
Figure 1.
(a) XRD pattern, (b, c) SEM imagines, (d) HRTEM image, (e–h) TEM elemental mapping images of CuO/NiO.
Constructing built-in electric field via CuO/NiO heterojunction for electrocatalytic reduction of nitrate at low concentrations to ammonia
Ying Chen , Xingyuan Xia , Lei Tian , Mengying Yin , Ling-Ling Zheng , Qian Fu , Daishe Wu , Jian-Ping Zou

The widespread nitrate (NO3−) pollution in water environment seriously endangers human health and environmental safety [1,2]. Electrocatalytic reduction of NO3− to industrially valuable ammonia (NRA) has been recognized as a "two birds with one stone" strategy to alleviate nitrate pollution and restore nitrogen cycle [3]. Recently, Cu-based catalysts have been found to be effective for electrocatalytic NRA due to their favorable NO3−-binding ability [4]. However, the residual NO3− after electroreduction treatment of nitrate-contaminated water by the reported Cu-based catalysts does not meet the drinking water standard [5]. In practice, the NO3− concentration in many polluted waters (such as industrial wastewater and groundwater) is typically low [2,6]. Therefore, it is essential to develop Cu-based catalysts with high activity and selectivity for electrocatalytic reduction of NO3− at low concentrations to NH4+.
There are two crucial bottlenecks for Cu-based catalysts to achieve efficient electrocatalytic reduction of NO3− at low concentrations to NH4+: ⅰ) The small concentration gradients of reactants (i.e., NO3− and its reaction intermediates) near electrocatalyst surface region leads to sluggish mass transfer [5,7]; ⅱ) the sluggish Volmer step (H2O-to-H*) on Cu-based catalysts leads to the insufficient supply of atomic hydrogen (H*) [2,8]. Therefore, enhancing mass transfer of reactants and H* provision is the key to the design of Cu-based catalysts for electrocatalytic reduction of NO3− at low concentrations to NH4+. Currently, defect engineering has been employed to regulate the electron-density distribution and surface properties of Cu-based catalysts, thereby strengthening the adsorption of reactants and H* [1,9]. Unfortunately, the occurrence of defects in Cu-based catalysts is mostly random and uncontrollable, which inevitably brings about the uncontrollable NO3− electroreduction pathway [10]. Thus, the controllable modification of Cu-based catalysts is advantageous for achieving high-efficiency electrocatalytic reduction of NO3− at low concentrations to NH4+.
Recent works demonstrate that heterostructure design is an attractive approach for the controllable modification of Cu-based catalysts [11-13]. When Cu-based catalyst and other semiconductor with different work functions (Φ) contact to form a heterojunction, the built-in electric field is spontaneously generated in the interface region, thereby accelerating electronic transfer at the interface [14-17]. This can lead to local charge polarization and asymmetrical charge distribution in the interface region, significantly enhancing the adsorption of reactants [18]. Hence constructing a built-in electric field via Cu-based heterojunction is expected to enhance the mass transfer of low concentration reactants during electrocatalytic NRA process. In a similar consideration, Sun et al. [7] reported that the built-in electric field formed between CuCl and TiO2 could effectively accumulate a higher concentration of NO3− near electrocatalyst surface region. Unfortunately, the high NO3− conversion and NH4+ selectivity are difficult to realize. The phenomenon is because the key problem of the insufficient H* supply is still ignored. As a result, the other semiconductor in the Cu-based heterojunction is required to enhance H* provision, thus guaranteeing the efficient electrocatalytic reduction of NO3− at low concentrations to NH4+.
In this work, inspired by nickel-based semiconductors with the superiority of H2O-to-H* [19,20], we choose CuO and NiO with different work functions to form a heterojunction. A built-in electric field formed between CuO and NiO was confirmed by X-ray photoelectron spectroscopy and ultraviolet photoemission spectroscopy. The electrocatalytic NRA performance of CuO/NiO under low NO3− concentration conditions, including NO3− conversion, the selectivity of products (i.e., NH4+, nitrite (NO2−), and dinitrogen (N2)), and FE, was evaluated and compared with those of pure CuO and NiO. Impacts of reaction potential, solution pH, and NO3− concentration on electrocatalytic NRA performance of CuO/NiO were also investigated. Density functional theory (DFT) calculations revealed that CuO/NiO with a built-in electric field is advantageous for enhancing mass transfer of reactants and H* provision. Moreover, combining CuO/NiO system and electrochlorination could effectively induce NO3− to N2 via a NO3−-NH4+-N2 route. The electrocatalytic stability of CuO/NiO was evaluated by consecutive cycle tests. The findings offer helpful guidance for the development and application of electrocatalytic NRA process for nitrate remediation with low concentration.
A two-step hydrothermal and calcination method was developed to synthesize CuO/NiO, as shown in Fig. S1 (Supporting information, and details in Experiment Section). The crystalline structures of the as-obtained catalyst were determined by X-ray diffraction (XRD). As shown in Fig. 1a, the diffraction peaks at 32.51°, 35.42°, and 38.90° correspond to the (110), (002), and (200) planes of CuO (JCPDS No. 48–1548), while the peaks at 37.25°, 43.29°, 62.88°, and 75.41° are assigned to the (111), (200), (220) and (311) planes of NiO (JCPDS No. 78–0429). The field-emission scanning electron microscopy (SEM) images unveil that the as-obtained catalyst has a flower-like microsphere structure (Figs. 1b and c). The phenomenon is due to the assistance of urea and ammonium fluoride in the hydrothermal process, which is consistent with the reported studies [21]. The high-resolution transmission electron microscopy (HRTEM) image reveals distinct lattice fringes with the lattice distance of 0.253 nm and 0.147 nm, which match well with that of the (002) plane of CuO and (220) plane of NiO, respectively (Fig. 1d). Besides, the corresponding elemental mapping results exhibit that Cu, Ni, and O are uniformly distributed in the flower-like microspheres (Figs. 1e–h). The above analysis confirms the successful synthesis of CuO/NiO via a facile hydrothermal and calcination method.
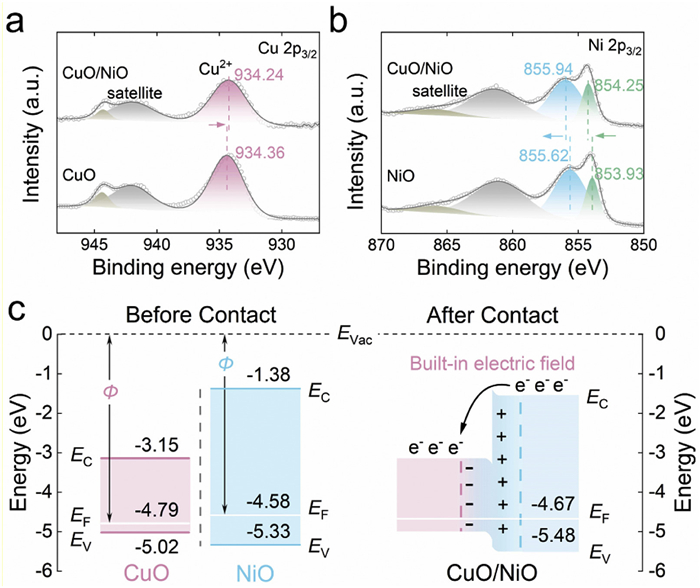
X-ray photoelectron spectroscopy (XPS) was employed to further unveil the chemical state of CuO/NiO. The Cu 2p3/2 spectrum consists of two satellite peaks and the characteristic peak of Cu2+ (934.24 eV) (Fig. 2a). Note that the characteristic Cu2+ peak of CuO/NiO shows a slight negative shift relative to that of pure CuO (934.36 eV). For Ni 2p3/2 spectrum, in addition to two satellite peaks, the peaks at 854.25 eV and 855.94 eV correspond to Ni2+ and Ni3+, respectively (Fig. 2b). Besides, the Ni 2p3/2 spectrum displays that the characteristic peaks of Ni2+ and Ni3+ in CuO/NiO have an obvious positive shift of 0.32 eV compared to those of pure NiO. These findings manifest that the electron transfer exists from NiO to CuO in CuO/NiO. To further determine the electronic interaction between CuO and NiO, ultraviolet photoemission spectroscopy (UPS) was performed. As shown in Figs. S2a and b (Supporting information), the work function values of CuO and NiO are quantified to be 4.79 eV and 4.58 eV, respectively. The difference in work function (ΔΦ) between CuO and NiO begets a driving force for electron transfer, directing electron flow spontaneously from NiO to CuO. Moreover, the Fermi level (EF) of CuO/NiO (−4.67 eV) is between EF of CuO (−4.79 eV) and EF of NiO (−4.58 eV), further testifying that the establishment of a built-in electric field in CuO/NiO (Figs. S2c in Supporting information). Furthermore, the valence band (EV) values of CuO, NiO, and CuO/NiO heterojunction are measured to be −5.02 eV, −5.33 eV, and −5.48 eV by UPS spectra, respectively. Based on the above results and the energy gap (Eg) of CuO (1.87 eV) and NiO (3.95 eV) obtained in the reported studies [22], the energy band diagram of CuO/NiO is constructed in Fig. 2c. When CuO and NiO contact, the self-driven electron donation from NiO to CuO induces the energy band bending of both, thus forming a built-in electric field at the heterojunction interface to accelerate charge transfer, thus accelerating electrocatalytic process [23,24].
The electrocatalytic NRA performance of CuO/NiO was assessed at room temperature using 0.05 mol/L Na2SO4 and low concentration NO3− (100 mg/L NO3−, ca. 22.6 mg/L NO3−-N) as the electrolyte. To preliminarily investigate the electrocatalytic activity of CuO/NiO, linear sweep voltammetry (LSV) tests were conducted with or without the addition of NO3−. As shown in Fig. 3a, the addition of NO3− leads to a noticeable increase in the reduction current density of CuO/NiO, implying that NO3− can be reduced by CuO/NiO [25-27]. The concentrations of NO3−, NO2−, and NH4+ were determined by UV–vis spectrophotometric method and quantified by the corresponding standard curves (Fig. S3 in Supporting information). Fig. 3b displays that NO3− conversion increases from 65.5% at −0.9 V to 100% (i.e., 8.87 NH4+-N mg cm−2 h−1) at −1.3 V, and then maintains the maximum value of 100% in the potential range of −1.3 to −1.5 V. The NH4+ selectivity increases with increasing negative potential from −0.9 to −1.3 V and reaches 100% in the potential range of −1.3 to −1.5 V. NH4+ is not detected in a 0.05 mol/L Na2SO4 electrolyte without NO3− by nuclear magnetic resonance (NMR) and UV–vis spectrophotometric methods (Fig. 3c and Fig. S4 in Supporting information). These results exclude the possible interference of trace ammonia from air or other contaminants and confirm the origin of NH4+ via electrocatalytic NO3− reduction [28,29]. It is worthwhile to point out that CuO/NiO exhibits superior NO3− conversion efficiency and NH4+ selectivity efficiency than many recently reported electrocatalysts (such as FeSAs/g-C3N4, CuCl/TiO2, Ag/GO/Ti, and Cu-SAC) under low NO3− concentration conditions (Table S1 in Supporting information). In addition, the corresponding FE of electrocatalytic NRA follows a volcano plot with the reaction potential. Specifically, the FE of NRA is as low as 4.3% at −0.9 V, and then gradually increases to the maximum value of 61.0% (at −1.3 V) with the increase of negative potential. This trend negatively correlates with the gradual decrease in the NO2− selectivity, demonstrating that NO2− is the intermediate product of electrocatalytic NRA process and can be further reduced to NH4+ at larger negative potentials. This is supported by the concentration-time curves of NO3−-N, NO2−-N, and NH4+-N at −1.3 V. With increasing reaction time, NO3−-N concentration gradually decreases, NO2−-N concentration follows a volcano-type trend, and NH4+-N concentration gradually increases (Fig. S5 in Supporting information). NMR was carried out to further determine the NH4+-yield and FE at −1.3 V. 1H NMR spectra of different NH4+-concentrations were shown in Fig. S5a (Supporting information). By plotting the integral area of NH4+ peaks against the concentration, the standard curve was obtained (Fig. S5b in Supporting information). As shown in Fig. 3c, the concentration of NH4+ and FE calculated by the 1H NMR spectroscopy and UV–vis spectrophotometric methods are nearly equal, evidently proving the accuracy of the two quantitative methods. In the potential range of −1.3 to −1.5 V, FE of NRA exhibits a decrease (34.8% – 45.3%), whereas NO3− conversion and NH4+ selectivity both remain unchanged (100%). The phenomenon could be attributed to the enhanced presence of competitive hydrogen evolution reaction at larger negative potentials [28]. Compared with some other electrocatalysts including CuCl/TiO2 (44.7% FE, 100 mg/L NO3−) [5], single-atom iron (~50% FE, 20 mg/L NO3−-N) [30], CuO/NiO exhibits higher FE (61.0%, 100 mg/L NO3−) for electrocatalytic reduction of NO3− at ultralow concentrations to NH4+ (Table S1). Considering that NO3− conversion, NH4+ selectivity, and FE are the important indexes of electrocatalytic NRA process, the applied potential at −1.3 V is chosen as the performing condition in the following experiments.

To determine the effect of the built-in electric field in CuO/NiO on electrocatalytic NRA performance, contrast experiments with pure CuO and NiO as catalysts for electrocatalytic NRA were carried out. As shown in Fig. 3d, CuO exhibits lower NO3− conversion (95.7%) and NH4+ selectivity (87.0%) than those of CuO/NiO. Moreover, the accumulation of N2 might be due to the insufficient H* supply of CuO [31]. In addition, pure NiO has no effect on NO3− electroreduction, which might be altered for several reasons, such as superior HER activity, and too weak adsorption of low concentration NO3− [22,32]. These results affirm that the built-in electric field formed between CuO and NiO facilitates electrocatalytic reduction of NO3− at low concentrations to NH4+. To further understand the intrinsic performance of CuO/NiO, the electrochemically active surface areas (ECSA) of these catalysts were evaluated by electrochemical double-layer capacitance (Cdl) method. As shown in Fig. S6 (Supporting information), the Cdl value of CuO/NiO heterojunction (0.87 mF/cm) is higher than that of CuO (0.47 mF/cm) and NiO (0.85 mF/cm). The result highlights the built-in electric field at CuO/NiO can augment ECSA and expose more active sites, thereby promoting electrocatalytic NRA process [33,34].
To further elucidate the reaction mechanism in the CuO/NiO–mediated electrocatalytic NRA process, electron spin resonance (ESR) with the assistance of dimethyl-1-pyrroline-N-oxide (DMPO) as the radical trapping reagent was conducted to detect the existence of H* [35]. Nine typical signals with intensity ratios of 1:1:2:1:2:1:2:1:1 associated with DMPO-H* are observed in the spectrum of CuO/NiO without adding NO3− (Fig. 3e). The signal intensity of DMPO-H* decreases when NO3− is added, indicating that the generated H* is consumed in the electrocatalytic NRA process. Moreover, H* quenching experiment using tertiary butanol (TBA) was further performed [36]. The amount of residual NO3− increases with increasing TBA dosage, also suggesting that H* participates in the electrocatalytic NRA process (Fig. S7 in Supporting information). Furthermore, CuO/NiO demonstrates remarkable NO3− conversion (> 95%) and NH4+-N selectivity (> 98%) in the pH range of 4–10 (Fig. S8 in Supporting information). The observation may be because CuO/NiO affords the built-in electric field for the enhanced adsorption of reactants, thereby weakening the influence of initial pH change on electrocatalytic NRA process. In addition, Fig. 3f shows that CuO/NiO can achieve NO3− conversion of 100% and NH4+ selectivity of 100% regardless of NO3− concentration (20–100 mg/L NO3−). The result further proves that CuO/NiO with the assistance of a built-in electric field can guarantee the efficient mass transfer of low concentration reactants during electrocatalytic NRA process. All of these results provide experimental evidence to support the aforementioned electrocatalytic NRA mechanism of CuO/NiO.
The DFT calculations are performed to validate the built-in electric field formed between CuO and NiO in terms of enhanced mass transfer of reactants. It is well known that NO3− adsorption is the first and most important step for electrocatalytic reduction of NO3− at low concentrations to NH4+ [37]. As shown in Figs. 4a-c, the calculated adsorption energy of NO3− on CuO/NiO heterojunction interface is −1.90 eV, which is much higher than that on pure CuO (−1.63 eV) and NiO (−1.40 eV). Considering that electrocatalytic NRA involves the tandem process of NO3− to NO2− and NO2− to NH4+, the adsorption of NO2− intermediate is a decisive step for NH4+ generation in electrocatalytic reduction of NO3− at low concentrations to NH4+. As shown in Figs. 4d-f, the calculated adsorption energy of NO2− on CuO/NiO heterojunction interface is −1.29 eV, which is much higher than that on pure CuO (−1.08 eV) and NiO (−0.82 eV). These results affirm that the built-in electric field formed between CuO and NiO can enhance the adsorption of reactants (NO3− and NO2−), contributing to a higher coverage of reactants on the electrocatalyst surface region.

In addition, projected partial density of states (PDOS) is performed to further verify and support the above discussion. It is observed that the energy difference (ΔE) between d band center (εd) of CuO/NiO and p band center (εp) of NO3− is identified to be 2.61 eV, which is lower than ΔE between εp of NO3− and εd of CuO (2.85 eV) or εd of NiO (3.13 eV) (Figs. 5a-c). These results confirm that the strong interaction between CuO/NiO and NO3− originates from the strong d-p orbital hybridization between CuO/NiO and NO3− [38,39]. Figs. 5d-f show that the hybridization between CuO/NiO and NO2− is also stronger than that between NO2− and CuO or NiO. The above calculations confirm that the built-in electric field formed between CuO and NiO indeed achieves the enhanced mass transfer of reactants.
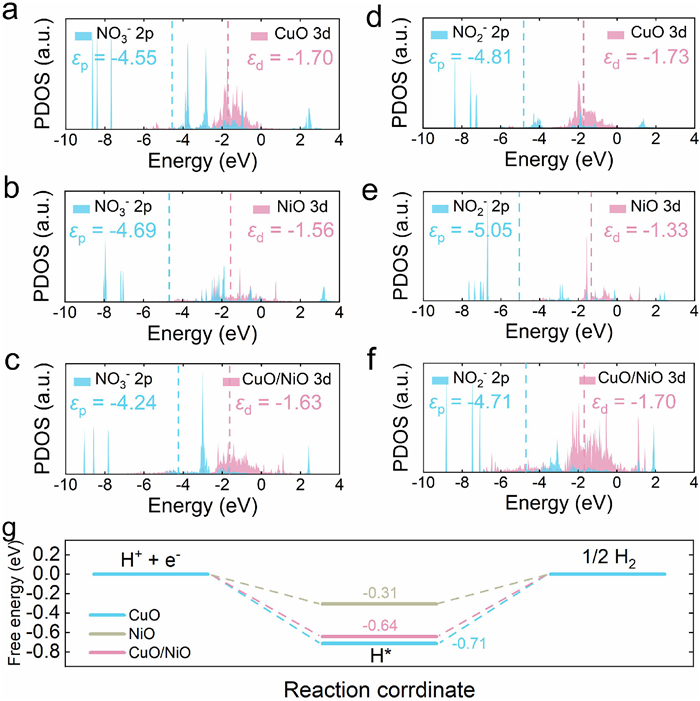
The free energy diagrams of hydrogen evolution reaction (HER) on CuO/NiO and pure CuO and NiO are investigated. As shown in Fig. 5g, the energy barrier of hydrogen evolution on CuO/NiO is calculated to be 0.64 eV, which is lower than that on CuO (0.71 eV). The result reveals the improvement of H2O-to-H* (Volmer step) on CuO/NiO, increasing the coverage of H* on interface sites. Besides, the energy barrier of hydrogen evolution on CuO/NiO is much higher than that on NiO (0.31 eV). It is known that NiO is one of the intensively investigated semiconductors for HER. The result manifests that the generated H* in CuO/NiO cannot be easily released to generate hydrogen, but can be used as a "proton warehouse" to promote electrocatalytic reduction of NO3− [18,32]. The simulation results are consistent with the experimental observations and verify the enhanced H* provision of CuO/NiO for electrocatalytic reduction of NO3−.
Considering the removal of NO3− pollution in real wastewater, electrocatalytic conversion of NO3− to N2 was investigated by combining CuO/NiO system and electrochlorination (Eqs. 1–4) [40,41]. As shown in Fig. 6a, when the ratio of NaCl and Na2SO4 is 1:4, the NH4+ selectivity decreases to 92.2%, and N2 is generated during the reaction. An increase in the NaCl concentration (i.e., the ratio of NaCl and Na2SO4 are 2:3 and 3:2) further decreases NH4+ selectivity (from 60.2% to 3.6%), while N2 selectivity significantly increases (from 39.8% to 96.4%). When the ratio of NaCl and Na2SO4 is 4:1, the degraded NO3− is completely converted to N2. Moreover, the intermediate byproducts, NO2− and NH4+, are not detected during the reaction, suggesting that NO3− is rapidly converted to N2 (Fig. 6b). Furthermore, no undesired products (such as chloramines) were generated after the reaction, manifesting that NO3− could be efficiently transformed into N2 in the coupling process of electrocatalytic NRA and electrochlorination (Fig. S10 in Supporting information) [34]. The residual chlorine concentration (1.1 mg/L) complied with the concentration limits specified by the World Health Organization (0.2–5.0 mg/L) [30]. In addition, the structural stability of CuO/NiO after undergoing the coupling process of electrocatalytic NRA and electrochlorination is verified by XRD measurement, demonstrating that chloride ions do not corrode the CuO/NiO electrode (Fig. S11 in Supporting information). The experiment study on the treatment of pickle wastewater further confirmed the durability and feasibility of CuO/NiO for efficient NO3−-N removal efficiency (92.6%) and long-term operation (Fig. S12 in Supporting information). These results demonstrate that CuO/NiO system can achieve NO3−-NH4+-N2 conversion of wastewater by combining electrocatalytic NRA and electrochlorination.
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
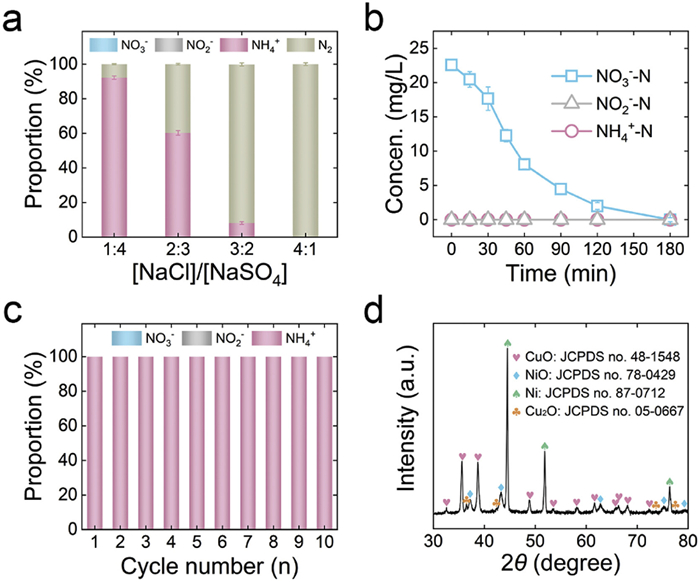
To further evaluate the potential of CuO/NiO for practical application, its electrocatalytic stability evaluation was conducted by consecutive cycle tests. As shown in Fig. 6c, the NO3− conversion and NH4+ selectivity remain 100% during ten cycles of 180 min electrocatalytic NRA reaction, indicating the excellent electrocatalytic stability of CuO/NiO. Besides, the leaching of Cu and Ni during cycle tests is not detected (Fig. S13 in Supporting information). Notably, a small content of NO2− is detected at 120 min of cycle 1 test, the decreased content of NO2− is detected at 120 min of cycle 2 test, and no NO2− within 120 min is detected from cycle 3 to cycle 10 (Fig. S14 in Supporting information). To ascertain the origin of this increment, XRD, TEM, and XPS analysis further characterized the CuO/NiO after cycle 1 test. The XRD patterns of the post-tested CuO/NiO were presented in Fig. 6d. Except for the diffraction peaks of CuO and NiO, the additional diffraction peaks in the XRD patterns are assigned to Cu2O (JCPDS No. 05–0667). TEM image further confirms the formation of Cu2O, which is evident from the distinct lattice fringe of 0.151 nm corresponding to (220) plane of Cu2O (Fig. S15a in Supporting information). XPS analysis was performed to further identify the chemical state of post-tested CuO/NiO. As shown in Fig. S15b (Supporting information), the peaks at 932.20 eV and 934.50 eV in the Cu 2p3/2 spectrum match well with Cu+ and Cu2+, respectively. Cu2+ is the main form of Cu species in the post-tested CuO/NiO. As shown in Fig. S15c (Supporting information), the intensity of Ni3+ peak of post-tested CuO/NiO increases compared to that of CuO/NiO before reaction. The XPS analysis of the post-tested CuO/NiO indicates that although partial CuO is reduced to Cu2O, the constructed CuO/NiO heterostructure is stable during electrocatalytic NRA process, wherein electron transfers from NiO to CuO [14,23]. It is reported that Cu2O is more beneficial than CuO for electrocatalytic NRA, which can explain the enhanced NO3− conversion obtained by post-tested CuO/NiO [42-44]. Dring 50 cycles tests, the NO3− conversion and NH4+ selectivity gradually decreased from 23 cycles to 29 cycles. The decline might be attributed to the reduction of partial CuO to metallic Cu during the consecutive cycle tests, which was comparatively unfavorable for electrocatalytic NRA [2,45]. The electrode was then calcined under the air atmosphere at 500 ℃ for 1 h. The NO3− conversion and NH4+ selectivity increased to 100% and remained almost unchanged from 30 cycles to 50 cycles. Given that coexisting ions and dissolved organics are typically present in real wastewater, the effect of these coexisting substances on electrocatalytic NRA performance is further investgated. As shown in Fig. S17 (Supporting information), the addition of CO32− and SO32− had no effect on the electrocatalytic NRA performance. However, the inclusion of metal ions (Fe2+, Mg2+, and Ca2+) decreased the NO3− conversion, NH4+ selectivity, and FE. The phenomenon could be attributed to the occupation of the active sites by the metal ions [46]. Humic acid had little effect on the conversion efficiency of NO3− to NH4+, which may be due to the strong complexation between humic acid and active sites. Therefore, CuO/NiO shows practical application in nitrate control in industrial wastewater/groundwater treatment.
In this work, we demonstrate that CuO/NiO electrocatalyst can enhance mass transfer of reactants and H* provision, achieving NO3− conversion of 100% and NH4+ selectivity of 100% under NO3− concentration conditions (100 mg/L NO3−, ca. 22.6 mg/L NO3−-N). The built-in electric field is spontaneously generated at CuO/NiO interface, efficiently enhancing the adsorption of reactants on heterojunction interface and hybridization between reactants and CuO/NiO, as revealed by UPS analysis and DFT calculations. The enhanced H* provision in CuO/NiO is also supported by the free energy diagram of HER. These experiments (such as combining electrocatalytic NRA and electrochlorination, stability evaluation) further show the potential of CuO/NiO in practical water applications. This work implies an effective approach for the design and manipulation of electrocatalysts to achieve electrocatalytic reduction of NO3− at low concentrations to NH4+. Further studies on the ammonia collection system are still needed [47].
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ying Chen: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft, Writing – review & editing. Xingyuan Xia: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. Lei Tian: Funding acquisition, Project administration, Software, Supervision, Validation, Writing – review & editing. Mengying Yin: Data curation, Formal analysis, Methodology, Resources, Validation, Visualization. Ling-Ling Zheng: Investigation, Methodology, Resources, Validation. Qian Fu: Data curation, Methodology, Supervision, Validation, Visualization. Daishe Wu: Conceptualization, Project administration, Resources, Validation, Writing – review & editing. Jian-Ping Zou: Formal analysis, Funding acquisition, Methodology, Project administration, Writing – review & editing.
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (Nos. 52170082, 51938007, 21906076, and 52300081), the Natural Science Foundation of Jiangxi Province (No. 20212ACB203008).
Supplementary material associated with this article can be found, in the online version, at doi:
D. Liu, L.L. Qiao, S.Y. Peng, et al., Adv. Funct. Mater. 33 (2023) 2303480. doi: 10.1002/adfm.202303480
W.D. Chen, X.Y. Yang, Z.D. Chen, et al., Adv. Funct. Mater. 33 (2023) 2300512. doi: 10.1002/adfm.202300512
Z. Shu, H.F. Chen, X. Liu, et al., Adv. Funct. Mater. 33 (2023) 2301493. doi: 10.1002/adfm.202301493
Y.F. Fu, S. Wang, Y. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202303327. doi: 10.1002/anie.202303327
Z.M. Song, Y. Liu, Y.Z. Zhong, et al., Adv. Mater. 34 (2022) 202204306.
F.Y. Chen, Z.Y. Wu, S. Gupta, et al., Nat. Nanotechnol. 17 (2022) 759–767. doi: 10.1038/s41565-022-01121-4
W.J. Sun, H.Q. Ji, L.X. Li, et al., Angew. Chem. Int. Ed. 60 (2021) 22933–22939. doi: 10.1002/anie.202109785
Y.T. Wang, P. Zhang, X.Y. Lin, et al., Sci. China Chem. 66 (2023) 913–922.
H.J. Wang, Y.A. Guo, C.J. Li, et al., ACS Appl. Mater. Interfaces 14 (2022) 34761–34769. doi: 10.1021/acsami.2c08534
Y.F. Gao, S. Liang, B.M. Liu, et al., Nat. Commun. 14 (2023) 2059. doi: 10.1038/s41467-023-37676-6
P. Chang, Y.H. Wang, Y.T. Wang, Y.Y. Zhu, Chem. Eng. J. 450 (2022) 137804. doi: 10.1016/j.cej.2022.137804
Y. Zhang, C.Q. Ma, X.J. Zhu, Adv. Energy Mater. 13 (2023) 2301492. doi: 10.1002/aenm.202301492
T.P.Y. Taraka, A. Gautam, S.L. Jain, S. Bojja, U. Pal, J. CO2 Util. 31 (2019) 207–214. doi: 10.1016/j.jcou.2019.03.012
L. Chen, H.Y. Wang, W.W. Tian, et al., Small 20 (2024) 2307252. doi: 10.1002/smll.202307252
W.X. Chen, W. Wei, F. Li, et al., Adv. Funct. Mater. 34 (2024) 2310690. doi: 10.1002/adfm.202310690
L. Chen, L.G. Yue, X.Y. Wang, et al., Small 19 (2023) 2206462. doi: 10.1002/smll.202206462
B.Y. Kim, J.H. Ahn, J.W. Yoon, et al., ACS Appl. Mater. Interfaces 8 (2016) 34603–34611. doi: 10.1021/acsami.6b13930
Y. Feng, L. Chen, Z.Y. Yuan, et al., Inorg. Chem. Front. 10 (2023) 5225–5243. doi: 10.1039/D3QI01113E
F. Zhou, G.H. Tao, J. Phys. Chem. C 127 (2023) 23180–23188. doi: 10.1021/acs.jpcc.3c05321
T.Y. Kou, M.P. Chen, F. Wu, et al., Nat. Commun. 11 (2020) 590. doi: 10.1038/s41467-020-14462-2
H.B. Ma, Z.W. Chen, Z.L. Wang, C.V. Singh, Q. Jiang, Adv. Sci. 9 (2022) 2105313. doi: 10.1002/advs.202105313
D. Yin, D. Chen, Y.X. Zhang, et al., Adv. Funct. Mater. 33 (2023) 2303803. doi: 10.1002/adfm.202303803
Y. Xu, Y.W. Sheng, M.Z. Wang, et al., J. Mater. Chem. A 10 (2022) 16883–16890. doi: 10.1039/D2TA02006H
S. Ni, H.G. Qu, H.F. Xing, et al., Chinese J. Chem. Eng. 41 (2022) 320–328. doi: 10.1016/j.cjche.2021.09.026
M.H. Jiang, Q. Zhu, X.M. Song, et al., Environ. Sci. Technol. 56 (2022) 10299–10307. doi: 10.1021/acs.est.2c01057
T.L. Ren, Z. Yu, H.J. Yu, et al., Appl. Catal. B: Environ. 318 (2022) 121805. doi: 10.1016/j.apcatb.2022.121805
C.L. Liu, G. Zhang, W. Zhang, Z.N. Gu, G.B. Zhu, P. Natl. Acad. Sci. U. S. A. 120 (2023) e2209979120. doi: 10.1073/pnas.2209979120
Y. Xu, K.K. Shi, T.L. Ren, et al., Small 18 (2022) 2203335. doi: 10.1002/smll.202203335
Y. Wang, S. Shu, M. Peng, et al., Nanoscale 13 (2021) 17504. doi: 10.1039/D1NR04962C
Q.A. Song, M. Li, X.S. Hou, et al., Appl. Catal. B: Environ. 317 (2022) 121721. doi: 10.1016/j.apcatb.2022.121721
Y. Li, J.X. Ma, Z.C. Wu, Z.W. Wang, Environ. Sci. Technol. 56 (2022) 8673–8681. doi: 10.1021/acs.est.1c05841
J. Zhou, M. Wen, R. Huang, et al., Energy Environ. Sci. 16 (2023) 2611–2620. doi: 10.1039/D2EE04095F
S.Q. Yang, X.L. Wang, D.Y. Jin, et al., Sep. Purif. Technol. 326 (2023) 124815. doi: 10.1016/j.seppur.2023.124815
L. Tian, X.Y. Xia, L.J. Zhou, et al., Appl. Catal. B: Environ. 340 (2024) 123260. doi: 10.1016/j.apcatb.2023.123260
K. Fan, W.F. Xie, J.Z. Li, et al., Nat. Commun. 13 (2022) 7958. doi: 10.1038/s41467-022-35664-w
L. Tian, L.S. Zhang, L.L. Zheng, et al., Angew. Chem. Int. Ed. 61 (2023) e202214145.
S. Zhang, M. Li, J.C. Li, Q. Song, X. Liu, Natl. Acad. Sci. U. S. A. 120 (2023) e2207080119. doi: 10.1073/pnas.2207080119
G.F. Chen, Y.F. Yuan, H.F. Jiang, et al., Nat. Energy 5 (2020) 605–613. doi: 10.1038/s41560-020-0654-1
W.J. Zhu, F. Yao, Q.F. Wu, et al., Energy Environ. Sci. 16 (2023) 2483. doi: 10.1039/D3EE00371J
L. Tian, P. Chen, X.H. Jiang, et al., Water Res. 209 (2022) 117890. doi: 10.1016/j.watres.2021.117890
L.H. Su, K. Li, H.B. Zhang, et al., Water Res. 120 (2017) 1–11. doi: 10.1016/j.watres.2017.04.069
H.B. Yin, Y. Peng, J.H. Li, Environ. Sci. Technol. 57 (2023) 3134–3144. doi: 10.1021/acs.est.2c07968
J.J. Zhou, Y.Q. Zhu, K.Y. Wen, et al., Environ. Sci. Technol. 58 (2024) 4824–4836. doi: 10.1021/acs.est.3c10936
J.J. Zhou, F. Pan, Q.F. Yao, et al., Appl. Catal. B: Environ. 317 (2022) 121811. doi: 10.1016/j.apcatb.2022.121811
T.H. Zhu, Q.S. Chen, P. Liao, et al., Small 16 (2020) 2004526. doi: 10.1002/smll.202004526
G.M. Jiang, M. Peng, L. Hu, et al., Chem. Eng. J. 435 (2022) 134853. doi: 10.1016/j.cej.2022.134853
Z.X. Liu, F. Shen, L. Shi, et al., Environ. Sci. Technol. 57 (2023) 10117–10126. doi: 10.1021/acs.est.3c03431

Figure 1 (a) XRD pattern, (b, c) SEM imagines, (d) HRTEM image, (e–h) TEM elemental mapping images of CuO/NiO.

Figure 2 (a) Cu 2p3/2 XPS spectra of CuO and CuO/NiO. (b) Ni 2p3/2 XPS spectra of NiO and CuO/NiO. (c) Band structures of CuO and NiO before contact (left) and energy band diagram of CuO/NiO (right).

Figure 3 (a) LSV curves of CuO/NiO in 0.05 mol/L Na2SO4 with and without 100 mg/L NO3−. (b) Product distribution of electrocatalytic NO3− reduction by CuO/NiO at different potentials. (c) 1H NMR spectra of the electrolyte (0.05 mol/L Na2SO4 with and without 100 mg/L NO3−) after the electrocatalytic NRA reaction. Inset of (c): the quantitative comparison of the NH4+ yield rates and FEs achieved by CuO/NiO based on the NMR and UV–vis spectrophotometric methods. (d) Comparison of product distribution of electrocatalytic NO3− reduction by CuO/NiO, CuO, and NiO. (e) Dimethyl-1-pyrroline-N-oxide spin-trapping ESR spectra of H* stemmed from CuO/NiO in the absence and presence of NO3−. (f) Effect of initial NO3− concentration on NO3− conversion and NH4+ selectivity of CuO/NiO.

Figure 4 Calculated adsorption energies of NO3− and NO2− on (a, d) CuO, (b, e) NiO, and (c, f) CuO/NiO.

Figure 5 PDOS of (a, d) CuO, (b, e) NiO, and (c, f) CuO/NiO with NO3− and NO2− adsorption. (g) Calculated Gibbs free energies for HER on CuO/NiO and pure CuO and NiO.

Figure 6 (a) Effect of the molar ratio of NaCl and Na2SO4 on NO3− conversion (the total molar concentration of NaCl and Na2SO4 was 0.05 mol/L). (b) Time-dependent changes in the concentrations of NO3−-N, NO2−-N, and NH4+-N of CuO/NiO in the molar ratios of NaCl and Na2SO4 of 4:1. (c) Product distribution of electrocatalytic NO3− reduction by CuO/NiO in cycle tests (each test lasting 180 min). (d) XRD pattern of CuO/NiO after cycle 1 test.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: