. Li, H. Wang, Z. Wang, et al., Drug Des. Dev. Ther. 14 (2021) 1897–1908. doi: 10.1039/d0fo02210a
X. He, G. Liu, X. Chen, Y. Wang, et al., Clin. Ther. 45 (2023) 655–661. doi: 10.1016/j.clinthera.2023.06.002
Y. Wang, H. Wu, Z. Han, H. Sheng, et al., Phytomedicine 99 (2022) 154034. doi: 10.1016/j.phymed.2022.154034
H. Hong, Q. Zou, Y. Liu, S. Wang, et al., ChemMedChem 16 (2021) 2381–2385. doi: 10.1002/cmdc.202100236
Y. Huang, R. Liu, Y. Wang, G. Liu, et al., Clin. Ther. 44 (2022) 945–956. doi: 10.1016/j.clinthera.2022.06.001
S. Zhao, L. Ma, Z. Chu, H. Xu, et al., Acta Pharm. Sin. 38 (2017) 445–458. doi: 10.1038/aps.2016.162
C. Zhao, S.A. Blaszczyk, J. Wang, Green Synth. Catal. 2 (2021) 198–215. doi: 10.1016/j.gresc.2021.03.003
C. Curti, L. Battistini, A. Sartori, et al., Chem. Rev. 120 (2020) 2448–2512. doi: 10.1021/acs.chemrev.9b00481
J. Cheng, Z. Huang, Y.R. Chi, Angew. Chem. Int. Ed. 52 (2013) 8592–8596. doi: 10.1002/anie.201303247
I. Nakanishi, S. Itoh, T. Suenobu, et al., Chem. Commun. 19 (1997) 1927–1928. doi: 10.1039/a704559j
I. Nakanishi, S. Itoh, T. Suenobu, et al., Angew. Chem. Int. Ed. 37 (1998) 992–994. doi: 10.1002/(SICI)1521-3773(19980420)37:7<992::AID-ANIE992>3.0.CO;2-P
J. Guin, S. Sarkar, S. Grimme, et al., Angew. Chem. Int. Ed. 47 (2008) 8727–8730. doi: 10.1002/anie.200802735
S.D. Sarkar, A. Studer, J. Am. Chem. Soc. 132 (2010) 1190–1191. doi: 10.1021/ja910540j
Y. Du, Y. Wang, X. Li, et al., Org. Lett. 16 (2014) 5678–5681. doi: 10.1021/ol5027415
B.S. Li, Y. Wang, R.S. Proctor, et al., Nat. Commun. 7 (2016) 12933–12940. doi: 10.1038/ncomms12933
X. Wu, Y. Zhang, Y. Wang, et al., Angew. Chem. Int. Ed. 56 (2017) 2942–2946. doi: 10.1002/anie.201611692
T. Ishii, Y. Kakeno, K. Nagao, et al., J. Am. Chem. Soc. 141 (2019) 3854–3858. doi: 10.1021/jacs.9b00880
T. Ishii, K. Ota, K. Nagao, et al., J. Am. Chem. Soc. 141 (2019) 14073–14077. doi: 10.1021/jacs.9b07194
J.L. Li, Y.Q. Liu, W.L. Zou, et al., Angew. Chem. Int. Ed. 59 (2020) 1863–1870. doi: 10.1002/anie.201912450
A.V. Bay, K.P. Fitzpatrick, G.A. González-Montiel, et al., Angew. Chem. Int. Ed. 60 (2021) 17925–17931. doi: 10.1002/anie.202105354
Q.Z. Li, Y.Q. Liu, X.X. Kou, et al., Angew. Chem. Int. Ed. 61 (2022) e202207824. doi: 10.1002/anie.202207824
W.D. Liu, W. Lee, H. Shu, et al., J. Am. Chem. Soc. 144 (2022) 22767–22777. doi: 10.1021/jacs.2c11209
S.C. Ren, X. Yang, B. Mondal, et al., Nat. Commun. 13 (2022) 2846–2855. doi: 10.1038/s41467-022-30583-2
N. Takemura, Y. Sumida, H. Ohmiya, ACS Catal. 12 (2022) 7804–7810. doi: 10.1021/acscatal.2c01964
C. Liu, Z. Zhang, L.L. Zhao, et al., Angew. Chem. Int. Ed. 62 (2023) e202303478. doi: 10.1002/anie.202303478
F. Su, J. Zou, X. Lv, et al., Angew. Chem. Int. Ed. 62 (2023) e202303388. doi: 10.1002/anie.202303388
S. Byun, M.U. Hwang, H.R. Wise, et al., Angew. Chem. Int. Ed. 62 (2023) e202312829. doi: 10.1002/anie.202312829
N.A. White, T. Rovis, J. Am. Chem. Soc. 136 (2014) 14674–14677. doi: 10.1021/ja5080739
N.A. White, T. Rovis, J. Am. Chem. Soc. 137 (2015) 10112–10115. doi: 10.1021/jacs.5b06390
Y. Zhang, Y. Du, Z. Huang, et al., J. Am. Chem. Soc. 137 (2015) 2416–2419. doi: 10.1021/ja511371a
X.Y. Chen, K.Q. Chen, D.Q. Sun, et al., Chem. Sci. 8 (2017) 1936–1941. doi: 10.1039/C6SC04135C
K. Zhao, D. Enders, Angew. Chem. Int. Ed. 5 (2017) 3754–3756. doi: 10.1002/anie.201700370
Z. Li, M. Huang, X. Zhang, et al., ACS Catal. 11 (2021) 10123–10130. doi: 10.1021/acscatal.1c02576
H. Choi, G.R. Mathi, S. Hong, et al., Nat. Commun. 13 (2022) 1776–1783. doi: 10.1038/s41467-022-29462-7
H.M. Huang, Gard.H. uño-Castro, C. Morrill, et al., Chem. Soc. Rev. 48 (2019) 4626–4638. doi: 10.1039/c8cs00947c
Z.Y. Song, K.Q. Chen, X.Y. Chen, et al., J. Org. Chem. 83 (2018) 2966–2970. doi: 10.1021/acs.joc.7b03161
Q. Chen, T. Zhu, P.K. Majhi, et al., Chem. Sci. 9 (2018) 8711–8715. doi: 10.1039/c8sc03480j
J. Charpentier, N. Früh, A. Togni, Chem. Rev. 115 (2015) 50–82.
H.W. Xiao, Z.Z. Zhang, Y.W. Fang, et al., Chem. Soc. Rev. 50 (2021) 6308–6319. doi: 10.1039/d1cs00200g
Z.P. Jiang, L.W. Tian, L. Shen, et al., Fitoterapia 130 (2018) 272–280. doi: 10.1016/j.fitote.2018.09.011
D.W. Li, X.P. Deng, X. He, et al., Phytochemistry 183 (2021) 112593–112600. doi: 10.1016/j.phytochem.2020.112593
T. Matsubara, K. Takahashi, J. Ishihara, et al., Angew. Chem. Int. Ed. 53 (2014) 757–760. doi: 10.1002/anie.201307835
Y. Hitotsuyanagi, I.H. Kim, T. Hasuda, et al., Tetrahedron 62 (2006) 4262–4271. doi: 10.1016/j.tet.2006.01.083
A.V. Marenich, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. B 113 (2009) 6378–6396. doi: 10.1021/jp810292n
Y. Zhao, D.G. Truhlar, Theor. Chem. Account. 120 (2008) 215–241. doi: 10.1007/s00214-007-0310-x
S. Grimme, J. Antony, S. Ehrlich, et al., J. Chem. Phys. 132 (2010) 154104–154122. doi: 10.1063/1.3382344
A. Schäfer, C. Huber, R. Ahlrichs, J. Chem. Phys. 100 (1994) 5829–5835. doi: 10.1063/1.467146
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
E.R. Johnson, S. Keinan, P. Mori-Sánchez, et al., J. Am. Chem. Soc. 32 (2010) 6498–6506. doi: 10.1021/ja100936w
D. Svatunek, K.N. Houk, J. Comput. Chem. 40 (2019) 2509–2515. doi: 10.1002/jcc.26023

Scheme 2 Scope of cinnamaldehyde 1. Reaction conditions: 1 (0.1 mmol), 2a (0.15 mmol), NHC pre-cat. G (0.01 mmol), DBU (0.02 mmol), 3 (0.15 mmol) were stirred in DMF (1 mL) at 60 ℃ under nitrogen for 12 h. dr was determined by 19F NMR analysis. a MeCN as solvent.

Scheme 3 Scope of olefin 2. Reaction conditions: 1a (0.1 mmol), 2 (0.15 mmol), NHC pre-cat. G (0.01 mmol), DBU (0.02 mmol), 3 (0.15 mmol) were stirred in DMF (1 mL) at 60 ℃ under nitrogen for 12 h. dr was determined by 19F NMR analysis.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:



 下载:
下载: