School of Chemistry and Chemical Engineering, Key Laboratory of Jiangxi Province for Environment and Energy Catalysis, Nanchang University, Nanchang 330031, China
b.
School of Resources and Environment, Key Laboratory of Poyang Lake Environment and Resource Utilization, Ministry of Education, Nanchang University. Nanchang 330031, China
Received Date:
19 October 2023 Accepted Date:
11 March 2024 Revised Date:
29 February 2024 Available Online:
15 December 2024
Abstract:
Efficient yield of 1O2 determines the photocatalytic degradation rate of antibiotics, but the regulatory mechanism for 1O2 selective generation in O2 activation is still lacking exploration. Herein, oxygen vacancy (OV) modification strategy of MIL-125 was successfully practiced to promote the selective generation of 1O2. Multiple characterizations including extended X-ray absorption fine structure (EXAFS) and electron paramagnetic resonance spectra (EPR) confirmed the formation of oxygen vacancy in OV-MIL-125. The synthesized OV-MIL-125 exhibited greatly enhanced 1O2 selective (~90%) and antibiotics removal rate in water with high mineralization rate. Dynamics analysis of excitons by transient-steady state fluorescence and phosphorescence, transient absorption spectra (TAS) revealed that oxygen vacancy greatly enhanced the intersystem crossing (ISC) of singlet exciton, promoting triplet exciton generation. Density functional theoretical (DFT) calculation also proved the reduced gap of intersystem (ΔEST) and the modulated highest occupied molecular orbital (HOMO)-lowest unoccupied molecular orbital (LUMO) population which was conducive to intersystem crossing process. Calculation of transition state further confirmed the lower energy barrier for π* orbital spin flip of O2 adsorbed on OV-MIL-125. The Dexter energy transfer involving triplet annihilation dominated the O2 activation mechanism to generate 1O2 instead of the charge transfer to generate O2•− which happened in MIL-125. This study provides new thinking for photocatalytic activation of molecular oxygen and is expected to guide the design of MOF-based catalysts for water treatment.
As a kind of emerging contaminants (ECs) in water, antibiotic is seriously threating human and ecosystem health due to their biological toxicity, environmental persistence and cultivation of drug-resistant. Advanced oxidation techniques based on photocatalysis [1-3], hydrogen peroxide activation [4] and persulfate activation [5,6] has attracted much attention for antibiotic removal. Among them, activation of molecular oxygen through photocatalysis to produce reactive oxygen species (ROS) has great potential in view of the economical and efficient superiority. The species and concentration of ROS directly affect the rate of organic degradation in a typical photocatalysis process. Superoxide radical (O2•−) and singlet oxygen (1O2) are common ROS through photoactivation of oxygen molecules. The 1O2 has higher redox potential and longer lifetime compared with O2•− which endowed the more higher oxidizing activity in theory [7,8]. Compared with the nucleophilicity of O2•−, the electrophilic oxidation of 1O2 make it more likely to attack the aromatic structure, which is conducive to the mineralization of ECs. 1O2 dominated oxidation of organic matter has also been frequently demonstrated in catalytic ozonation [9], electrocatalysis [10], persulfate-based water purification [11,12], and photodynamic therapy [13-15]. The regulation of oxygen activation to selectively generate 1O2 has been be a significant research hotspot in photodegradation of organics. However, the regulatory mechanism with general applicability for 1O2 selective generation in O2 activation is still lacking exploration.
The formation of O2•− and 1O2 undergo two different mechanisms which involves charge transfer and energy transfer respectively [16]. The electron transfer from carrier (singlet exciton) formed in electronic transition to ground state oxygen (triplet state) is the classical process for O2•− generation [17]. The deexcitation caused by the recombination of photoelectron and hole lead to the very short lifetimes of singlet excitons which limits reaction rate. While, the generation of 1O2 undergoes the Dexter energy transfer process accompanied in triplet exciton-triplet oxygen annihilation (TTA) [18]. Singlet excitons transformed into triplet excitons through intersystem crossing (ISC) and the intersystem gap (ΔEST) determines the possibility of this process [19]. It's worth noting that the lifetime triplet exciton is commonly higher than singlet exciton [20,21]. In this regard, approaches to promoted ISC process is vitally important to design efficient photocatalyst based on achievement of high utilization rate of photogenerated carriers and high 1O2 yield. The conversion rate of singlet exciton to triplet exciton is closely related to the structure and electronic state of the semiconductor [22-24]. Introduction of heavy metal [25,26] or halogen [27] and designing applicable charge donor–acceptor structure were explored to accelerate the ISC of excited state [28]. However, these strategies require complicated design and synthesis.
Metal organic frameworks (MOFs) are porous coordination polymers composed of inorganic secondary building units and organic linkers. MOFs and their derived materials have been researched widely in photocatalytic degradation of organic pollutants [29-32]. The great structural adjustability provides proximal placement of the functional entities for efficient intermolecular energy transfer [33]. The photocatalytic performance of MOFs can be greatly improved by constructing Z-scheme heterostructure [34,35]. Wang et al. enhanced light response and electron transfer of NH2-MIL-125 through quantum dots modify [36] and ligand grafting [37] for effective antibacterial property. Its immense specific surface area and void ratio bring huge preferential adsorption capacity for O2 [38], which is conducive to the collision between triplet state species. Moreover, the efficient ligand to metal charge transfer (means high singlet exciton yield) [39] and antenna effect [40] leads to the high photoexcitation performance. Additionally, the available spin-orbit coupling provided by inherent metal also provide advantages to ISC enhancement [33]. With the above considerations in mind, MOFs seem ideal structure for photocatalysis triplet excitons yield and 1O2 generation.
To achieve efficient and regulable 1O2 generation with high selective for antibiotic removal, herein, a very simple oxygen vacancy (OV) modification strategy of MIL-125 was successfully practiced in molecular oxygen activation under visible light. The synthesized catalyst OV-MIL-125 exhibited greatly enhanced 1O2 selective (~90%) and antibiotics removal rate in water with high mineralization rate. The formation of OV through ligand replacement-thermal treatment of MIL-125 was detected by extended X-ray absorption fine structure (EXAFS), electron paramagnetic resonance spectra (EPR), X-ray photoelectron spectroscopy (XPS) and Raman spectra. Dynamics analysis of excitons by transient-steady state fluorescence and phosphorescence, transient absorption spectra (TAS) revealed the modulated transition modes of excited states involving enhanced intersystem crossing (ISC) in OV-MIL-125 compared with that in MIL-125. DFT calculation proved the reduced ΔEST and the modulated HOMO-LUMO population which was conducive to ISC of singlet to triplet exciton, meanwhile, the Dexter energy transfer involving annihilation between triplet exciton and ground state O2 was also increased. Calculation of transition state further confirmed the lower energy barrier for π* orbital spin flip of O2 adsorbed on OV-MIL-125. Thus, the ISC process and energy transfer dominated the O2 activation mechanism to generate 1O2 in OV-MIL-125 instead of the charge transfer to generate O2•− happened in MIL-125.
The synthesis of OV-MIL-125 went through ligand replacement-heat treatment two step reaction as illustrated in Fig. 1a and the detail experimental was provided in Texts S1 and S2 (Supporting information). The microstructure and morphology of OV-MIL-125 significantly changed compared with the original MIL-125. The synthesized OV-MIL-125 was round cake shaped with the diameter of 800 nm under SEM view and the orientation of the lattice plane changed to irregular compared with MIL-125 which closed to the polyhedron (Figs. 1b-d). The lattice plane orientation of 2%NH2-MIL-125 was in between MIL-125 and OV-MIL-125. Additionally, the OV-MIL-125 had more abundant pore structure reflected by the dispersed higher transmittance in TEM images (Figs. 1e-h). This was consisted with the N2 absorption-desorption and aperture analysis (Figs. S3a and b, Table S1 in Supporting information), the pore volume corresponding to ~4 nm was significantly increased in OV-MIL-125 which was more convenient for the mass transfer of oxygen molecules. The appearance of these pore could be attributed to the partial destruction of MOFs skeleton by defect [41]. Nevertheless, the OV-MIL-125 remained the same crystal phase of typical MIL-125 according to the XRD pattern of MIL-125 and OV-MIL-125 (Figs. S4, S5a and b in Supporting information). The HR-TEM image of OV-MIL-125 (Fig. 1i) also indicated the Ti specie existed in Ti-O cluster (8 Å × 8 Å, which was the theoretical size in MIL-125) rather than aggregation of titanium oxides which was consisted with XRD pattern. The EDS elemental mapping of OV-MIL-125 and 2%NH2-MIL-125 showed the uniform distribution of Ti, O, C, N without aggregation (Fig. 1j, Figs. S6a and b in Supporting information).
Figure 1
Figure 1.
(a) Scheme of structural evolution of OV-MIL-125 during synthesis (the grey, black, and red balls respectively present Ti, C, and O atoms). (b-d) SEM images of the MIL-125, 2%NH2-MIL-125 and OV-MIL-125. (e, f) TEM images of the MIL-125, 2%NH2-MIL-125 and OV-MIL-125. (h, i) HR-TEM images of OV-MIL-125. (j) EDS elemental mapping of OV-MIL-125.
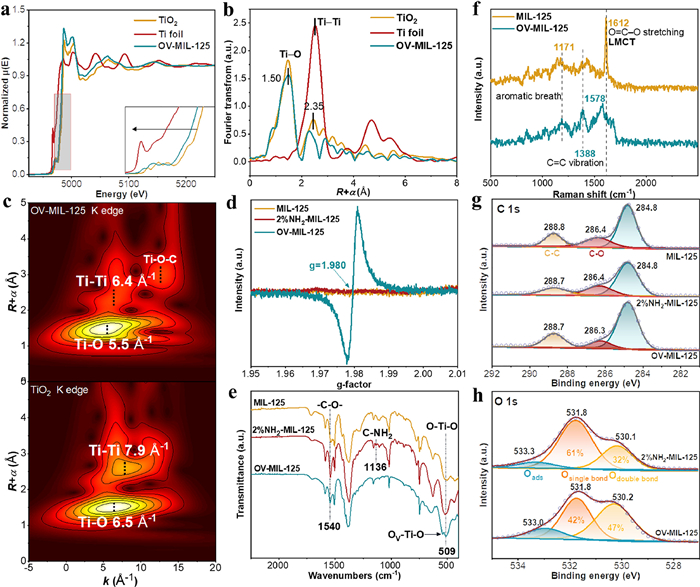
The Ti K-edge XAFS provided the accurate coordination information. The absorption edge of OV-MIL-125 shifted to lower energy compared to TiO2 (Fig. 2a), suggesting the decrease of oxidation state of Ti in OV-MIL-125. In consideration of the same oxidation state (+4) of TiO2 and MIL-125 [42], the oxygen vacancy (OV) might form around Ti atom in OV-MIL-125 which could be account for the reduction by the remaining single electrons on OV. The band at ~1.50 and ~2.35 Å in the χ(R) spectra of Ti K-edge EXAFS (Fig. 2b) corresponded to Ti–O and Ti-Ti shell, respectively. The asymmetric peak at 1.50 Å indicating two different lengths of Ti–O shell corresponded to Ti–O–Ti and Ti–O–C scattering path which could be further confirmed by the wavelet transform of Ti k3-weighted EXAFS. The additional intensity peak at ~13 Å−1 with R + α = 3.0 Å accounted for the Ti–O–C scattering path (Fig. 2c). The above results were consisted with k2-weighted fitting in k space and R space (Fig. S7 in Supporting information) and the structural parameters obtained from Ti K-edge EXAFS fittings for OV-MIL-125 was provided in Table S2 (Supporting information). The R value of 1.915, 2.403, 2.657, and 3.014 Å accounted for Ti−OI, Ti−OII, Ti−Ti and Ti−C shell, respectively. Importantly, excluding Ti−OI, the coordination numbers of Ti−OII, Ti−Ti and Ti−C were lower than the theoretical value (2,1 and 2 severally). This suggested the OV might appeared at the carboxyl connecting with Ti atom (As shown in the structure model of OV-MIL-125 in Fig. 2b).
Figure 2
Figure 2.
(a) Ti K-edge XANES spectra of OV-MIL-125. The insert shows partial enlarged detail. (b) Fourier transforms k2-weighted χ(R) spectra of Ti K-edge in OV-MIL-125. The insert shows the structure model of OV-MIL-125. (c) Wavelet transform of Ti k3-weighted EXAFS for OV-MIL-125 and TiO2. EPR spectra (d) and FTIR spectra (e) of MIL-125, 2%NH2-MIL-125 and OV-MIL-125. (f) Raman spectra of MIL-125 and OV-MIL-125. C 1s (g) and O 1s (h) XPS spectra MIL-125, 2%NH2-MIL-125 and OV-MIL-125.
Detailed chemical structure analysis confirmed the defect in OV-MIL-125 as speculated above. The EPR spectra of OV-MIL-125 appeared a very distinct symmetrical peak with the g value of 1.980 (Fig. 2d) corresponding to Ti3+ caused by the loss of adjacent O atom [43], which was consitsed with XAFS analysis. This signal was not found in MIL-125 and 2%NH2-MIL-125. The formation of oxygen vacancy was in connection with -NH2 loss according to further FTIR and Raman spectra. As shown in Fig. 2e, the peak at 1540 and 509 cm−1 corresponded to the stretching of C—O in carboxyl and bending of O–Ti–O respectively [44]. The peak splitting of O–Ti–O also accounted for the OV–Ti–O bending. A new peak at 1136 cm−1 corresponded to the stretching of C—NH2 in 2%NH2-MIL-125 and this peak decreased in OV-MIL-125 suggesting the losing of -NH2. Concomitantly, the peak of C—O decreased obviously indicating the formation of OV might derived for the O extraction by -NH2. Thermogravimetric analysis supported above conjecture for the more weight loss of 2%NH2-MIL-125 at ~300 ℃ compared with -MIL-125 (Fig. S8 in Supporting information) and C—O and C—NH2 reduced simultaneously with the increase of heat treatment temperature (Fig. S9 in Supporting information). The relative amount of OV derived from n%NH2-MIL-125 precursor reflected in EPR spectra (Fig. S10 in Supporting information) showing the 2% ligand replacement was the optimal amount for OV generation. The oxygen vacancy in OV-MIL-125 could be further confirmed by XPS and Raman spectra. The relative amount of N and O element in OV-MIL-125 respectively reduced and increased compared with 2%NH2-MIL-125 according to XPS survey and N 1s XPS (Figs. S11 and S12, Table S3 in Supporting information). The peak at ~286.4 eV (Fig. 2g) corresponding to the C in C—O bond decreased obviously in OV-MIL-125, proving the OV formed through the O spillage in C—O bond. O 1s XPS provided the proportion of O in different chemical species. The peaks at ~530, 532 and 533 eV (Fig. 2h) were ascribed to O atoms in double bond, single bond and adsorbed oxygen, respectively [43]. The distinct binding energy of Oads peak in O 1s spectra for 2%NH2-MIL-125 and OV-MIL-125 could be attributed to the different chemical environment or bonding conditions of O atom in the adsorbed O2 due to the formation of oxygen vacancy. The slight shift of binding energy of C—O bonds in C 1s spectra for OV-MIL-125 might be caused by the change in C—O bond length. This could be further confirmed by the adsorption model of O2 on 2%NH2-MIL-125 (O—O bond 1.248 Å) and OV-MIL-125 (O—O bond 1.248 Å) according to DFT calculation (Fig. S13 in Supporting information). The distinct reduce of O in single bond of OV-MIL-125 (42%) compared with 2%NH2-MIL-125 (61%) suggested the C—O or Ti-O bond breaking. Meanwhile, the formation of OV leaded to increase of chemisorbed oxygen according to the correspond peak area which would be conducive to molecular oxygen activation. Based on the above structural analysis, structures of MIL-125, 2%NH2-MIL-125 and OV-MIL-125 were optimized by DFT calculation (Fig. S2 in Supporting information) which also matched with Raman spectra. A new peak appeared at 1388 cm−1 for OV-MIL-125 (Fig. 2f) suggesting C=C formation due to O losing in O=C—O. The strong scattering at 1612 cm−1 was attributed to asymmetric stretching of -O=C—O- caused by the LMCT (ligand to metal charge transfer) [39,45]. While, this scattering frequency shifted to 1578 cm−1 and the intensity decreased significantly. This indicated that the energy transfer of excited state through non-radiative processes (intersystem crossing or internal conversion) instead of LMCT was enhanced in OV-MIL-125 due to OV formation.
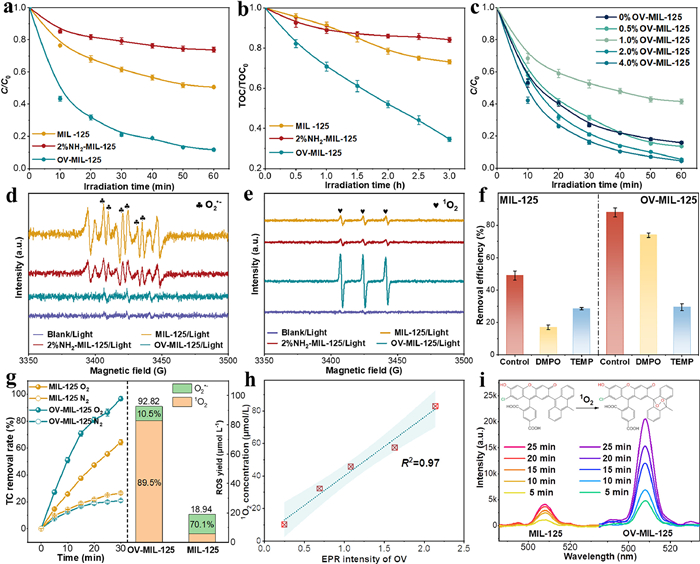
The formation of OV in OV-MIL-125 greatly improved the photocatalytic activity for antibiotic degradation. The removal rate of TC by OV-MIL-125 exceeded 78% at 30 min at reached ~90% at 60 min which was much higher than that by MIL-125 and 2%NH2-MIL-125 (Fig. 3a, the adsorption quantity of TC by different catalysts were provided in Fig. S14 in Supporting information). Importantly, the mineralization rate of TC in OV-MIL-125/Light system was quite high with the TOC removal rate exceeding 65% at 3 h (Fig. 3b). The removal rates of different target antibiotics all reached 80% within 60 min and the corresponding first order reaction constants were 0.026–0.041 min−1 (Figs. S15a-f in Supporting information) which demonstrating the wide applicability of OV-MIL-125. To confirm the key role of OV in antibiotic degradation, the photocatalytic activity of OV-MIL-125 with different OV concentration was tested (Fig. 3c). The removal rate of TC was positively correlated with the OV concentration (reflected by EPR signal in Fig. S10). The performance was relatively stable after repeated use and the removal efficiency of TC after six cycles remained 88% (Fig. S16a in Supporting information). The effect of several common anions and humic acid in water on the removal rate of TC was shown in Fig. S16b (Supporting information) and the TC removal rate was less affected by the initial pH of reaction system (Fig. S16c in Supporting information). More than 85% TC could be removed under acidic (pH 2) or alkaline (pH 10) condition. To optimize working condition of OV-MIL-125, the effects of catalyst dose and TC concentration on TC removal rate were also evaluated (Fig. S17 in Supporting information). The marginal effect of OV-MIL-125 dose for TC removal rate under 10 mg/L TC condition appeared at 300–400 mg/L. The above results exhibited the immense potential in ECs removal by OV modification of MIL-125.
Figure 3
Figure 3.
Removal rate of TC (a) and TOC removal rate (b) during TC degradation by different catalysts. (c) TC degradation rate by OV-MIL-125 with different relative concentration of oxygen vacancy. Spin-trapping EPR spectra for O2•− (d) and 1O2 (e) in different photocatalytic system. (f) Effect of DMPO and TEMP for TC removal efficiency in MIL-125 and OV-MIL-125 system. (g) 1O2 yield in MIL-125 and OV-MIL-125 systems and TC removal rate under aeration with O2 and N2. (h) The correlation between 1O2 concentration and OV concentration. (i) Time-dependent 1O2 yield in MIL-125 and OV-MIL-125 systems through SOSG fluorescence assay.
To detect the reactive oxygen species such as •OH, 1O2 and O2•− in different photoreaction system, the spin-trapping EPR was performed. The detection of O2•− and •OH were conducted after tertiary butanol and p-benzoquinone addition respectively to shield mutual influence. The population of ROS in OV-MIL-125 system changed obviously due to the participation of OV compared with that in MIL-125 and 2%NH2-MIL-125 systems. The triplet signal with six peaks in Fig. 3d was the characteristic of DMPO-O2•−. This signal was very weak in OV-MIL-125 system while was relatively strong in MIL-125 and 2%NH2-MIL-125 systems suggesting the very low O2•− concentration induced by OV-MIL-125. Inversely, the 1O2 concentration was much higher in OV-MIL-125 system compared that in MIL-125 and 2%NH2-MIL-125 systems according to the TEPM-1O2 signal intensity of three peaks with same magnetic field spacing (Fig. 3e). The DMPO-•OH signals were not detected both in MIL-125 and OV-MIL-125 systems (Fig. S18a in Supporting information). EPR detection of TEPM-1O2 at different sampling times confirmed the continuous generation of 1O2 in OV-MIL-125 system (Fig. S18b in Supporting information). To compare the actual contribution of O2•− and 1O2, DMPO and TEMP were added in MIL-125 and OV-MIL-125 systems as quenching agent for O2•− and 1O2 respectively to test their effect on TC removal rate. The removal efficiency of TC decreased from 89% to 25% after the addition of TEMP in OV-MIL-125 system which suggested that 1O2 was the dominating ROS for TC removal, while, the contribution O2•− in MIL-125 system was higher than 1O2 (Fig. 3f). The TC removal rate decreased significantly under aeration with O2 and N2, indicating the degradation effect derived from O2 activation (Fig. 3g). EPR spin quantum number quantification of O2•− and 1O2 also proved the higher ROS total quantity (92.82 µmol/L) and the higher 1O2 selectivity (89.5%) which was much higher than that in MIL-125 system (29.9%) (Fig. 3g). Moreover, the 1O2 yield by OV-MIL-125 with different OV concentration was detected which exhibiting a significant positive correlation with EPR signal intensity of OV (Fig. 3h). SOSG fluorescence assay was used to quantify the real-time concentration of 1O2 in MIL-125 and OV-MIL-125 systems (Fig. 3i), the 1O2 generation in OV-MIL-125 was much faster compared with MIL-125.
To reveal the dynamics of excitons in OV-MIL-125, series of photoelectric tests was performed which proving the pivotal role of OV in the transition of excitons to triplet state. As shown in Fig. 4a, the optical absorption property of OV-MIL-125 at visible light range was relatively higher compared with MIL-125 and 2%NH2-MIL-125. The new adsorption edge of 2%NH2-MIL-125 was attributed to introduction of impurity levels by-NH2 [46]. The formation of OV shrank band gap the apparent band gap of OV-MIL-125 to 1.91 eV (Fig. S19a in Supporting information) and decreased the electrochemical impedance (Fig. S19b in Supporting information). The shranked band gap could be further confirmed by Mott-Schottky curve and XPS valence band spectra. XPS valence band spectra (Fig. 4b) exhibited the higher valence band position (1.71 eV) of OV-MIL-125. The Mott-Schottky curve reflected the flat band which was usually more positive 0.1–0.2 eV than conduction band for n-type semiconductor. It could be inferred from Fig. 4c that the OV-MIL-125 have lower conduction band position compared with MIL-125. However, the photocurrent of OV-MIL-125 was the smallest among above catalysts (Fig. 4d). This might be due to the lowered charge carrier separation efficiency or enhanced recombination of carrier. The formation of photocurrent goes through photoexcited electron transition, electron-hole pair (charge carrier) separation and carrier migration processes. The separation of electron-hole pair is the premise of carrier migration to form current. On the other hand, the separated charge carrier can recombine which will reduce carrier migration and photocurrent. Direct recombination of electron-hole pairs leads to fluorescence-emission, while, the intersystem crossing of electron-hole pairs to triplet exiton and the deexcitation of triplet exiton leads to phosphorescence emission. Given the weaker fluorescence emission (Fig. 4g) of OV-MIL-125 which caused by carrier recombination, the photogenerated charge carriers tended to bind into excitons rather than to separate in OV-MIL-125 which could be supported by the higher slope of Mott-Schottky curve (Fig. 4b) [16,47]. The ill-defined step of absorption edge also indicated the higher exciton binding energy in OV-MIL-125 [48,49]. In contrast, the photocatalytic mechanism in MIL-125 and 2%NH2-MIL-125 tended to rely on carrier mobility (charge transfer). Since the process of O2 directly transition to 1O2 is spin-forbidden, the pretty high 1O2 production in OV-MIL-125 must undergo the energy transfer between triplet exciton and ground state O2. The transient and steady-state fluorescence (FL) and phosphorescence (pH) spectra revealed the transition from singlet exciton to triplet exciton due to the formation of OV in OV-MIL-125. The FL emission band in steady-state FL of MIL-125, 2%NH2-MIL-125 and OV-MIL-125 appeared at ~420 nm (Fig. 4g). However, the FL intensity of OV-MIL-125 was significantly weak than that of MIL-125 and 2%NH2-MIL-125. FL emission is caused by the relaxation and transition between singlet exciton (Sn+m → Sn → S0). The relative low FL of 2%NH2-MIL-125 could attributed to the introduction of impurity levels which increased the vibration relaxation. This weaker FL proved that the deexcitation mode through transitions between singlet in OV-MIL-125 was reduced and the intersystem crossing (ISC) between singlet and triplet exciton was raised. The steady-state pH also supported the above conclusion. The pH intensity of OV-MIL-125 was much higher and the pH emission band shifted to 490 nm compare with that of MIL-125 (~693 nm). pH emission is caused by the transition from triplet exciton to singlet exciton (Sn → Tn → S0).
Figure 4
Figure 4.
Optical spectroscopy (a), Mott-Schottky curve (b), XPS valence band spectra (c), photocurrent (d), transient fluorescence spectra (e), and transient phosphorescence spectra (f) of MIL-125, 2%NH2-MIL-125 and OV-MIL-125. (g) Steady-state fluorescent and phosphorescence spectra of MIL-125, 2%NH2-MIL-125 and OV-MIL-125, λ(excitation) = 325 nm. (h) Ultralow temperature fluorescence spectra of MIL-125, 2%NH2-MIL-125 and OV-MIL-125 at 2.0 K, λ(excitation) = 325 nm. λ(detection) of transient fluorescence was 425 nm.
The strong pH suggested the high yield of triplet exciton in OV-MIL-125 which indicating the enhanced ISC process due to OV formation. The fluorescence lifetime was the statistic of the average residence time of excitons tested by transient FL spectra (Fig. 4e) which uncover the different dynamic processes of transition. The fluorescence lifetime of OV-MIL-125 (Tave = 1.8 ns) was obviously shorter than of MIL-125 (Tave = 2.5 ns). The fitted results showed the triple exponential attenuation of MIL-125, while the exciton attenuation showed double exponential of OV-MIL-125 with τ1 = 0.32 and τ2 = 3.19 ns. The missing attenuation mode might be attributed to the ISC process which competed with spontaneous FL emission through non-radiative mode. This could also account for the decrease of FL lifetime of OV-MIL-125. The longer pH lifetime (Tave = 6.0 µs) further confirmed the enhanced ISC process of OV-MIL-125 compared with MIL-125 (Tave = 4.1 µs) (Fig. 4f). Commonly, the ISC rate was negatively correlated with the energy gap between singlet and triplet exciton (ISC energy, ΔEST). The energy gap between FL and pH emission could be approximated as ΔEST, which was sharply decreased in OV-MIL-125 (0.39 eV) compared with MIL-125 (1.23 eV). Ultralow temperature (2.0 K) FL spectra further confirmed the shrunken ISC energy gap (Fig. 4h). The red-shift of emission wavelength between room temperature and ultralow temperature FL spectra in OV-MIL-125 was 35 nm (corresponding energy shift = 0.225 eV) which was smaller than that in MIL-125 and 2%NH2-MIL-125. The above results proved that the transition mode of OV-MIL-125 obviously changed from singlet transition to ISC process due to the formation of OV. Such effect promoted the generation of long lifetime triplet exciton, thus increase 1O2 yield in photoreaction with O2.
The transient absorption spectra (TAS) of provided direct evidence for the existence of triplet exciton in OV-MIL-125. As shown in Fig. 5a, the negative absorption modulation (∆A) at the range of 400–460 nm with the delay time of 0.43 ps could be attributed to transition between singlet exciton (Sn → S0) through stimulated radiation (SE), which corresponded to the FL emission of OV-MIL-125 at 425 nm (Fig. 4g). Commonly, the transition between singlet state takes place in very fast timescale (femtosecond) and the transition between triplet state takes place in relatively long time (nanosecond). The positive ∆A at the range of 520–570 nm with the delay time of 0.1 ps corresponded to excited state absorption (ESA) of singlet exciton (S1 → Sn) given such short timescale. Distinctly, a strong positive ∆A appeared at 500–560 nm with long delay time (1.03 ns and 5.81 ns) caused by the ESA of triplet exciton (T1 → Tn and Tn → Tn+m). While, barely long-lifetime adsorption weas detected in the TAS of MIL-125 instead of a series of singlet exciton with extreme short lifetime (Fig. 5b). The very weak positive ∆A might be due to the offsetting of singlet ESA and SE. The normalized ∆A of OV-MIL-125 and MIL-125 at various delay times were provided in Figs. S20a and b in Supporting information. The kinetic fitting of the S1 → Sn adsorption decay (caused by the deexcitation of S1 state) confirmed the enhanced ISC process in OV-MIL-125. As shown in Fig. 5c, the ESA of S1 exhibited triple exponential decay with the τ1 = 0.61 τ2 = 5.43 and τ3 = 13.9 ps, which corresponded to the transition mode of S1 through vibration relaxation, ISC and fluorescence emission respectively. By contrast, the ESA decay of S1 only showed double-exponential kinetic with the τ1 = 0.32 ps and τ2 = 19.7 ps (Fig. 5d), which suggested the much weaker ISC process compared with OV-MIL-125.
Figure 5
Figure 5.
Two-dimensional contour map of OV-MIL-125 (a) and MIL-125 (b). TA dynamics monitored and fitted results at different wavelength for OV-MIL-125 (c) and MIL-125 (d). (e) Frontier orbital population of MIL-125 and OV-MIL-125. Red, white, gray and silver ball-and-stick represent O, H, C and Ti atoms, respectively. (f) PDOS of MIL-125 and OV-MIL-125 adsorbed with O2. (g) LDOS of O2 adsorbed on MIL-125 and OV-MIL-125.
The spin flip of a ground-state oxygen molecule to form 1O2 requires two successive processes: intersystem crosssing (ISC) and Dexter energy transfer which was illustrated in Fig. S21 (Supporting information). The Dexter energy transfer is essentially the exchange of photoelectron with different spin (triplet exciton-triplet oxygen annihilation). This two processes control the rate of 1O2 generation according to the Arrhenius equation (Text S8 in Supporting information) [50,51] which is related to the ∆EST and the overlap integral of electron wave function between donor and acceptor. To better understand the exciton dynamics and 1O2 generation, density functional theory calculation (DFT) was carried out. The population of frontier orbital indicating the different trend of exciton between OV-MIL-125 and MIL-125. The HOMO and LUMO of MIL-125 distributed on the ligand and Ti–O cluster respectively (Fig. 5e) and the spatial separation of HOMO and LUMO suggested the typical excitation mode involving ligand to metal charge transfer (LMCT). While, the HOMO and LUMO of OV-MIL-125 both spread over the ligand with a short distance. The relative shorter distance between HOMO and LUMO electron cloud was conducive to binding the carriers to form exciton instead of charge separation [52]. This was consisted with the photocurrent intensity (Fig. 4d). Additionally, the proximity of HOMO and LUMO would induce the intraligand charge transfer (ILCT) which is favorable for 1O2 formation [53,54]. In terms of the occupy energy, the gap between HOMO (−3.95 eV) and LUMO (−3.90 eV) of OV-MIL-125 was much narrow (0.05 eV) than 2.54 eV of MIL-125 (HOMO = −7.31 eV, LUMO = −4.77 eV). This also account for the stronger light absorptivity and indistinct absorption edge of OV-MIL-125 (Fig. 4a). In principle, ∆EST (the intersystem energy gap between singlet and triplet state) should decrease upon decreasing the exchange interaction integral of the HOMO and LUMO wavefunctions [19]. However, when the overlap and distance between HOMO and LUMO reach to some degree, internal conversion (IC, Sn→S1) became the key factor to reduce ∆EST. This could account for the shrinked ∆EST in photoexcited OV-MIL-125. Meanwhile, the energy of different excited states was calculated by time dependent density function (TD-DFT). The energy of S1 and T1 state were modulate by OV in OV-MIL-125 (S1 = 2.621 eV, T1 = 2.600 eV) compare with that in MIL-125 (S1 = 3.215 eV, T1 = 2.698 eV) and 2%NH2-MIL-125 (Table S4 in Supporting information). The ∆EST (ES1 − ET1) greatly reduced to 0.021 eV due to the formation of OV compared with MIL-125 which indicating the high ISC rate in OV-MIL-125. The oscillator strength (fos) of S1 state in MIL-125, 2%NH2-MIL-125 and OV-MIL-125 was 0.374, 0.112 and 0.043, respectively. The smaller fos indicated that the transition probability of S1 → S0 in OV-MIL-125 was lower than that in MIL-125, which would increase the ISC probability of S1 → T1.
DOS (density of states) analysis for different models adsorbed with O2 indicated the charge transfer tendency. The optimized adsorption model was provided in Fig. S13 which showed the higher oxygen adsorption energy on OV-MIL-125. PDOS of different catalysts (Fig. S22 in Supporting information) suggested the reduced band gap (0.94 eV) due to the formation of OV compared with MIL-125 (1.70 eV). Impurity level appear between CB and VB in 2%NH2-MIL-125 which was consisted with its optical spectroscopy. After O2 adsorption, an impurity level appeared at 1.25 eV in the PDOS for MIL-125 attributed to the 2p orbital of O2 (Fig. 5f). This would be conducive to electron transfer to absorbed O2 which could also be confirmed by the LUMO distributing on adsorbed O2 (Fig. S23 in Supporting information). For OV-MIL-125 adsorbed with O2, the DOS of O 2p orbital (Fig. 5g) crossed Fermi surface which was conducive to Dexter excitation transfer to form 1O2 through triplet annihilation.
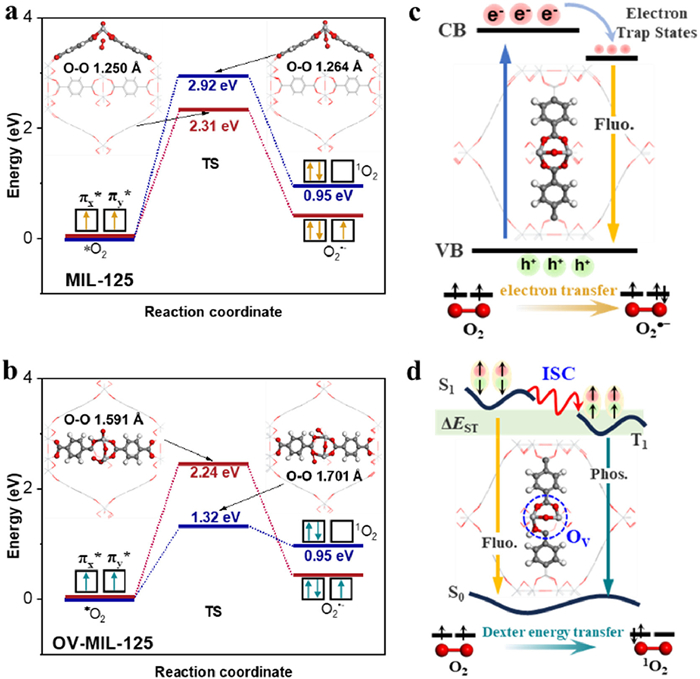
Dexter excitation transfer requires the overlap between the wave functions of donor and acceptor of electron with different spin. The overlap between HOMO of OV-MIL-125 and LUMO of OV-MIL-125 adsorbed with O2 agreed with the above condition (Fig. 5e and Fig. S22). The relative large distribution of HOMO (−4.08 eV) on adsorbed O2 and lagre distribution of LUMO (−4.05 eV) on organic ligand with OV indicating the flow of transient electrons tend to be from adsorbed O2 to triplet excitation unit, which was matched with Dexter energy transfer. The rate of Dexter excitation transfer (Text S8 in Supporting information) was positive correlation with the distance between donor and acceptor [50]. The larger overlap of wave functions of HOMO and LUMO could reduce the distance between elecrton donor and acceptor which was convenience for the return of electron with reversal spin. The reduced distance between O2 and substrate (Fig. S13) also indicated the enhanced 1O2 yield. Moreover, the spin density distribution and magnetic moment of chemisorbed O2 on different catalysts was calculated. The spin density mainly distributed on chemisorbed oxygen molecules on MIL-125 (Fig. S24 in Supporting information), while the O2 absorbed on OV-MIL-125 had small spin density indicating the same spin of two electrons on 2pπ* orbital tend to spin cancellation. The magnetic moment of free O2 molecules was 2.0 µB and the magnetic moment of O2 absorbed on MIL-125 decreased to 1.56 µB (Table S5 in Supporting information). In contrast, the magnetic moment decreased to 0.09 µB after the interaction between O2 and OV-MIL-125, suggesting the easier spin-flip process of O2 to generate 1O2 on OV-MIL-125 compared with MIL-125. To further confirmed the effect of OV introduction on mechanism of O2 activation, the transient states (TS) and the energy of the reaction coordinates during O2 activation to form O2•− and 1O2 on catalytic interface were located and calculated. As shown in Figs. 6a and b, the O2 adsorbed on MIL-125 (optimized model was shown in Fig. S13) need to cross the energy barrier of 2.92 eV to achieve the spin flip of the electrons in π* orbital. This activation energy was higher than that (2.31 eV) for the reduction of absorbed O2 to form O2•−, indicating that O2 was more likely to activated into O2•−. While, the activation energy for O2 adsorbed on OV-MIL-125 were obviously lower. Moreover, the energy barrier (1.32 eV) to generate 1O2 (spin flip of the electrons in π* orbital) was much lower than that (2.24 eV) to form O2•−. The transition mode and O2 activation mechanism for MIL-125 and OV-MIL-125 were illustrated in Figs. 6c and d, respectively.
Figure 6
Figure 6.
Energy of reaction coordinate during O2 activation to form O2•− and 1O2 on MIL-125 (a) and OV-MIL-125 (b). Scheme of transition mode and O2 activation mechanism for MIL-125 (c) and OV-MIL-125 (d). The TS and *O2 represent transient state and O2 adsorbed with catalyst interface, the inset was the optimized model of corresponding TS.
In summary, a novel oxygen vacancy (OV) modification strategy for MIL-125 was successfully practiced by ligand replacement-thermal treatment. Efficient 1O2 generation with high selective (~90%) and enhanced removal rate for multiple antibiotics were achieved by OV-MIL-125 under visble light. Multiple characterizations including EXAFS, EPR, XPS, and Raman spectra confirmed the formation of OV in OV-MIL-125. The OV played key role in increasing 1O2 yield and selectivity in O2 activation which accounted for efficient antibiotics degradation. Photoelectric analysis including transient-steady state fluorescence and phosphorescence, transient absorption spectra (TAS) combined with DFT calculation revealed the transformation of transition mode in OV-MIL-125. The intersystem crossing process was promoted owing to the modulated HOMO-LUMO and ISC energy gap which increased the content of triplet exciton. Thus, the Dexter energy transfer involving triplet annihilation dominate the O2 activation mechanism to generate 1O2 instead of the charge transfer to generate O2•− happened in MIL-125. This study demonstrates the structure-activity relationship between OV and 1O2 generation and provided new thinking for photocatalytic activation of molecular oxygen. In view of the high efficiency for 1O2 generation, the strategy of oxygen vacancy modification is expected to guide the design and synthesis of MOF-based catalysts for water treatment.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 22276086 and 21976078), the Natural Science Foundation of Jiangxi Province (Nos. 20202ACB213001 and 20232BAB213029), all of which are greatly acknowledged by the authors.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109761.
[1]
S. Zhou, L. Jiang, H. Wang, et al., Adv. Funct. Mater. 33 (2023) 2307702. doi: 10.1002/adfm.202307702
C.L. Liu, R.L. Zhang, C.S. Lin, et al., J. Am. Chem. Soc. 139 (2017) 12474–12479. doi: 10.1021/jacs.7b05157
[54]
W. Huang, X. Wang, W. Zhang, et al., Appl. Catal. B: Environ. 273 (2020) 119087. doi: 10.1016/j.apcatb.2020.119087
Figure 1
(a) Scheme of structural evolution of OV-MIL-125 during synthesis (the grey, black, and red balls respectively present Ti, C, and O atoms). (b-d) SEM images of the MIL-125, 2%NH2-MIL-125 and OV-MIL-125. (e, f) TEM images of the MIL-125, 2%NH2-MIL-125 and OV-MIL-125. (h, i) HR-TEM images of OV-MIL-125. (j) EDS elemental mapping of OV-MIL-125.
Figure 2
(a) Ti K-edge XANES spectra of OV-MIL-125. The insert shows partial enlarged detail. (b) Fourier transforms k2-weighted χ(R) spectra of Ti K-edge in OV-MIL-125. The insert shows the structure model of OV-MIL-125. (c) Wavelet transform of Ti k3-weighted EXAFS for OV-MIL-125 and TiO2. EPR spectra (d) and FTIR spectra (e) of MIL-125, 2%NH2-MIL-125 and OV-MIL-125. (f) Raman spectra of MIL-125 and OV-MIL-125. C 1s (g) and O 1s (h) XPS spectra MIL-125, 2%NH2-MIL-125 and OV-MIL-125.
Figure 3
Removal rate of TC (a) and TOC removal rate (b) during TC degradation by different catalysts. (c) TC degradation rate by OV-MIL-125 with different relative concentration of oxygen vacancy. Spin-trapping EPR spectra for O2•− (d) and 1O2 (e) in different photocatalytic system. (f) Effect of DMPO and TEMP for TC removal efficiency in MIL-125 and OV-MIL-125 system. (g) 1O2 yield in MIL-125 and OV-MIL-125 systems and TC removal rate under aeration with O2 and N2. (h) The correlation between 1O2 concentration and OV concentration. (i) Time-dependent 1O2 yield in MIL-125 and OV-MIL-125 systems through SOSG fluorescence assay.
Figure 5
Two-dimensional contour map of OV-MIL-125 (a) and MIL-125 (b). TA dynamics monitored and fitted results at different wavelength for OV-MIL-125 (c) and MIL-125 (d). (e) Frontier orbital population of MIL-125 and OV-MIL-125. Red, white, gray and silver ball-and-stick represent O, H, C and Ti atoms, respectively. (f) PDOS of MIL-125 and OV-MIL-125 adsorbed with O2. (g) LDOS of O2 adsorbed on MIL-125 and OV-MIL-125.
Figure 6
Energy of reaction coordinate during O2 activation to form O2•− and 1O2 on MIL-125 (a) and OV-MIL-125 (b). Scheme of transition mode and O2 activation mechanism for MIL-125 (c) and OV-MIL-125 (d). The TS and *O2 represent transient state and O2 adsorbed with catalyst interface, the inset was the optimized model of corresponding TS.


 DownLoad:
DownLoad:



 下载:
下载:







 下载:
下载:
