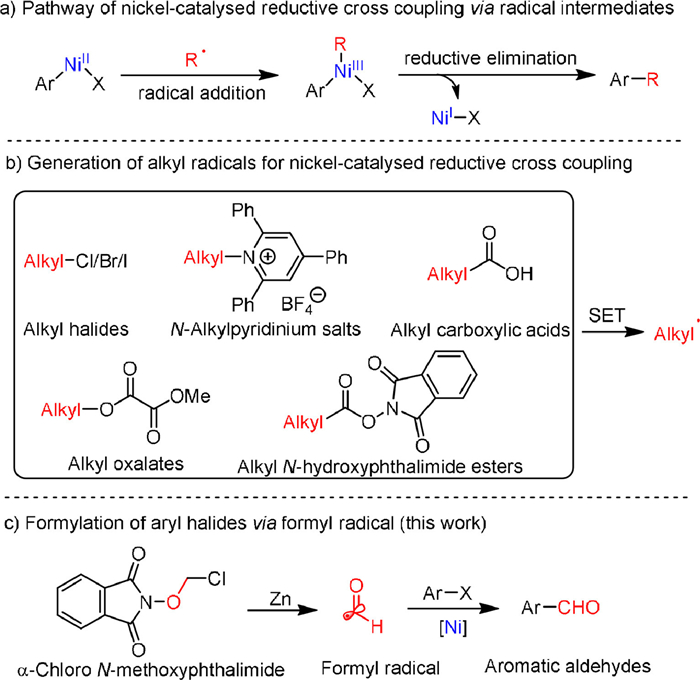
Scheme 1.
Nickel-catalyzed reductive cross coupling via radical intermediates for the formation of C–C bonds.
Nickel-catalyzed reductive formylation of aryl halides via formyl radical
Xiao-Bo Liu , Ren-Ming Liu , Xiao-Di Bao , Hua-Jian Xu , Qi Zhang , Yu-Feng Liang

Earth-abundant nickel-catalyzed reductive cross coupling of aryl halides with electrophiles has gained substantial attentions because of the useful role in organic synthesis for accurate construction of a wide variety of C—C bonds [1–30]. Radical intermediates are always involved in the reactions, generally proceed via the initial oxidative addition and reduction to give nickelII intermediate, followed by radical addition to afford aryl-alkyl nickelIII species, which could give the product through reductive elimination (Scheme 1a) [31–34]. A range of alkyl electrophiles, such as alkyl halides [11,35–53], N-alkylpyridinium salts [54–64], alkyl carboxylic acids [65–68], alkyl oxalates [69–76], and alkyl N-hydroxyphthalimide esters [77–81] could be employed as the sources of alkyl radicals through single electron transfer (SET) process in nickel-catalyzed reductive cross coupling reactions (Scheme 1b). Despite these considerable achievements, to the best of our knowledge, the nickel-catalyzed reductive formylation of aryl halides via formyl radical intermediate still remains undeveloped.
Considering the importance of aromatic aldehydes, to data, a diversity of methods for the formylation of aryl halides using transition metal catalysts have been developed [81–85]. Earlier studies on the palladium/rhodium-catalyzed formylation of aryl halides with CO and H2 have yielded impressive results [86–91]. For example, Beller and coworkers clarified the catalytic cycle of the formylation of aryl bromides catalyzed by palladium for industrial application [86]. However, the inherent drawbacks of the reported methods, such as the need for toxic and flammable CO, high reaction temperatures, and precious metal complexes as accelerators, have led chemists to explore cleaner and cheaper ways to synthesize aromatic aldehydes. Inspired by the nickel-catalyzed reductive cross coupling of aryl halides via radical intermediates, we envisioned that aromatic aldehydes could be prepared if a formyl radical is present in the catalytic conditions. Herein, we disclose a nickel-catalyzed reductive formylation of aryl halides with α–chloro N-methoxyphthalimide as the source of formyl radical to access functionalized aromatic aldehydes (Scheme 1c). In terms of practicality and usability, this protocol is easy to handle, scalable and proceeds smoothly with excellent tolerance of functional groups.
Recently, Chen and coworkers demonstrated the generation of formyl radical from α–chloro N-methoxyphthalimide under photoredox conditions [92]. Encouraged by this elegant example, we initiated our studies by monitoring the formylation of p-iodotoluene 1 with α–chloro N-methoxyphthalimides 2 as the formyl source under nickel-catalyzed reductive conditions. A collection of salient results is summarized in Table 1 (see Supporting information for further and exhaustive list ofattempts). Upon extensive investigation, we were pleased to find that the reaction using NiBr2.bpy as the catalyst, Zn as the reducing agent, KI as the additive, in DMA at room temperature generated formylation product 3 in 92% yield (entry 1). Several NiII catalysts were systematic screened, such as NiCl2.phen, NiBr2.(PPh3)2, and NiBr2.DME, suggesting that NiBr2.bpy performed as the best catalyst, and in situ formation of NiBr2.bpy or without bpy resulted in lower yields (entries 2–6). The replacement of chloro with fluoro in substrate 2 led to significant decrease of the yield (entry 7). In addition, the attempts to use α-trimethylsilyl N-methoxyphthalimide, N-formylsaccharin, S-phenyl methanethioate, phenyl formate or acetic formic anhydride as the formylation reagents failed to afford any desired product (entry 8). Furthermore, the addition of KI could increase the yield, while NaI, LiBr and MgCl2 failed to improve the reaction efficiency (entries 9–12). Raising the temperature would significantly decrease the yield because of undesired homo-coupling and dehalogenation of p-iodotoluene (entries 13 and 14). Notably, zinc powder worked as a better reducing agent than manganese and iron (entries 15 and 16). Next, the solvent effects were carefully investigated. The reactions in dioxane, DCE, DMSO and NMP gave lower yields than in DMA (entries 17–20). For the use of unactivated aryl bromide as substrate under standard conditions, the yield is only 14%, which could be improved to 36% after proper optimization (entries 21 and 22). As expected, the control experiments demonstrated that the presence of both Ni catalyst and reductant were mandatory for the formation of product (entry 23).
With the optimal reaction conditions in hand, we then examined the generality of this transformation (Scheme 2). The formylation reactions proceeded well for electron-donating groups and generated the desired products bearing OMe (4), NMe2 (5), NHBoc (6), NHAc (7), OAc (8), Ph (9). Good yields were obtained regardless of substitution on the ortho (21, 22), meta (25) or para (3–12) position. Similarly, a wide range of electron-withdrawing groups (i.e., halogens, cyanide, trifluoromethyl, ester, methylsulfonyl) were effectively handled on the ortho (23, 24), meta (26–28) and para (14–20) positions. Interestingly, with minor adjustments, the range of substrates can be extended to bromoaromatics (16, 17, 19). In addition, the reaction showed excellent tolerance to sensitive groups. The OTf (10), OAc (8), Bpin (12) and TMS (18) units were all well tolerated and the corresponding products were isolated in satisfactory yields, showing a notable selectivity of our cross coupling. Iodine on electron-rich heterocycles such as naphthalene (29, 30) and carbazole (32) also participated well in this conversion. Moreover, Furan also did not affect the reaction (31). Significantly, alkenyl bromides with electron-withdrawing or electron-donating groups can also reacted smoothly to furnish the target cinnamaldehydes with excellent E/Z selectivity (33–35). Unfortunately, alkyl halides and aryl sulfonates could not be transformed into the desired formylation products.

Of particular note, substrates containing unprotected phenolic hydroxyl, primary amino and alcoholic hydroxyl, which were difficult substituents in transition metal-catalyzed cross-couplings, were compatible well in this reductive formylation, further demonstrating the high compatibility of this transformation with diverse functional groups (Scheme 3a). The robustness and synthetic significance of the catalytic protocol were also shown on the late-stage functionalization of derivatized biologically relevant scaffolds (Scheme 3b). As an illustration, derivatives of levulose and amino acids (tyrosine, aspartic acid) reacted smoothly to deliver the corresponding products in moderate yields (43, 44, 47). Additionally, highly lipophilic scaffold such as Vitamin E derivative proved to be suitable as well, providing the desired aldehyde in 58% yield (45). Derivatives of cholesterol, eugenol and the natural product menthol were also well compatible with this reaction (46, 48, 49), with the olefin groups tolerated. Finally, to evaluate the practicality of this approach, a gram-scale experiment was carried out. To our delight, the desired product was successfully constructed in 77% yield (Scheme 3c).

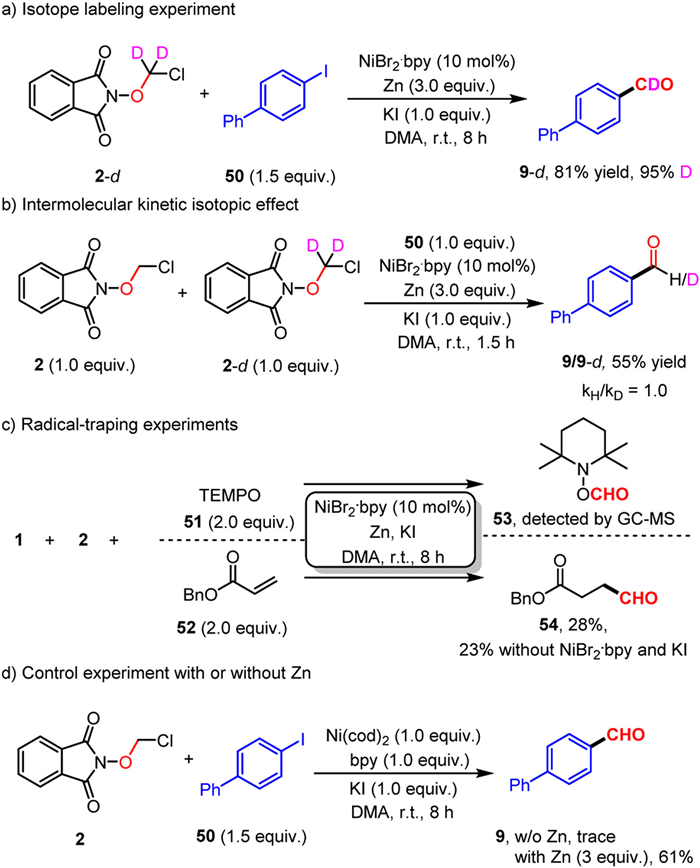
To gain more insight into this reaction, a series of mechanistic studies were conducted (Scheme 4). The isotope-labeling experiments provided product with up to 95% deuterium incorporation (Scheme 5a), indicating that the aldehyde hydrogen of the product comes from ClCH2O unit. Moreover, intermolecular isotope competition experiments show that the elimination of HCl is not the rate-determining step in this transformation (Scheme 5b) [93,94]. The capture of formyl radical with the use of radical scavengers (TEMPO, 51) and Michael acceptor (benzyl acrylate, 52) under the standard conditions confirmed the radical process in this reaction (Scheme 5c) [95–97]. Furthermore, the radical adduct 53 could still be detected and addition product 54 could be obtained in 23% yield in the absence of nickel catalyst and KI, implying that zinc powder plays an essential role in the reduction of α–chloro N-methoxyphthalimide to generate formyl radical, while nickel catalyst is useless in this process. Additionally, the control experiments with stoichiometric of Ni(0) with or without of zinc afforded the desired product in trace and 61% yield respectively, further suggesting that Ni(0) alone failed to promote the formation of formyl radical (Scheme 5d) [74,98].
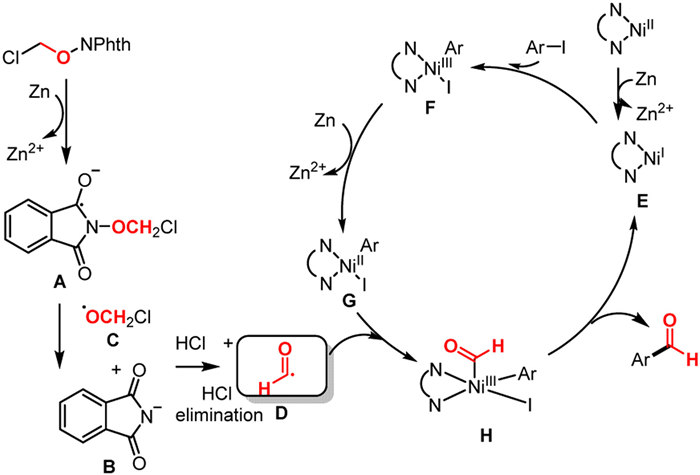
To further elucidate the reaction mechanism, detailed DFT calculations have been carried out. In the presence of Zn powder, the original catalyst L-Ni(II)Br2 can be reduced to the form of low valent Ni. As shown in Fig. 1, the Gibbs free energy change in the process of reducing to Ni(0) catalyst cat2 is 34.2 kcal/mol, which is difficult to carry out under the current reaction conditions at room temperature. Relatively, the energy change for the process of reducing to Ni(I) catalyst cat1 (LNi(I)Br) is −23.5 kcal/mol, which is the favorable process. Therefore, Ni(I) is the form of active catalyst. The I atom in p-iodotoluene 1 then complexes with cat1 to obtain Int1, and the intermediate undergoes oxidative addition transition state TS1 to obtain the Ni(III) intermediate Int2, with an energy barrier of 14.6 kcal/mol (cat1→TS1). The obtained intermediate Int2 was then reduced by Zn powder to form a Ni(II) intermediate, which prepared for the subsequent formyl radical addition. In this process, both halogen atoms (Br and I) of Int2 can be reduced. The Ni(II) intermediates Int3 and Int5 were obtained by removing Br or I, respectively, with energies of −43.3 and −41.7 kcal/mol, respectively. In the subsequent formyl radical addition step, we hope to obtain the Ni(III) intermediate for Ni−C bonding, but during the structure optimization process, the formyl group and toluene group are automatically bonded to form Ni(I) intermediates Int4 and Int6, respectively, with corresponding energies of −99.3 and −98.5 kcal/mol, respectively. This shows that the C—C bonding step is extremely advantageous in terms of kinetics and thermodynamics. Finally, product 3 was generated, and the recyclable Ni(I) active catalysts cat1 and cat3 were obtained, with free energies of −103.3 and −101.4 kcal/mol, respectively. Accordingly, the Ni−Br involved mechanism (black lines) is more favorable than that of Ni−I (dark green lines), L-Ni(I)Br is therefore the advantageous form of active catalyst.

The generation process of formyl radical is also examined in detail. As shown in the upper right corner of Fig. 1, the substrate α–chloro N-methoxyphthalimide 2 reacts with zinc powder via single electron transfer (SET) to afford the anionic radical I, which can be performed at room temperature with free energy change of 14.7 kcal/mol. After SET, both the N—O bond and the C═O bond in substrate 2 were elongated. Subsequently, I underwent an N═O cleavage process to obtain phthalimido anion and α-chloromethoxy radical II, which was relatively easy and had an energy change of −26.6 kcal/mol. Subsequently, the α-chloromethoxy radical II undergoes the HCl geminal elimination to provide the formyl radical III. The transition state of HCl elimination is TS2, and the corresponding energy barrier is 5.4 kcal/mol (II→TS2), and the energy change of this elimination step is −11.0 kcal/mol. Based on this, the process of formyl radical formation with the participation of Zn powder as a reducing agent is available.
Overall, the reaction may undergo a reaction mechanism of active Ni(I) catalyst formation, substrate complexation, oxidative addition, reduction of Ni(III) to Ni(II) with the participation of Zn, and synergistic formyl radical addition-elimination (Scheme 5). Among them, the generated formyl radical undergoes the SET, the N—O bond dissociation and HCl elimination steps could occur with Zn's participation. The additive KI could activate Zn to improve the efficiency of the reaction. On the other hand, due to the poor activity of aryl bromides in this system, the addition of KI could in situ generate the reactive aryl iodides electrophiles. Under reaction conditions, the above process is thermodynamically and kinetically feasible.
In summary, a unique radical approach for the construction of structurally diverse aromatic aldehydes via nickel-catalyzed reductive coupling of aryl halides with α–chloro N-methoxyphthalimide as the formylation reagent has been established. The procedure exploits the in-situ generation of a key formyl radical from α-chlorine elimination. The present system is characterized by a wide substrate scope, mild reaction conditions, and excellent functional group compatibility. The late stage functionalization of structural complex molecules highlighted the utility of this protocol. The gram scale synthesis further demonstrated the usability and applicability of this strategy. DFT calculations were conducted to illustrate the catalytic radical pathway. Further efforts to extend this protocol to deliver aliphatic aldehydes and ketones and detailed mechanistic studies are ongoing in our group.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 22001147), Taishan Scholars Project of Shandong Province (No. tsqn202103027), Distinguished Young Scholars of Shandong Province (Overseas) (No. 2022HWYQ-001), and Qilu Youth Scholar Funding of Shandong University.
Supplementary material associated with this article can be found, in the online version, at doi:
C.E.I. Knappke, S. Grupe, D. Gärtner, et al., Chem. Eur. J. 20 (2014) 6828–6842. doi: 10.1002/chem.201402302
T. Moragas, A. Correa, R. Martin, Chem. Eur. J. 20 (2014) 8242–8258. doi: 10.1002/chem.201402509
D.J. Weix, Acc. Chem. Res. 48 (2015) 1767–1775. doi: 10.1021/acs.accounts.5b00057
J. Gu, X. Wang, W. Xue, H. Gong, Org. Chem. Front. 2 (2015) 1411–1421. doi: 10.1039/C5QO00224A
E. Richmond, J. Moran, Synthesis 50 (2018) 499–513. doi: 10.1055/s-0036-1591853
J. Liu, Y. Ye, J.L. Sessler, H. Gong, Acc. Chem. Res. 53 (2020) 1833–1845. doi: 10.1021/acs.accounts.0c00291
K.E. Poremba, S.E. Dibrell, S.E. Reisman, ACS Catal. 10 (2020) 8237–8246. doi: 10.1021/acscatal.0c01842
X. Pang, P.F. Su, X.Z. Shu, Acc. Chem. Res. 55 (2022) 2491–2509. doi: 10.1021/acs.accounts.2c00381
Q. Pan, Y. Ping, W. Kong, Acc. Chem. Res. 56 (2023) 515–535. doi: 10.1021/acs.accounts.2c00771
Y. Liu, P. Li, Y. Wang, Y. Qiu, Angew. Chem. Int. Ed. 62 (2023) e202306679. doi: 10.1002/anie.202306679
J.L. Hofstra, A.H. Cherney, C.M. Ordner, S.E. Reisman, J. Am. Chem. Soc. 140 (2018) 139–142. doi: 10.1021/jacs.7b11707
L.J. Oxtoby, Z.Q. Li, V.T. Tran, et al., Angew. Chem. Int. Ed. 59 (2020) 8885–8890. doi: 10.1002/anie.202001069
F. Cong, X.Y. Lv, C.S. Day, R. Martin, J. Am. Chem. Soc. 142 (2020) 20594–20599. doi: 10.1021/jacs.0c11172
X.W. Chen, J.P. Yue, K. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 14068–14075. doi: 10.1002/anie.202102769
T.J. DeLano, S.E. Dibrell, C.R. Lacker, et al., Chem. Sci. 12 (2021) 7758–7762. doi: 10.1039/D1SC00822F
S. Guven, G. Kundu, A. Weβels, et al., J. Am. Chem. Soc. 143 (2021) 8375–8380. doi: 10.1021/jacs.1c01797
F. Wang, Y. Chen, W. Rao, L. Ackermann, S.Y. Wang, Nat. Commun. 13 (2022) 2588–2598. doi: 10.1038/s41467-022-30256-0
Q. Wang, Q. Sun, Y. Jiang, et al., Nat. Synth. 1 (2022) 235–244. doi: 10.1038/s44160-022-00024-5
Y.Z. Li, N. Rao, L. An, et al., Nat. Commun. 13 (2022) 5539–5547. doi: 10.1038/s41467-022-33159-2
Z. Zhu, J. Xiao, M. Li, Z. Shi, Angew. Chem. Int. Ed. 61 (2022) e202201370. doi: 10.1002/anie.202201370
Y. Dai, F. Wang, S. Zhu, L. Chu, Chin. Chem. Lett. 33 (2022) 4074–4078. doi: 10.1016/j.cclet.2021.12.050
L. Xi, L. Du, Z. Shi, Chin. Chem. Lett. 33 (2022) 4287–4292. doi: 10.1016/j.cclet.2022.01.077
Y. Wang, Z. Zhao, D. Pan, et al., Angew. Chem. Int. Ed. 61 (2022) e202210201. doi: 10.1002/anie.202210201
X. Hu, I. Cheng-Sánchez, S. Cuesta-Galisteo, C. Nevado, J. Am. Chem. Soc. 145 (2023) 6270–6279. doi: 10.1021/jacs.2c12869
Z.M. Su, J. Twilton, C.B. Hoyt, et al., ACS Cent. Sci. 9 (2023) 159–165. doi: 10.1021/acscentsci.2c01324
X. Ying, Y. Li, L. Li, C. Li, Angew. Chem. Int. Ed. 62 (2023) e202304177. doi: 10.1002/anie.202304177
W. Guan, L. Lu, Q. Jiang, et al., Angew. Chem. Int. Ed. 62 (2023) e202303592. doi: 10.1002/anie.202303592
C.S. Day, A. Rentería-Gómez, S.J. Ton, et al., Nat. Catal. 6 (2023) 244–253. doi: 10.1038/s41929-023-00925-4
J. Zhou, D. Wang, W. Xu, Z. Hu, T. Xu, J. Am. Chem. Soc. 145 (2023) 2081–2087. doi: 10.1021/jacs.2c13220
L. Wan, Y. Tong, X. Lu, Y. Fu, Chin. Chem. Lett. 35 (2024) 109283. doi: 10.1016/j.cclet.2023.109283
S.Z. Tasker, E.A. Standley, T.F. Jamison, Nature 509 (2014) 299–309. doi: 10.1038/nature13274
J. Diccianni, Q. Lin, T. Diao, Acc. Chem. Res. 53 (2020) 906–919. doi: 10.1021/acs.accounts.0c00032
N. Lalloo, C.E. Brigham, M.S. Sanford, Acc. Chem. Res. 55 (2022) 3430–3444. doi: 10.1021/acs.accounts.2c00496
Q. Lin, E. Spielvogel, T. Diao, Chem 9 (2023) 1295–1308. doi: 10.1016/j.chempr.2023.02.010
S. Biswas, D.J. Weix, J. Am. Chem. Soc. 135 (2013) 16192–16197. doi: 10.1021/ja407589e
L.L. Anka-Lufford, K.M.M. Huihui, N.J. Gower, L.K.G. Ackerman, D.J. Weix, Chem. Eur. J. 22 (2016) 11564–11567. doi: 10.1002/chem.201602668
S.Z. Sun, R. Martin, Angew. Chem. Int. Ed. 57 (2018) 3622–3625. doi: 10.1002/anie.201712428
Y. Fang, T. Rogge, L. Ackermann, S.Y. Wang, S.J. Ji, Nat. Commun. 9 (2018) 2240–2250. doi: 10.1038/s41467-018-04646-2
H. Yin, J. Sheng, K.F. Zhang, et al., Chem. Commun. 55 (2019) 7635–7638. doi: 10.1039/C9CC03737C
S.J. He, J.W. Wang, Y. Li, et al., J. Am. Chem. Soc. 142 (2020) 214–221. doi: 10.1021/jacs.9b09415
P. Fan, C. Zhang, L. Zhang, C. Wang, Org. Lett. 22 (2020) 3875–3878. doi: 10.1021/acs.orglett.0c01121
D.J. Charboneau, E.L. Barth, N. Hazari, M.R. Uehling, S.L. Zultanski, ACS Catal 10 (2020) 12642–12656. doi: 10.1021/acscatal.0c03237
H.Y. Tu, F. Wang, L. Huo, et al., J. Am. Chem. Soc. 142 (2020) 9604–9611. doi: 10.1021/jacs.0c03708
D. Sun, G. Ma, X. Zhao, C. Lei, H. Gong, Chem. Sci. 12 (2021) 5253–5258. doi: 10.1039/D1SC00283J
Z. Li, W. Sun, X. Wang, et al., J. Am. Chem. Soc. 143 (2021) 3536–3543. doi: 10.1021/jacs.0c13093
P. Zheng, P. Zhou, D. Wang, et al., Nat. Commun. 12 (2021) 1646. doi: 10.1038/s41467-021-21947-1
X. Li, X. Gao, C.Y. He, X. Zhang, Org. Lett. 23 (2021) 1400–1405. doi: 10.1021/acs.orglett.1c00058
N.W.J. Ang, L. Ackermann, Chem. Eur. J. 27 (2021) 4883–4887. doi: 10.1002/chem.202005449
R.F. Turro, M. Brandstätter, S.E. Reisman, Angew. Chem. Int. Ed. 61 (2022) e202207597. doi: 10.1002/anie.202207597
W. Hu, Z. Lin, C. Wang, Org. Lett. 24 (2022) 5751–5755. doi: 10.1021/acs.orglett.2c02199
Y. Gong, L. Su, Z. Zhu, Y. Ye, H. Gong, Angew. Chem. Int. Ed. 61 (2022) e202201662. doi: 10.1002/anie.202201662
H. Wang, P. Zheng, X. Wu, Y. Li, T. Xu, J. Am. Chem. Soc. 144 (2022) 3989–3997. doi: 10.1021/jacs.1c12424
D. Wang, L. Ackermann, Chem. Sci. 13 (2022) 7256–7263. doi: 10.1039/D2SC02277J
W.T. Zhao, W. Shu, Sci. Adv. 9 (2023) eadg9898. doi: 10.1126/sciadv.adg9898
J. Yi, S.O. Badir, L.M. Kammer, M. Ribagorda, G.A. Molander, Org. Lett. 21 (2019) 3346–3351. doi: 10.1021/acs.orglett.9b01097
H. Yue, C. Zhu, L. Shen, et al., Chem. Sci. 10 (2019) 4430–4435. doi: 10.1039/C9SC00783K
S. Ni, C.X. Li, Y. Mao, et al., Sci. Adv. 5 (2019) eaaw9516. doi: 10.1126/sciadv.aaw9516
S.Z. Sun, C. Romano, R. Martin, J. Am. Chem. Soc. 141 (2019) 16197–16201. doi: 10.1021/jacs.9b07489
S. Plunkett, C.H. Basch, S.O. Santana, M.P. Watson, J. Am. Chem. Soc. 141 (2019) 2257–2262. doi: 10.1021/jacs.9b00111
J. Wang, M.E. Hoerrner, M.P. Watson, D.J. Weix, Angew. Chem. Int. Ed. 59 (2020) 13484–13489. doi: 10.1002/anie.202002271
Y. Jin, J. Wu, Z. Lin, Y. Lan, C. Wang, Org. Lett. 22 (2020) 5347–5352. doi: 10.1021/acs.orglett.0c01592
J.T.M. Correia, V.A. Fernandes, B.T. Matsuo, et al., Chem. Commun. 56 (2020) 503–514. doi: 10.1039/C9CC08348K
J. Fu, W. Lundy, R. Chowdhury, et al., ACS Catal. 13 (2023) 9336–9345. doi: 10.1021/acscatal.3c01939
J. Wang, L.E. Ehehalt, Z. Huang, et al., J. Am. Chem. Soc. 145 (2023) 9951–9958. doi: 10.1021/jacs.2c11552
J.L. Douthwaite, R. Zhao, E. Shim, et al., J. Am. Chem. Soc. 145 (2023) 10930–10937. doi: 10.1021/jacs.2c11563
A. Noble, S.J. McCarver, D.W.C. MacMillan, J. Am. Chem. Soc. 137 (2015) 624–627. doi: 10.1021/ja511913h
J. Luo, J. Zhang, ACS Catal. 6 (2016) 873–877. doi: 10.1021/acscatal.5b02204
H. Huang, X. Li, C. Yu, et al., Angew. Chem. Int. Ed. 56 (2017) 1500–1505. doi: 10.1002/anie.201610108
Y. Hioki, M. Costantini, J. Griffin, et al., Science 380 (2023) 81–87. doi: 10.1126/science.adf4762
M. Gao, D. Sun, H. Gong, Org. Lett. 21 (2019) 1645–1648. doi: 10.1021/acs.orglett.9b00174
Y. Ye, H. Chen, J.L. Sessler, H. Gong, J. Am. Chem. Soc. 141 (2019) 820–824. doi: 10.1021/jacs.8b12801
P. Guo, K. Wang, W.J. Jin, et al., J. Am. Chem. Soc. 143 (2021) 513–523. doi: 10.1021/jacs.0c12462
X. Pang, X.Z. Shu, Chin. J. Chem. 41 (2023) 1637–1652. doi: 10.1002/cjoc.202200769
K.M.M. Huihui, J.A. Caputo, Z. Melchor, et al., J. Am. Chem. Soc. 138 (2016) 5016–5019. doi: 10.1021/jacs.6b01533
L. Huang, A.M. Olivares, D.J. Weix, Angew. Chem. Int. Ed. 56 (2017) 11901–11905. doi: 10.1002/anie.201706781
J. Wang, B.P. Cary, P.D. Beyer, S.H. Gellman, D.J. Weix, Angew. Chem. Int. Ed. 58 (2019) 12081–12085. doi: 10.1002/anie.201906000
Z. Li, K.F. Wang, X. Zhao, et al., Nat. Commun. 11 (2020) 5036. doi: 10.1038/s41467-020-18834-6
L.M. Kammer, S.O. Badir, R.M. Hu, G.A. Molander, Chem. Sci. 12 (2021) 5450–5457. doi: 10.1039/D1SC00943E
D.C. Salgueiro, B.K. Chi, I.A. Guzei, P. García-Reynaga, D.J. Weix, Angew. Chem. Int. Ed. 61 (2022) e202205673. doi: 10.1002/anie.202205673
E.M. DeCicco, S. Berritt, T. Knauber, S.B. Coffey, J. Hou, J. Org. Chem. 88 (2023) 12329–12340. doi: 10.1021/acs.joc.3c01072
Y. Gao, B. Zhang, J. He, P.S. Baran, J. Am. Chem. Soc. 145 (2023) 11518–11523. doi: 10.1021/jacs.3c03337
A. Schoenberg, I. Bartolet, R.F. Heck, J. Org. Chem. 39 (1974) 3318–3326. doi: 10.1021/jo00937a003
W. Wu, W. Su, J. Am. Chem. Soc. 133 (2011) 11924–11927. doi: 10.1021/ja2048495
T. Ueda, H. Konishi, K. Manabe, Angew. Chem. Int. Ed. 52 (2013) 8611–8615. doi: 10.1002/anie.201303926
B. Zhao, R. Shang, W.M. Cheng, Y. Fu, Org. Chem. Front. 5 (2018) 1782–1786. doi: 10.1039/C8QO00253C
S. Klaus, H. Neumann, A. Zapf, et al., Angew. Chem. Int. Ed. 45 (2006) 154–158. doi: 10.1002/anie.200502697
A. Brennfãhrer, H. Neumann, M. Beller, Synlett 16 (2007) 2537–2540.
A.G. Sergeev, A. Spannenberg, M. Beller, J. Am. Chem. Soc. 130 (2008) 15549–15563. doi: 10.1021/ja804997z
S. Korsager, R.H. Taaning, T. Skrydstrup, J. Am. Chem. Soc. 135 (2013) 2891–2894. doi: 10.1021/ja3114032
K. Natte, A. Dumrath, H. Neumann, M. Beller, Angew. Chem. Int. Ed. 53 (2014) 10090–10094. doi: 10.1002/anie.201404833
Z. Liu, Z. Yang, B. Yu, et al., Org. Lett. 20 (2018) 5130–5134. doi: 10.1021/acs.orglett.8b02027
D. Liu, K. Yang, D. Fang, et al., Angew. Chem. Int. Ed. 62 (2023) e202213686. doi: 10.1002/anie.202213686
C.T. Wang, P.Y. Liang, M. Li, et al., Angew. Chem. Int. Ed. 62 (2023) e202304447. doi: 10.1002/anie.202304447
J. Sheng, X. Cheng, CCS Chem. 6 (2024) 230–240. doi: 10.31635/ccschem.023.202302835
J. Choi, P. Martín-Gago, G.C. Fu, J. Am. Chem. Soc. 136 (2014) 12161–12165. doi: 10.1021/ja506885s
L. Lombardi, A. Cerveri, R. Giovanelli, et al., Angew. Chem. Int. Ed. 61 (2022) e202211732. doi: 10.1002/anie.202211732
G. Bertoli, Á.M. Martínez, J.F. Goebel, et al., Angew. Chem. Int. Ed. 62 (2023) e202215920. doi: 10.1002/anie.202215920
R.F. Turro, J.L.H. Wahlman, Z.J. Tong, et al., J. Am. Chem. Soc. 145 (2023) 14705–14715. doi: 10.1021/jacs.3c02649

Scheme 1 Nickel-catalyzed reductive cross coupling via radical intermediates for the formation of C–C bonds.

Scheme 2 Scope of substrates. Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol, 1.5 equiv.), NiBr2.bpy (10 mol%), Zn (3 equiv.) and KI (1 equiv.) in DMA (1 mL) at room temperature (25 ℃) for 8 h under a nitrogen atmosphere. Isolated yields. a With TBAI (0.2 mmol), Py (0.4 mmol) instead of KI in dioxane: DMA (4:1) at 35 ℃. b At 10 ℃. c 2 h.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:




 下载:
下载: