Department of Neurosurgery and Institute of Neurosurgery, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University, Chengdu 610041, China
b.
West China School of Basic Medical Sciences&Forensic Medicine, Sichuan University, Chengdu 610041, China
c.
Department of Hematology, West China Hospital, Sichuan University, Chengdu 610041, China
xianggao@scu.edu.cn (X. Gao). 1 These authors contributed equally to this work.
Received Date:
29 January 2024 Accepted Date:
14 March 2024 Revised Date:
13 March 2024 Available Online:
15 September 2024
Abstract:
FMS-like tyrosine kinase 3 (FLT3) is a viable and important therapeutic target for acute myeloid leukemia (AML). FLT3 internal tandem duplication (FLT3-ITD) mutations have been identified in approximately 30% of AML patients, and are associated with unfavorable prognosis, higher risk of relapse, drug resistance, and poor clinical outcome. Even FLT3 inhibitors have demonstrated promising efficacy, they cannot cure AML or even significantly extend the lives of patients with FLT3-ITD mutations. This is partly because of poor water solubility, insufficient membrane penetration and short half-life of small molecule inhibitors. Besides, the presence of enzymes like CYP3A4 in bone marrow accelerate the elimination and metabolism of FLT3 inhibitors, resulting in low plasma concentrations and side effects. Here we report the erythrocyte membrane-camouflaged FLT3 inhibitor nanoparticles to enhance FLT3-ITD AML treatment. Briefly, we physically coextruded red blood cell (RBC) membrane vesicles with nanoparticles derived from FLT3 inhibitor F30 to obtain F30@RBC-M, which exhibited comparable potent FLT3-ITD inhibitory effects compared to free F30in vitro, while displaying a higher potent antitumor efficacy in xenograft models due to the prolonged circulation properties. Furthermore, administration of F30@RBC-M significantly extended the survival of mice in a transplanted mouse model than F30 free drug. These findings suggest that RBC membrane-coated nanoparticles derived from FLT3 inhibitors hold promise as a tool to enhance the therapeutic efficacy to treat FLT3-ITD AML.
Acute myeloid leukemia (AML) is a complex hematological malignancy characterized by abnormal differentiation and uncontrolled proliferation of immature myeloid cells [1,2], which accounts for approximately 60%–80% of adult acute leukemia. FMS-like tyrosine kinase 3 (FLT3) is a receptor of tyrosine kinase that is expressed in hematopoietic stem/progenitor cells (HSC) in the bone marrow and plays a critical role in promoting the survival, proliferation, and differentiation of multipotent HSC cells [3–5]. FLT3 is overexpressed in leukemic cells and its activation FLT3 ligand can promote cell growth and inhibit apoptosis. It is found that approximately 30% of AML patients carry FLT3 mutations, which are associated with higher incidence of relapse and shorter overall survival [6–8]. FLT3 internal tandem duplication (FLT3-ITD) mutation in the juxtamembrane (JM) domain is the most frequent one that occurs in approximately 25% of AML patients and point mutations in the kinase domain accounts for around 5% [9–11]. FLT3-ITD mutation acts as a driven factor that is associated with substantial adverse outcomes and contributes to a greater leukemic burden in AML, and the infrequently occurring point mutation in the kinase domain is linked to acquired resistance, relapse, and reduced overall survival rates [12,13]. These mutations lead to the constitutive activation of the FLT3 receptor and subsequent activation of its downstream signaling pathways [14], including Janus kinase/signal transducers and activators of transcription 5 (JAK/STAT5), phosphoinositide 3-kinases/protein kinase B (PI3K/AKT), and mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK/ERK) [10,15–18]. These mutations ultimately result in increased proliferation and decreased apoptosis of AML blasts. Due to the overexpression and mutation of FLT3 receptor in leukemic blasts, FLT3 is a key therapeutic target for AML, and FLT3 inhibitors against FLT3 kinase activity has been proposed and evaluated as an effective therapeutic approach for the treatment of AML.
Despite numerous FLT3 inhibitors have been developed and demonstrated promising results in clinical trials, they are inadequate in achieving complete remission or significantly extending the overall survival rate of AML patients [19–21]. For instance, the first generation of FLT3 tyrosine kinase inhibitors are multi-kinase inhibitors, including sorafenib [22–24], midostaurin [25–28], lestaurtinib [29,30], tandutinib [31–33] and dovitinib [34], displayed broad inhibition of various kinase targets and produced undesired inhibitory efficacy in the treatment of AML. Besides, due to less specificity and weak inhibitory activity against FLT3 kinase, poor tolerability, suboptimal potency, and/or resistance mechanisms, the first generation of FLT3 inhibitors have low efficacy and high toxicity associated with their high dose usage in clinic, which limit their use in AML treatment or combination therapies. The second generation of FLT3 inhibitors, including quizartinib [35–38], gilteritinib [39–41] and crenolanib [42], displayed improved selectivity for FLT3 and higher inhibitory activity against this target. Gilteritinib has already been approved for the treatment of relapsed/refractory AML patients with FTL3-ITD [43]. However, the Phase Ⅲ ADMIRAL Trial results showed that among the AML patients, who received this treatment, only 37% survived longer than one year [9,26]. In addition, despite their improved specificity for FLT3 and higher potency, the secondary FIT3 inhibitors still exhibit off-target effects on other kinases, leading to side effects, including diarrhea (37%), anemia (34%), increased aspartate aminotransferase (26%), grade 3 gratrointestinal toxicities, fluid retention and even myelosuppression and hair depigmentation when higher doses of quizatinib were used [44–48]. These side effects limit the maximum dose usage, and subsequently, induce insufficient drug concentrations reaching target cells.
One of the most important reasons for the unideal therapeutic effects and side effects of current FLT3 inhibitors is poor pharmacokinetics properties, including poor water solubility, inadequate membrane penetration properties and low plasma concentration. Furthermore, FLT3 inhibitors with suboptimal pharmacokinetics properties appeal to have a reduced bioavailability and a shorter half-life, leading to rapid elimination and metabolism in plasma. It becomes necessary to upregulate the dosage or increase of administration frequency in order to achieve effective plasma concentrations. However, higher plasm concentration may bring with a higher risk of inducing drug resistance and off-target toxicity [49,50]. On the other hand, despite the optimized pharmacodynamic and pharmacokinetic profiles of the second generation FLT3 inhibitors, such as quizartinib, they induce limited apoptosis and low rate of clearance of AML blasts in the bone marrow microenvironment. This is because of high concentrations of CYP3A4 enzymes present in bone marrow stromal cells which result in the metabolic breakdown of FLT3 inhibitors and provide chemoprotection to FLT3-ITD mutant AML blasts [39,51–53]. Furthermore, due to the rapid movement of AML blasts in the bloodstream, the penetration of drugs through the cell membrane and into the target cells is reduced. Consequently, an adequate FLT3 inhibitor concentration required for therapeutic effects may not be achieved. As a result, when these FLT3 inhibitors are administered as a single agent, their weak inhibitory activity may lead to clinically unimpressive outcomes. Additionally, the suboptimal pharmacodynamic and pharmacokinetic properties of these inhibitors have hindered the development and clinical use of novel potential potent FLT3 targeting candidates [54,55]. Altogether, the drawbacks of current FLT3 inhibitors, including inappropriate pharmacokinetics, solubility, and biodistribution as well as limited selectivity and a lack of effectiveness, challenge us to design and develop better therapeutic approaches for AML patients with FLT3-ITD.
Nanoparticle-based drug delivery systems have been successfully applied in both experiments and clinic to improve the efficacy and reduce side effects of small molecule compounds/drugs [56]. The red blood cell (RBC)-membrane-coated biomimetic nanoparticle delivery system represents a novel and promising approach to extend therapeutic molecules' blood circulation time and enhance drugs therapeutic effects in vivo, which was initially described by Zhang et al. in 2011 [57]. This strategy involves the physical extrusion of the RBC-membrane-derived vesicles and poly(lactic-co-glycolic acid) (PLGA) nanoparticles to obtain the bilayer RBC membrane-camouflaged polymeric nanoparticles.
The RBC-encapsulated biomimetic nanoparticles combined the natural advantages of the RBC membrane as the outer shell and the properties of artificial inner core material. To start with, the RBC membrane is an ideal source for fabricating biomimetic nanoparticles due to the abundance of RBCs in blood circulation and their ease of isolation from donors. Secondly, RBC membrane possesses inherent cargo transportation advantages since it transport oxygen and other nutrients in the blood circulation throughout the body. More importantly, the presence of numerous protein markers, such as CD47, on the RBCs surface act as "don't-eat-me" signals, preventing phagocytosis of the nanoparticles by macrophage cells. Hence, nanoparticles that coated with RBCs membrane are capable of achieving prolonged circulation and an extended half-life. Additionally, RBC membrane-coated nanoparticles can effectively address specific challenges in drug delivery, such as limited water solubility, poor biocompatibility, or notable adverse effects. Therefore, RBC membrane-coated biomimetic nanoparticles have been demonstrated as a proficient drug delivery system, exhibiting enhanced delivery efficiency of anticancer drugs and improved antitumor activity in comparison to free drugs. This technology has been widely applicated in drug delivery, cancer treatment, vascular disease management, immune modulation, and detoxification, etc. [58–61].
Therefore, we hypothesize that the utilization of biomimetic strategy involving the encapsulation of FLT3 inhibitors using RBM membranes could effectively extend the systematic circulation time and half-life of FLT3 inhibitors, thus overcoming the limitations associated with poor pharmacokinetic properties, achieving effective drug delivery and enhancing therapeutic efficacy against AML.
We previously designed a novel FLT3 inhibitor, compound F30, which showed potent FLT3 inhibitory activity in FLT3-ITD positive AML cells, including MV4-11 and MOLM-13 [62]. Here, we firstly synthesized compound F30 (Fig. 1a). And then we produced F30@RBC-M. Next, the particles sizes of nanoparticle cores, RBC-M vesicles and F30@RBC-M were detected by using dynamic light scattering (DLS), whose average diameter was 31, 128, 116 nm, respectively (Figs. 1b and c). Furthermore, transmission electron microscopy (TEM) analysis was applied to detect the morphology of the particles. Samples were negatively stained with phosphotungstic acid. As displayed in Fig. 1d, the image exhibited a spherical shell-core structure as expected, and the particle's diameter was approximately 120 nm, which is similar with that detected using DLS. At last, the surface zeta potential was assessed using laser scattering microscopy (Fig. 1e). Results showed that the zeta potential of nanoparticle cores, RBC-M vesicles and F30@RBC-M was −18.91, −23.57, −31.92 mV, respectively. These results indicate that we have successfully generated F30@RBC-M particles.
Figure 1
Figure 1.
Synthesis of FLT3 inhibitor F30 and structural characterization of F30@RBM-M. (a) The synthesis of FLT3 inhibitor F30. (b) Size intensity curves of F30@RBC-M, RBC-M vesicles and nanoparticle cores. (c) Particle sizes of F30@RBC-M, RBC-M vesicles, nanoparticle cores. The sizes were measured using DLS. (d) TEM image of F30@RBC-M. The white solid core is F30 nanoparticles, the spherical shell is the RBC-M vesicle, the samples were negatively stained with uranyl acetate. (e) Zeta potential of F30@RBC-M, RBC-M vesicles and nanoparticle cores. Bars represent means ± SD (n = 3).
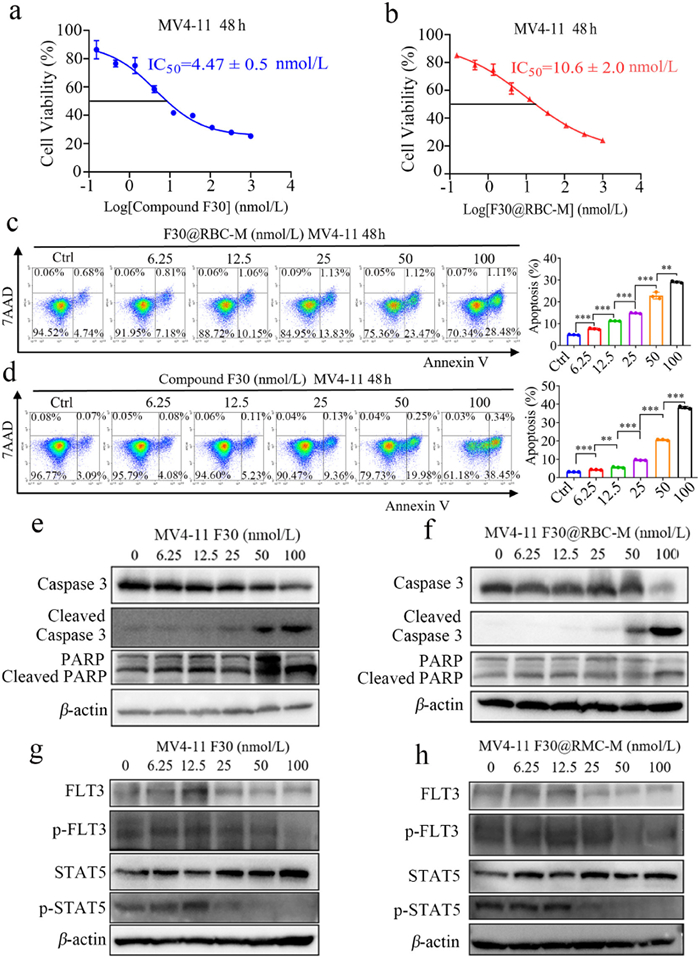
Next, we evaluated antiproliferation activity of F30@RBC-M in vitro against MV4-11 and MOLM-13 cells by MTS assay. As shown in Figs. 2a and b and Fig. S3 (Supporting information), after treatment for 48 h, both F30 and F30@RBC-M dose-responsively decreased the cell viability of MV4-11 and MOLM-13 cells, with the 50% growth inhibition concentration (IC50) values were 4.47 ± 0.5 and 10.6 ± 2 nmol/L, respectively for MV4-11 cells, 8.36 ± 1.05 and 15.44 ± 2.57 nmol/L, respectively for MOLM-13 cells. It has to be noted that the IC50 values of F30@RBC-M for MV4-11and MOLM-13 cells were higher than those of F30. It is because that it takes time for F30@RBC-M to release compound F30 from the RBC membrane and mPEG-PCL nanoparticles. We detected the antiproliferative effect of F30@RBC-M and compound F30 after 48 h treatment. However, compared with the free compound F30, the incubation time of compound F30 in F30@RBC-M was actually shorter than that of free compound F30. As expected, the RBC-M coating did not show statistically significant decrease of inhibitory reduction. Additionally, we also assessed antiproliferative activity of compound F30 and F30@RBC-M using CellTrace CFSE Cell Proliferation Kit. CFSE is a fluorescent prob that can be used to label live cells and is commonly used to investigate cells proliferation. As shown in Fig. S4 (Supporting information), compound F30 or F30@RBC-M displayed significant antiproliferative activity in a dose-dependent manner in MV4-11 and MOLM-13 cells.
Figure 2
Figure 2.
Anti-AML cell activity of compound F30 and F30@RBC-M. (a, b) The anti-proliferation activity of test compounds against MV4-11 cells. (c, d) Flow cytometry analysis of compound F30 or F30@RBC-M induced MV4-11 cells apoptosis. The cells were incubated with compound F30 or F30@RBC-M at indicated concentrations for 48 h, and the assessment was conducted following standard protocols. (e, f) Dose-dependent manner of compound F30 and F30@RBC-M induced apoptosis of MV4-11 cells. Cells were exposed to compound F30 or F30@RBC-M at indicated concentrations for 48 h. (g, h) Western blot analysis of the effects of compound F30 and F30@RBC-M on FLT3, STAT5 and p-STAT5 in MV4-11 cells. Data are presented as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
The effects of F30 and F30@RBC-M induced apoptosis on MV4-11 and MOLM-13 cells were evaluated using a flow cytometer. MV4-11 and MOLM-13 cells were treated with F30 free drug or F30@RBC-M at indicated concentrations for 48 h, which were then incubated with PE Annexin V in a buffer containing 7-amino-actinomycin (7-AAD). We found that both F30 free drug and F30@RBC-M induced apoptosis of MV4-11 and MOLM-13 cells in a dose-dependent manner (Figs. 2c and d, Fig. S5 in Supporting information). The MV4-11 apoptotic rate induced by F30 at 50 and 100 nmol/L was 24.5% and 29.6%, and F30@RBC-M incubation exhibited similar results in both MV4-11 and MOLM-13 cells, suggesting RBC-M coating did not significantly affect F30 induced apoptosis of MV4-11 and MOLM-13 cells.
To further explore the molecular mechanisms underlying F30@RBC-M-induced apoptosis in MV4-11 cells, we subsequently detected the activation of caspase 3 and cleavage of poly(ADP-ribose) polymerase (PARP). The results showed that both F30@RBC-M and F30 obviously increased the expression of cleaved caspase 3 in a concentration dependent manner (Figs. 2e and f and Fig. S6 in Supporting information). In addition, F30@RBC-M treatment also obviously increased expression levels of cleaved PARP (Figs. 2e and f, Fig. S6). Caspase 3 exerts a crucial role in cells apoptosis. These results indicate that F30@RBC-M may activate caspase 3 to increase the cleavage form of PARP to induce MV4-11 cell apoptosis. Afterwards, we assessed the effects of F30@RBC-M on FLT3 and its downstream signaling pathway in both MV4-11 and MOLM-13 cells. Cells were treated with different concentrations of F30@RBC-M for 48 h, and Western blot analysis was then used to evaluate the phosphorylation status of FLT3 and STAT5. As shown in Figs. 2g and h, both F30@RBC-M and F30 exhibited a dose-dependent inhibition of p-FLT3 and its downstream p-STAT5. It has to be noted that p-FLT3 and p-STAT5 were completely inhibited by 50 nmol/L of F30@RBC-M. A similar result was also observed in MOLM-13 cells (Fig. S7 in Supporting information). It has been shown that FLT3-ITD triggers constitutive STAT5 phosphorylation, and STAT5 exerts a crucial role in FLT3-ITD signal transduction for cell expansion and survival. Our results indicate that F30@RBC-M can prevent MV4-11 and MOLM-13 cell proliferation by inhibiting FLT3 and its downstream STAT5 phosphorylation. In summary, the above experiments demonstrate that F30 has a strong kinase inhibitory effect on FLT3-ITD AML cells, and the encapsulation of the RBC membrane does not significantly reduce its in vitro inhibitory and antiproliferative activity.
Accumulating evidence has revealed that self-recognition and the ability of homologous RBCs to avoid macrophages phagocytosis is arbitrated by numerous membrane proteins residing on the cell surface. Specifically, CD47 is such a "don't eat me" marker to escape immune clearance and is a crucial determinant for achieving prolonged circulation time of nanoparticles that encapsulated with RBCs. Therefore, western blot analysis was applied to ascertain the exist of CD47 in RBC-M shell. As shown in Fig. S8 (Supporting information), CD47 was still present at the red blood surface at a similar density to that of native RBCs.
In order to evaluation the in vivo distribution and extended circulation profiles of RBC-M NPs, we established a MV4-11 xenograft model. All animal procedures were conducted in accordance with the guidelines and regulations approved by the Animal Experimental Ethics Committee of State Key Laboratory Biotherapy (SKLB), Sichuan University (Chengdu, China). Animal welfare is adequately guaranteed, with efforts made to provide adequate housing, nutrition, and veterinary care, and to minimize animal stress and pain.
The MV4-11 xenograft model was established via subcutaneously injecting MV4-11 cells into immunodeficient NCG female mice on their right flank. Once the tumor size reached 400–500 mm3, mice were randomly divided into three groups and were intravenously administered Ce6@RBC-M, chlorin e6 (Ce6) nanoparticles and Ce6 free compound at a dose of 5 mg/kg. Imaging assays were used to investigate the tissue distribution of Ce6 at different time points, including 1, 4, 8, 12, and 24 h (Fig. S9a in Supporting information). Twenty-four hours after administration, the mice were sacrificed, tumors, the heart, liver, spleen, lung and kidney were analyzed. As shown in Fig. S9a, the fluorescence intensity of Ce6 was found to be approximately equivalent among the three groups 1 h after administration, which indicates that there is no significant difference in the initial dosage administration. However, 24 h after administration, the mice in the Ce6@RBC-M group displayed statistically higher chemiluminescence intensity than groups treated with Ce6 or Ce6 nano (Fig. S9b in Supporting information), which illustrated that RBC membrane coating can actually prolonged circulation time of its coated nanoparticles. Furthermore, major organs including the heart, liver, spleen, lung and kidney in Ce6@RBC-M group also displayed significantly higher chemiluminescence intensity than that in the rest of two groups (Figs. S9c and d in Supporting information). Besides, tumors in Ce6@RBC-M group displayed significantly higher chemiluminescence signals compared to Ce6 nano group and Ce6 group (Figs. S9e and f in Supporting information). Overall, treatment of mice with Ce6@RBC-M resulted in significantly higher fluorescence intensity in major organs and tumors compared to free Ce6 and Ce6 nano. This indicates that the coating of Ce6 with RBCs prolonged its circulation and prevented its rapid metabolism and elimination, thereby achieving a longer duration of therapeutic effect.
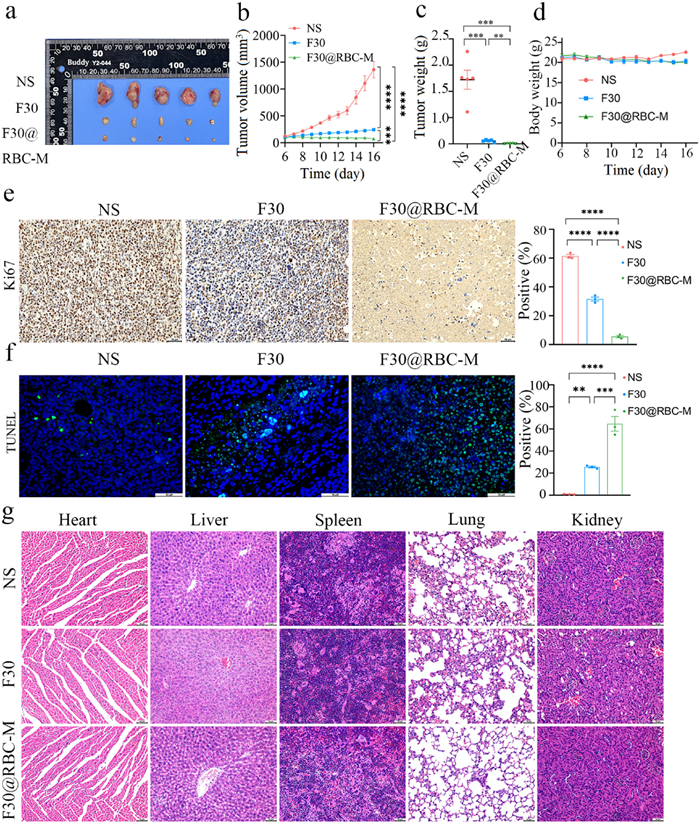
To further evaluate the in vivo antitumor efficacy, we built a MV4-11 xenograft model by subcutaneously injecting MV4-11 cells. As shown in Figs. 3a and b, over the course of 11 days of continuous treatment, the tumor volume in the vehicle group grew to an average size of 1362 mm3. While the administration of compound F30 at a dosage of 40 mg/kg exhibited a remarkable antitumor effect, with a tumor growth inhibition (TGI) of approximately 83%. Furthermore, the F30@RBC-M treatment induced a significant more potent in vivo antitumor inhibition than F30 treatment, resulting in a TGI of 95% (Figs. 3b and c). A similar result was also observed in the MOLM-13 xenograft model (Fig. S10 in Supporting information). The results indicate that compound F30 is a potent FLT3 inhibitor, and F30@RBC-M displayed stronger anti-AML activity in vivo. Figs. 3e and f showed that F30@RBC-M was more potent to induce apoptosis of tumor cells and inhibit cell proliferation that compound F30. A similar result was also observed in the MOLM-13 xenograft model (Figs. S10–S12 in Supporting information).
Figure 3
Figure 3.In vivo anti-AML efficacy of F30@RBC-M in the MV4-11 xenograft model. (a, b) Tumor volume. Mice were administrated with normal saline (NS), F30 free drug or F30@RBC-M at the dosage of 40 mg kg−1 day−1. Tumor volume was monitored every day. (c) Tumor weight. (d) Mice body weight. (e) Ki67 staining of tumor tissues. (f) Terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling (TUNEL) staining of tumor tissues. (g) Hematoxylin and eosin staining of major organs after treatment with normal saline, F30 free drug or F30@RBC-M. Data are presented as mean ± SEM (n = 3). **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bar: 50 µm. NS, normal saline.
Additionally, hematoxylin and eosin (H&E) staining revealed no obvious morphological changes (Fig. 3g, Fig. S13 in Supporting information), suggesting that F30@RBC-M treatment does not induce apparent toxicity to normal cells from the above organs. The blood biochemical assays showed that important biochemical indicators such as ALT, AST, ALP, LDL and ALB were within the normal range for the groups (Fig. S14 in Supporting information), which further demonstrated that F30@RBC-M treatment does not induce severe hepatic or renal toxicity.
At last, to further evaluate the anti-AML efficacy of F30@RBC-M in vivo, we established an intravenous-transplanted mouse model by injecting MV4-11 cells transduced with luciferase intravenously. Leukemic cell engraftment was evaluated using bioluminescence imaging (BLI). BLI analysis showed the signal intensities of all groups were comparable on day 4 (Figs. 4a and b, Fig. S15 in Supporting information), indicating no significant differences in the number of inoculated cells among the groups, and that their growth rates were roughly equivalent. Then, on day 5, by tail vein injection, one third of the mice were administered compound F30 (40 mg/kg) daily, another one third were treated with F30@RBC-M (40 mg/kg) daily, and the remaining were treated with normal saline, which served as the vehicle control. As illustrated in Fig. 4b and Fig. S15, on day 24, a notable enhancement of the total bioluminescent signal was detected in the control group; nevertheless, the F30@RBC-M group exhibited a statistically significant weaker BLI signal compared with the group treated with compound F30. Over the whole course of treatment, mice in the group administered with F30@RBC-M exhibited an increase in survival rate compared to control groups (Fig. 4c). The results indicate that the administration of F30@RBC-M enhanced the anti-AML efficacy of FLT3 inhibitor F30, resulting in a reduced leukemic burden. Finally, the toxicity of F30@RBC-M in the transplanted mouse model was also assessed. As shown in Fig. 4d, no significant body weight loss or obvious adverse effects were observed among all treatment groups throughout the treatment.
Figure 4
Figure 4.
Antileukemic effects of F30@RBC-M in a MV4-11 luciferase-transduced transplanted mouse model. (a) Fluorescence images of all groups on day 4, 10, 18, and 24. (b) Quantification of the bioluminescent signal intensity. (c) Survival of mice. (d) Changes of mice body weight after treatment. Data are presented as mean ± SEM (n = 5). *P < 0.05, **P < 0.01. ns, not significant.
In summary, we successfully fabricate the RBC membrane coated nanoparticles F30@RBC-M. The RBC-M coating endows compound F30 with prolonged circulation and prevents phagocytosis of the nanoparticles by macrophage cells. RBC-M coating does not induce a decrease in the antiproliferative activity of compound F30. F30@RBC-M treatment exhibited significantly more potent antitumor activity in xenograft models, enhancing anti-AML efficacy in the transplanted model without producing remarkable adverse effects. The findings demonstrate that RBC membrane-coated FLT3 inhibitor biomimetic nanoparticles could potentially enhance the therapeutic efficacy in the treatment of FLT3-ITD acute myeloid leukemia.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 32222046, China), the Sichuan Science and Technology Program (Nos. 2022NSFSC0823, 2023NSFSC193, 2022NSFSC0793, China) and the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (No. ZYJC21022, China).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109779.
[1]
V. Madan, H.P. Koeffler, Haematologica 106 (2021) 26–38.
Figure 1
Synthesis of FLT3 inhibitor F30 and structural characterization of F30@RBM-M. (a) The synthesis of FLT3 inhibitor F30. (b) Size intensity curves of F30@RBC-M, RBC-M vesicles and nanoparticle cores. (c) Particle sizes of F30@RBC-M, RBC-M vesicles, nanoparticle cores. The sizes were measured using DLS. (d) TEM image of F30@RBC-M. The white solid core is F30 nanoparticles, the spherical shell is the RBC-M vesicle, the samples were negatively stained with uranyl acetate. (e) Zeta potential of F30@RBC-M, RBC-M vesicles and nanoparticle cores. Bars represent means ± SD (n = 3).
Figure 2
Anti-AML cell activity of compound F30 and F30@RBC-M. (a, b) The anti-proliferation activity of test compounds against MV4-11 cells. (c, d) Flow cytometry analysis of compound F30 or F30@RBC-M induced MV4-11 cells apoptosis. The cells were incubated with compound F30 or F30@RBC-M at indicated concentrations for 48 h, and the assessment was conducted following standard protocols. (e, f) Dose-dependent manner of compound F30 and F30@RBC-M induced apoptosis of MV4-11 cells. Cells were exposed to compound F30 or F30@RBC-M at indicated concentrations for 48 h. (g, h) Western blot analysis of the effects of compound F30 and F30@RBC-M on FLT3, STAT5 and p-STAT5 in MV4-11 cells. Data are presented as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3In vivo anti-AML efficacy of F30@RBC-M in the MV4-11 xenograft model. (a, b) Tumor volume. Mice were administrated with normal saline (NS), F30 free drug or F30@RBC-M at the dosage of 40 mg kg−1 day−1. Tumor volume was monitored every day. (c) Tumor weight. (d) Mice body weight. (e) Ki67 staining of tumor tissues. (f) Terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling (TUNEL) staining of tumor tissues. (g) Hematoxylin and eosin staining of major organs after treatment with normal saline, F30 free drug or F30@RBC-M. Data are presented as mean ± SEM (n = 3). **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bar: 50 µm. NS, normal saline.
Figure 4
Antileukemic effects of F30@RBC-M in a MV4-11 luciferase-transduced transplanted mouse model. (a) Fluorescence images of all groups on day 4, 10, 18, and 24. (b) Quantification of the bioluminescent signal intensity. (c) Survival of mice. (d) Changes of mice body weight after treatment. Data are presented as mean ± SEM (n = 5). *P < 0.05, **P < 0.01. ns, not significant.


 DownLoad:
DownLoad:


 下载:
下载:





 下载:
下载:
