Figure 1.
Typical OSDAs used for the synthesis of known chiral zeolites.

Zeolitic molecular sieves are a type of porous crystalline material mainly composed of aluminosilicates, with structural characteristics including regular channel construction, relatively large specific surface area and adjustable size of accessible cavities [1-3]. Being widely used in chemical industrial production, zeolites have exerted great functions in adsorption [4], separation [5] and catalytic reactions [6,7]. The general structure of classic zeolites is mainly based on [TO4] (T = Si, Al, etc.) tetrahedra as basic structure units, which are bridged by oxygen atoms at common vertices to form composite building units (CBUs) of different shapes, thereby constructing diverse polyhedrons in three-dimensional space [8]. These polyhedrons are usually cavity-containing cages, such as α-cages, β-cages, ast cages, faujasite cages [9], normally possessing high symmetries involving mirror planes or inversion centers [10]. However, since the 1980s, researchers have started to pay attention to its contrary, namely chiral zeolites. It is assumed that the interplay between chirality and the physical and chemical properties of porous materials may open up a new growing point in the fields of enantioselective separation and asymmetric catalysis [11-14].
In the past three decades, certain kinds of chiral organic structure-directing agents (OSDAs) have been designed and utilized in the hydrothermal synthesis of zeolitic materials (Fig. 1) [15], successfully making the chirality induction and transfer from small organic molecules to the long-range rigid framework of inorganic composites [16], thus creating a series of novel chiral zeolites (Table 1). Advances have been achieved in multiple leaps from the formation of racemic intergrown chiral conglomerates [17,18] to that of a racemic mixture of homochiral crystals [19-22], and finally to some recent progress in acquiring enantiomeric-enriched chiral zeolites hitherto [23-25]. At the same time, the development of characterization methods for solid materials, especially the application of high-resolution transmission electron microscopy (HRTEM) [26,27] and three-dimensional electron diffraction (3D ED) [28] has, to some extent, solved the problem of determining the absolute configuration of polycrystalline solid materials with chiral zeolite as a representative. Based on these breakthroughs, chiral zeolites have preliminarily shown their possibilities and prospects in enantioselective gas adsorption [23] and catalytic organic reactions [14,23,24,29]. However, behind these rudimentary attempts, there are still many outstanding problems and challenges in this field. This review will start the tour with a brief introduction of the previously reported chiral zeolite structures and then proceed into several aspects of rational design, structure-directed synthesis, absolute configuration determination, and catalytic applications of chiral zeolites.
Chirality is a commonly existing natural phenomenon that describes an object or structure's property in three-dimensional space being unable to match its mirror image through proper rotation and translation [30]. The understanding of chirality in history started right from crystals and has progressed through observation of crystal shapes and apparent optical properties to their internal structure and space group symmetry [31]. Based on the knowledge of crystal symmetry, the chiral zeolites we explore can be classified into two types: structures with inherent chirality on chiral space groups and structures with chiral asymmetric units grafted onto achiral Sohncke space groups [8,32]. Listed in Table 1 are some kinds of chiral zeolites already included in the coding list of the Structure Commission of the International Zeolite Association (IZA-SC) [33], which indeed covers these two types in various symmetries.
Specifically, chiral crystals are formed exclusively in structures if their space group contains only proper rotation operations. Among the 230 crystallographic space groups, only 65 of them, referred to as Sohncke groups, can keep chirality [43,44]. Sohncke groups can be divided into two categories: the first comprises 22 chiral space groups (i.e., 11 pairs of enantiomeric relationships) with all containing one of each pair of enantiomorphic screw axes (31 or 32, 41 or 43, 61 or 65, 62 or 64) where strictly excludes the existence of mirror planes and roto-inversions. Belonging to this type of chiral space group is a sufficient but not necessary condition for crystals to have chirality. For example, chiral zeolites encoded *BEA, OSO, STW, CZP, -ITV, and LTJ belong to this category. The second type refers to the crystallization within the remaining 43 achiral Sohncke space groups. These space groups lack the second type of symmetry operation and also do not possess a chiral screw axis. The chirality of crystals in this group is contingent on the chiral asymmetric unit they originate from. Included in this category are the chiral zeolites encoded JRY, GOO, VNI, VFI, and ABW in Table 1.
Besides symmetrical properties, it is of equal importance to focus on the pore size of such chiral zeolites especially when expecting them to perform shape-selective adsorption or catalytic applications [45-48]. It must be ensured that the structural chirality is presented in confined cavities of a certain size. Ideally, these cavities or channels shall present chirality aiming at selective recognition toward adsorbate guest molecules or reaction substrates, products, intermediates, or transition states [49,50]. Moreover, if the cavity size is too small or the pore channels are too narrow (e.g., d < 4 Å), it becomes difficult to accommodate the reaction system or through which substrates and products would diffuse. According to the definitions of microporous and mesoporous materials [51], the chiral zeolites we are discussing herein belong to the former, and among the larger ones are *BEA, OSO, STW, VFI, and -ITV, while the latter two have pore sizes ~10 Å, large enough to approach mesopores [22,35]. However, it's worth noting that the chirality of some zeolites, such as VPI-5 (the type material of VFI), comes from the guest water molecule filled in the pore which loses chirality upon dehydration and thus no longer meets our demand.
In addition, over the past twenty years, there have been significant advancements in developing zeolite-like porous materials with chirality. Noteworthy examples are the chiral mesoporous silicates introduced by Che et al., which employed twisted micelle assemblies or nanoclusters as templates [52-54], as well as chiral nematic mesoporous materials [55] and chiral nanoceramics [56]. Additionally, researchers have successfully produced chiral metal-organic frameworks (cMOFs) [57-59] and chiral covalent organic frameworks (cCOFs) [60-64] by incorporating asymmetric organic structural fragments during the construction process of traditional MOFs or COFs. These materials have been individually characterized and have demonstrated certain capabilities in enantiomeric separation. Such development is worth learning from for the study of chiral zeolites though, there are already comprehensive reviews and summaries available on this topic, so we will not delve further into details.
In the following subsections, we will provide a brief overview of three typical chiral zeolites, i.e., *BEA, STW and -ITV, explain the emergence of chirality in their structural construction, and discuss some of the key points in their OSDA-directed synthesis.
Zeolite beta is the first zeolite discovered to exhibit chiral channels and is considered to be one of the most complex intergrown zeolitic structures [65,66]. The discovery of this material can be traced back to 1967 by Mobil Oil Corporation and its unique properties have been of interest to researchers ever since. Tetraethylammonium hydroxide was used as an OSDA during its synthesis, while its adsorption and other physicochemical properties suggested that the zeolite has a large pore structure. However, due to its highly disordered intergrown structure, the broadened peaks of powder X-ray diffraction make it a significant challenge to determine its proper structure. It was not until 1988 that Newsam et al. [17] and Higgins et al. [18] independently solved this problem by making use of advanced techniques such as electron diffraction and HRTEM together with computer simulation methods.
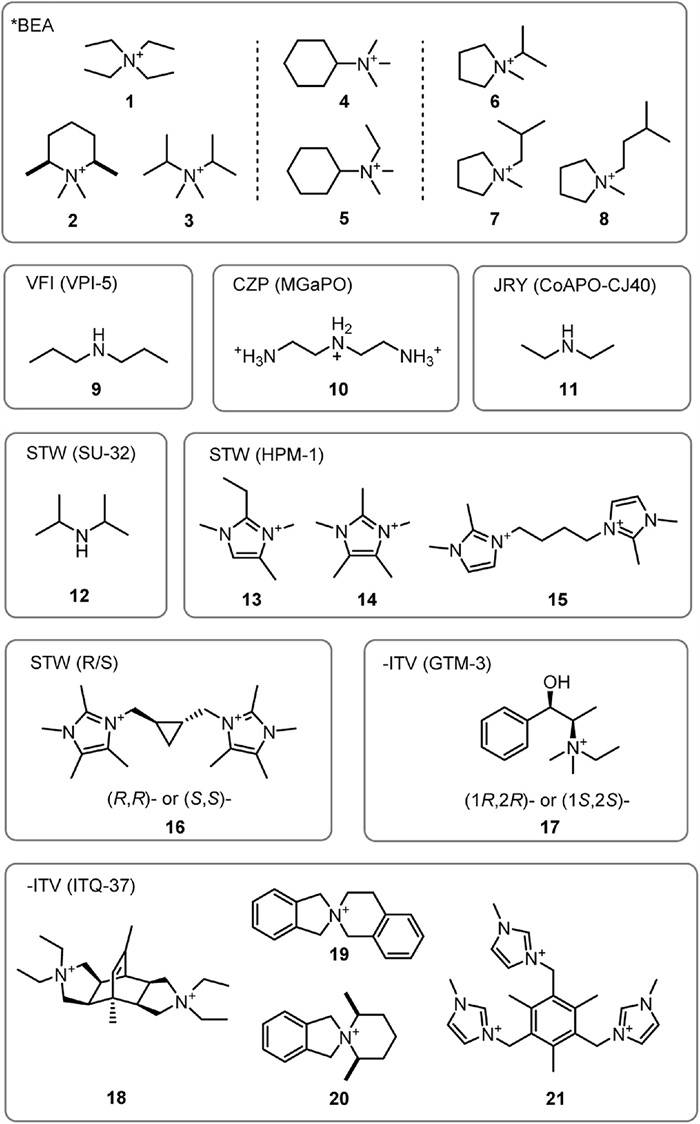
Zeolite beta has two polymorphs, known as A and B, which are very similar in construction. They are made up of the same centrosymmetric layered structural units that are stacked differently along the c-axis. Polymorph A is formed when the layered units are spirally stacked along the c-axis with the 41 or 43 screw axes, exhibiting a symmetry of P4122 or P4322 which are of mirror image. When observed through the direction perpendicular to the c-axis, the 12-membered ring (12-MR) channels are alternately stacked in a zig-zag form denoted as a-b-a-b (Fig. 2). Polymorph B, conversely, is formed when the layered units are stacked leftwards and rightwards alternately along the c-axis, exhibiting a symmetry of C2/c with chirality no longer existing. When observed perpendicular to the c-axis, the 12-MR channels are stacked alternately as a-b-c-a-b-c. Both polymorphs are very similar in structure and framework energy, so they exist in nearly equal proportions in traditional zeolite beta materials and have not been observed as pure ordered phases of either one so far [33].

Nonetheless, zeolite beta has already been widely used as a catalyst in industrial processes [67,68]. Its relatively large channel diameter (d ~ 6 Å) and solid Brønsted or Lewis acid properties upon doping of metal elements (e.g., Al, Sn, Ti) make it ideal for various reactions, including Friedel–Crafts alkylation [69], acylation [70], cracking isomerization [71], esterification [72], and more. Therefore, to realize potential asymmetric catalysis, it appears more urgent to develop zeolite beta with homochirality. The first step towards achieving this goal is to prepare zeolite beta enriched with polymorph A. In 2015, Xu et al. reported the highest known enrichment results so far [65]. They used a highly concentrated fluoride route in combination with traditional tetraethylammonium hydroxide and several other quaternary ammonium hydroxides as OSDAs (1–3 in Fig. 1), achieving 65%–70% enrichment of polymorph A by controlling the water–silica ratio down to about n(H2O)/n(SiO2) = 0.3. Subsequent characterization led to the speculation that triethylamine, produced by Hoffmann degradation of tetraethylammonium cation during the dehydration process at 80 ℃, may play an important role in the enrichment of polymorph A. Since then, Tian et al. have reported progress in a concentrated hydrofluoric acid system (with modified OSDAs 4–5 in Fig. 1) [73]; Yan et al. improved the synthesis using an acid-assisted approach (with modified OSDAs 6–8 in Fig. 1) [74] and certain alcohol additives [75]. However, there have been no further advances so far toward synthesizing an enantiomeric-enriched polymorph A of zeolite beta, leaving a bigger challenge ever since.
STW is a type of hexagonal germanosilicate zeolitic molecular sieve with chiral 61 or 65 screw axis symmetry and a medium-sized channel structure within a space group of P6122 or P6522 respectively. It is also STW-structured zeolites that played significant roles in achieving two breakthroughs in the synthesis of racemic chiral single crystals and chiral enantiomeric-enriched zeolites for the first time [20,23].
In 2008, Zou et al. reported the synthesis of the first chiral germanosilicate zeolite with STW topology named SU-32 [20]. By using a non-chiral diisopropylamine (DIPA) as OSDA (12 in Fig. 1), they obtained an SU-32 sample with a silicon-to-germanium ratio of Si/Ge = 4.72:5.28. The construction pattern of SU-32 is similar to that of *BEA, that is, consisting of a layered structure of 4-membered, 5-membered, and 12-membered rings, which are mutually connected through rotation and displacement, forming a three-dimensional framework structure. However, the rotation angle between layers of SU-32 is 60° instead of 90° as that in *BEA, which allows for the construction of a helical channel with either 61 or 65 screw axes (Fig. 3). The adjacent 12-MRs are not completely aligned, but dislocations are generated during the rotation process. Therefore, a shrinkage to 10-MR actually limits the size of the channel aperture. When the adjacent layered spiral stacking of SU-32 becomes centrosymmetric stacking, a non-chiral SU-15 zeolite structure is generated with a space group of C2/m, similar to the pattern of zeolite beta polymorph B. Interestingly, unlike the preference of racemic intergrowth of *BEA enantiomers, SU-32 almost crystallizes as a single crystal with enantiomeric purity. Its absolute configuration can be determined through single crystal X-ray diffraction (XRD) testing, and the Flack parameter is close to 0. It is speculated that a perfect single chiral SU-32 crystal has an energetic advantage over racemic intergrown twinnings with stacking defects, which may explain why SU-32 crystallizes almost exclusively as a single enantiomer.
One of the drawbacks of germanosilicate zeolite SU-32 is its relatively low silicon-to-germanium ratio (Si/Ge = 0.89 < 1), which leads to low hydrothermal tolerance and structural instability. Toward this problem, in just four years, Camblor et al. successfully synthesized a pure silicon STW, named HPM-1 [21], using 2-ethyl-1,3,4-trimethylimidazolium salt as OSDA (13 in Fig. 1). HPM-1 maintained the chiral nature of either P6122 or P6522 and demonstrated excellent thermal stability and high water tolerance. In 2014, Davis et al. continued to investigate structural directing agents for such zeolites and suggested that 1,2,3,4,5-pentamethylimidazolium salt (14 in Fig. 1) could also be used to efficiently synthesize pure silicate HPM-1 [76].
In 2017, Davis et al. made a significant breakthrough in the synthesis of enantiomeric-enriched STW [23]. Their work exploited the computational-assisted design of a suitable chiral OSDA, namely a pair of tetramethylimidazolium dication joined by a chiral trans-1,2-disubstituted cyclopropane linker (16 in Fig. 1). Combined with an amount of 10% pure silica racemic STW as seed crystals, they successfully produced enantiomeric-enriched R/S-STW with the highest Si/Ge ratio of 20:1, as well as enantiomeric-enriched aluminogermanosilicate R/S-STW with a Si/Ge ratio of 2:1 and Si/Al ratio of 100:1. By using the modified HRTEM method of rotating crystalline sample developed by Terasaki and Ma et al. [26], they determined the space group of a single crystal in the R/S-STW powder samples, distinguished their chiral absolute configurations from P6122 and P6522, and confirmed the correspondence between R/S-OSDA with R/S-STW (Fig. 4). They also semi-quantitatively determined the enantiomeric enrichment degree by measuring the absolute configurations of polycrystals, represented by an enantiomeric ratio of approximately 5:1. The synthesized chiral aluminogermanosilicate STW samples also exhibited enantioselective adsorption and catalytic effects right as expected.

Inspired by this study, researchers have been trying to improve the synthesis method of STW [77]. As the STW structure contains numerous double-4-ring (d4r) units with a relatively high strain, it was inevitable to use a higher proportion of germanium or otherwise concentrated fluoride ions to stabilize the silicate framework during the synthesis. However, this also poses a risk of exposure to harmful synthetic reagents such as hydrofluoric acid. Therefore, Iyoki et al. [78] and Du et al. [79] have successively found fluoride-free synthetic routes towards STW. They have also studied the host–guest binding mechanism between the zeolite framework and OSDA molecules. Meanwhile, Davis et al. attempted to synthesize enantiomeric-enriched STW using several chiral amino acids as additives to perform a co-template effect [80]. However, they unintentionally discovered a better crystallization condition for racemic STW. This helped them to obtain experimental evidence of host–guest binding modes through synchrotron radiation XRD.
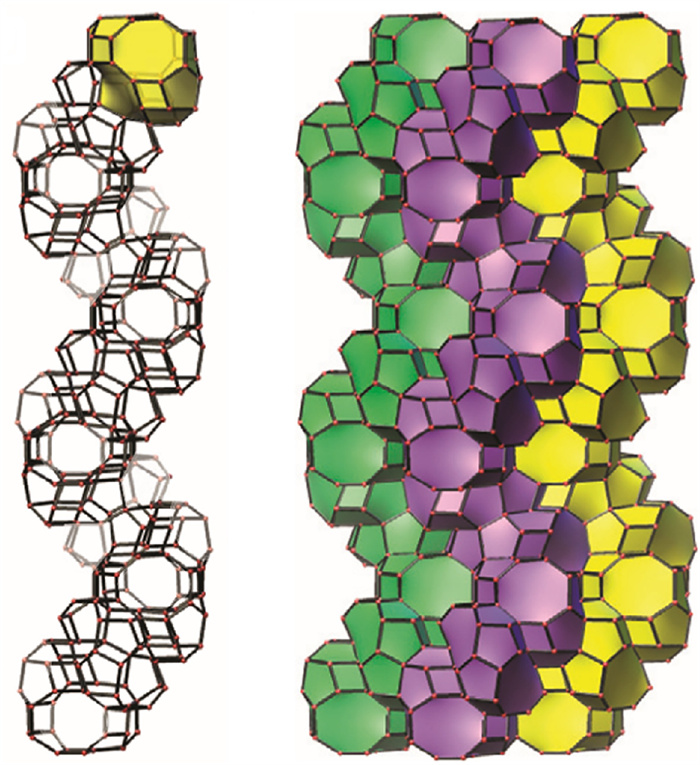
In 2009, Zou et al. reported the discovery of a mesoporous germanosilicate zeolite with a chiral channel structure and named it ITQ-37 [22]. Shortly afterward, the framework was assigned with a three-letter code -ITV. It belongs to the space group of either P4132 or P4332. ITQ-37 is the first three-dimensional germanosilicate zeolitic molecular sieve with a 30-membered ring, whose pore size even comes close to the standard of mesoporous material, and a skeleton density as low as only 10.3 T atoms per 1000 Å3. To synthesize this kind of chiral zeolite, a bridged-cyclic quaternary ammonium derivative containing four chiral centers (18 in Fig. 1) has been used as OSDA. The structure of ITQ-37 contains unique lau cages [4264] and two double 4-rings (one D4R1 and three D4R2) that form the nodes of the srs network structure. Every ten such structural units form a 30-MR mesopore in the ITQ-37 structure and the centers of adjacent 30-MRs are connected to form another set of srs network, in which the spiral channel system is present (Fig. 5). This spiral channel demonstrates a very large open skeleton structure that can accommodate a variety of guest molecules, making it expected to excel in the enantioselective application of larger substrates. However, the OSDA used in this report is merely a mesomeric molecule so the resulting synthesized zeolitic material lacks definite enantiomeric enrichment after all. Therefore, its enantioselective application scenarios cannot be confirmed yet.
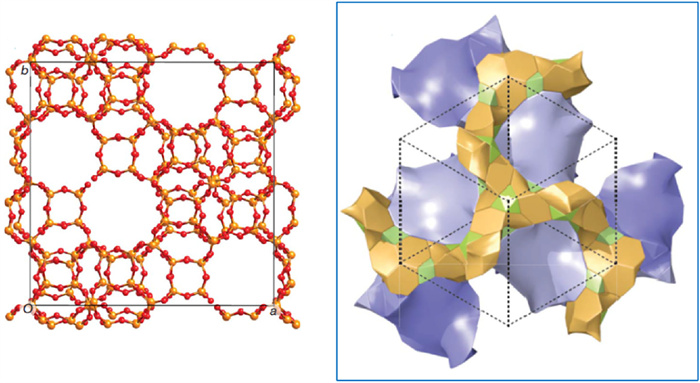
In the following years, several research groups including Yu et al. [81], Du et al. [82], and Jiang et al. [83] have respectively succeeded in the synthesis of ITQ-37 using various types of quaternary ammonium salts or imidazolium salt dimer as OSDAs (19–21 in Fig. 1). Nonetheless, all the OSDAs harnessed in these experiments were achiral organic molecules and no enantiomeric enrichment was achieved.
Fortunately, it was not long before Gómez-Hortigüela et al. successfully boosted chiral zeolites with -ITV topology to enantiomeric enrichment [24]. They used a chiral pseudoephedrine derivative called EMPS (17 in Fig. 1) to achieve this enrichment on -ITV zeolite with a silicon-to-germanium ratio of 2.6–2.8. The process was carried out under careful control of hydrothermal temperature (being kept below 110 ℃ or even down to 60 ℃), as high temperatures can cause OSDAs to decompose or racemize, while low temperatures require strong interaction between OSDA and the germanosilicate framework of zeolite to undergo nucleation and crystallization processes. Under such circumstances, the enantiomeric enrichment of -ITV-type zeolite was acquired and named GTM-3. However, the crystal structure of such zeolites is extremely sensitive to electron beam exposure, making it unable to definitively characterize their absolute configuration and enantiomeric excess value. Alternatively, indirect studies were conducted using powder XRD and solid-state NMR under comparison of seed-assisted synthesis. The definitive evidence of enantiomeric enrichment came from the reaction results of its catalytic asymmetric reactions. A repeatable catalytic ee value of up to 30% undoubtedly confirmed the presence of the catalyst's enantiomeric enrichment.
Much focus has been paid to the rational design towards the synthesis of zeolitic molecular sieves and chiral ones were no exception [84,85]. Although the exact process of how zeolite forms is not yet fully understood, some guiding principles have been established for synthesizing chiral molecular sieves under hydrothermal conditions, which are brought about by OSDAs. Ever since chiral zeolites were discovered, scientists have been attempting to design suitable chiral OSDAs to create chiral pores in the shapes of corresponding zeolites. In 1992, M. E. Davis summarized some guidelines for using chiral OSDAs and proposed certain conditions necessary for synthesizing chiral zeolites based on those guidelines [86]. They suggest that, firstly, to differentiate between a pair of enantiomers of chiral zeolites, it is crucial to create a difference in the interaction between a pair of chiral OSDA enantiomers and the chiral zeolitic framework. This difference in interaction shall be significant in terms of energy, which is a decisive factor of chiral OSDAs. Secondly, OSDAs should possess the following specific characteristics [14]:
1. OSDA itself must be chiral;
2. OSDA must be of sufficient size to interact with the zeolite framework, thereby determining the chirality of the framework;
3. OSDA must remain stable under hydrothermal synthetic conditions, maintaining both covalent stability and avoiding racemization;
4. OSDA needs to have a certain degree of rigidity and try to maintain its conformation unchanged during the synthesis process;
5. OSDA cannot rotate in the pore structure, otherwise the chiral induction effect will be lost.
These principles have been a crucial guide in the research on the synthesis of chiral molecular sieves with the use of chiral OSDA. However, numerous experiments have been reported that zeolites synthesized using chiral OSDAs are still intrinsically achiral, like SSZ-24 (P6/mcc, [87]) and CIT-5 (Imma, [88]) synthesized using N-methylsparteinium, CIT-1 (C2/m, [89]) and ZSM-12 (C2/c, [90]) using N-substituted myrtanylammonium salts, probably due to breaking of any one of the rules 2–5 above [12].
To create effective prediction methods for the structure of chiral zeolites, theoretical computations and molecular dynamics simulations have been applied to guide practical experimental synthesis. The theoretical computation and simulation of chiral zeolites mainly consists of three interrelated parts: (1) Rational design of plausible novel chiral zeolite structures; (2) virtual screening and predicting candidate chiral OSDAs based on known chiral zeolite framework structures; (3) prediction of the zeolite framework structures upon given chiral OSDAs that may be induced for synthesis.
In 2003, Yu et al. developed an algorithm aiming at the prediction of novel chiral zeolitic frameworks [91]. The algorithm introduced the concept of a "forbidden zone", allowing for the artificial setting of the desired zeolitic pore structure, and then assembling the framework by placing T atoms outside the forbidden zone. This method allows for adjusting the size of pores, the number of independent T atoms, the symmetry position of independent atoms, the size of crystal cells, and space group symmetry. As a result, chiral zeolite structures with a single helical axis symmetry can be directly designed. As depicted, the yellow tetrahedron in Fig. 6a represents the [TO4] unit, and the green cylinder represents the pre-designed forbidden zone. Using the given 61 spiral axis symmetry operation, the program can automatically generate the corresponding helical channel structure.
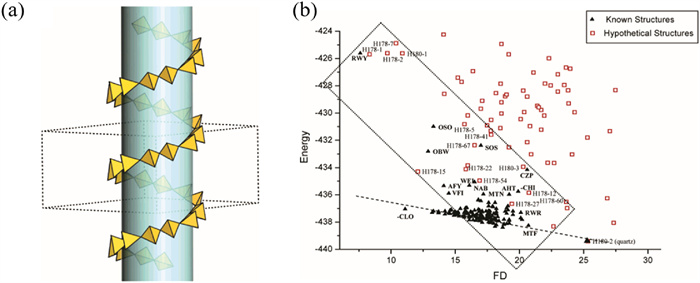
Once a set of target structures has been obtained through simulation, it becomes necessary to verify their actual reliability. In this regard, two possible indicators that can be used to measure the probability are proposed: framework energy and framework density (FD). By comparing the simulated target structure (red square in Fig. 6b) and the skeleton structures of 161 zeolite structures (black triangle in Fig. 6b) and fitting the approximate possible range, it is possible to determine the reliability of the prediction results to some extent.
Besides the prediction of novel zeolite structures, the other aspect is to predict candidate OSDAs. The main objectives are as follows [92]:
1. Determine the synthesizability, hydrothermal stability, and racemization energy barrier of the OSDA itself;
2. Dock the OSDAs into their right position in the framework and calculate the binding energy towards zeolitic frameworks, especially the relative difference in the energy of a pair of enantiomers. This helps us understand the basic relationship between OSDA and the chiral zeolites they induce.
3. Simulate the role of OSDA in nucleation and crystal growth processes, particularly in inducing symmetry breaking and chirality generation during the formation of CBUs.
4. Perform virtual high-throughput screening of new chiral OSDAs or predict better chiral OSDAs for the preparation of known chiral zeolite structures.
Currently, there have been a few successful reports of applying molecular simulation results to the experimental direction of synthesis towards zeolite beta. Corma et al. used GULP (General Utility Lattice Program) to perform calculations with the aim of synthesizing one of the three polymorphs of zeolite beta—BEC [93]. They screened nine different potential OSDAs from a series of molecules and successfully applied them in experiments to obtain the desired target structure. The predicted OSDA was found to exhibit the best reactivity among the OSDA candidates.
In 2019, Deem et al. proposed a simulation method for synthesizing the polymorph A enrichment of zeolite beta [94]. The method consists of three parts:
1. De novo design: synthetic accessibility of candidate molecules is addressed by screening known chemical reaction libraries and small molecule substances through a genetic algorithm;
2. Scoring function: candidate OSDAs are scored based on factors such as stability under hydrothermal synthesis conditions, number of degrees of freedom in twist angle, and molecular volume;
3. Optimization algorithm: this further calculates the interaction energy between OSDA and molecular sieve under the optimal configuration using the non-bonded Lennard-Jones potential function.
Using this computation method, Deem et al. studied the synthesis of *BEA by predicting 212 candidate OSDAs under the condition that the interaction *BEA is less than −15 kJ/molSi and the energy difference with polycrystalline C is greater than 2 kJ/molSi.
Almost at the same time, Deem et al. further proposed a machine-learning strategy for predicting OSDA for *BEA [95]. They trained neural networks on 4781 candidate OSDAs and filled in chemically feasible OSDA types through evolutionary algorithms. Finally, 469 OSDAs were proposed with stabilization energies below −17 kJ/molSi, all of which were superior to the known OSDAs for synthesizing beta zeolites. Unfortunately, there have been no further reports on whether these predicted OSDAs can be practically used to realize the synthesis of zeolite beta.
Nevertheless, experimental syntheses of other types of zeolites directed by computational design have been reported in recent years [96,97]. Some examples are ITE [98] and SSZ-43 [99]. Advanced computational techniques like data mining, generative neural networks [100], and deep learning generative models [101] have also been used for OSDA calculations in zeolite synthesis. These methods may provide inspiration for the rational design of chiral zeolites as well.
The study of chiral zeolites requires a thorough understanding of determining the absolute configuration of synthesized samples, which is essential for in-depth learning of guest-to-host chirality transfer during the structure-directing process and exploring subsequent enantioselective applications. Absolute configuration for a chiral single crystal can be generally determined through laboratory-level single-crystal X-ray diffractometers [44]. Anomalous scattering occurs due to the partial absorption of X-ray by tested substances, resulting in energy level transitions, which can lead to the breaking in the intensity of Friedel pairs. Chiral crystals with enantiomers exhibit opposite intensity differences, thus allowing for the differentiation in chirality. Unfortunately, obtaining single crystals of sufficient size is often difficult, especially for zeolitic materials. Therefore, new methods for absolute configuration characterization of chiral zeolites need to be contrived.
In 2017, Ma et al. reported two methods for determining the absolute configuration of chiral zeolites based on electron crystallography [26], successfully achieving chirality confirmation of nano chiral crystals. The first method involves using the HRTEM technique. Considering that the principle of HRTEM is to project the periodic array onto a plane and thus lose its 3D chiral features, but the screw axis in chiral crystals concatenates operations of both translation and rotation, it is possible to artificially rotate the crystal sample along the rotative component of the screw axis and that the image will exhibit its translative counterpart. Capturing at least two high-resolution images of the same area and comparing their different translation directions before and after, there will be observable differences to distinguish the absolute configuration of the crystal.
After the proposal of this method, it immediately applied to the first enantiomeric-enriched chiral zeolite STW case mentioned in Section 2.2 (Fig. 7), contributing to the identification of its proper space group and counting its enantiomeric excess amount.
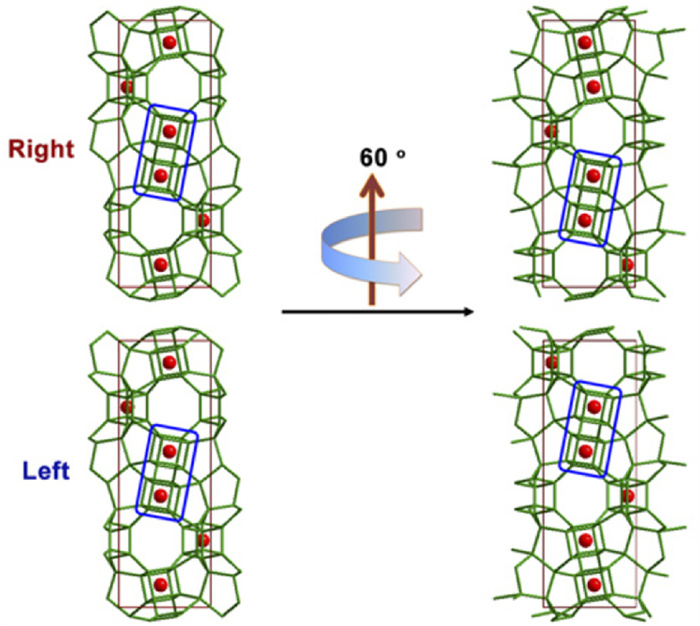
Due to the limitations of difficulty in location comparison and the influence of contrast transfer function (CTF) on crystal HRTEM imaging, Ma et al. further improved this method on an aberration-corrected scanning transmission electron microscopy (STEM) [27]. They changed the sample rotation angle to half of the screw axis angle, reducing the step of comparing the translation direction. Instead, the absolute configuration of the crystal can be identified solely by the specific atomic tilting arrangements in STEM images (Fig. 8).
Since the late 2000s, three-dimensional electron diffraction (3D ED) techniques have been prosperously developed, which refers to methods such as precession electron diffraction tomography (PEDT) [102,103], rotation electron diffraction (RED) [104,105], microcrystal electron diffraction (MicroED) [106], and their updated version for continuous data collection, etc. These techniques proved to be especially useful in the structural analysis of porous materials [107] such as zeolites [108], MOFs [109,110] and COFs [111,112] that are sensitive to electron beams [113]. Kinematic electron diffraction works similarly to normal X-ray diffraction when extracting diffraction peak intensities for structural analysis and refinement. However, when it comes to chiral crystals, the structure factor and diffraction intensities of a pair of enantiomers are identical, making it impossible to distinguish the structural attribution of enantiomers. Therefore, this is where dynamical effect of electron diffraction comes in. What electron beam differs from X-ray is that the interaction between electron and matter is much stronger so that the scattered beam is prone to undergo a secondary scattering, which is known as the dynamical effect [114]. This effect can cause the diffraction peak intensity to no longer follow the kinematic Bragg diffraction conditions, making it difficult for accurate intensity fitting or analysis of systematic absences [115], but a favorable side is the potential to break the center inversion symmetry in diffraction reciprocal space of a chiral crystal, allowing us to identify its absolute configuration [116,117].
In the 2017 report by Ma et al., the second method was to observe the dynamical effects in a precession electron diffraction (PED) experiment [26]. A series of PED patterns are collected along a specific band axis, and the zeroth-ordered Laue zone (ZOLZ) diffraction pattern reflects the plane symmetry properties of the crystal, while the high-ordered Laue zones (HOLZs) demonstrate a more significant dynamical effect, resulting in distinguishable enantiomorphic pairs. Meanwhile, dynamical intensities of diffraction patterns can be simulated by the Bloch-wave method, hence a comparison between the actually observed intensities and the simulation results in HOLZs could lead to the assignment of correct absolute configurations of a tested sample.
Not much later, Brázda et al. reported the application of the PEDT technique to determine the absolute configuration of chiral organic crystals consisting of only light atoms [118]. Dynamical diffraction intensities were compared with the Bragg diffraction intensities in the reciprocal space to obtain the excitation error function and then a double-peaked rocking curve was plotted. By performing a meticulous dynamical refinement, they were able to compare the difference in discrepancy R-factor caused by agreed or disagreed chiral structures, thus distinguishing the absolute configuration of the crystal. Later in 2023, Ma et al. reported a new method for determining the absolute structure of chiral organic nanocrystals using zone-axis electron diffraction [119]. They employed low-dose conditions for minimizing beam damage with the least data collection to adapt to the need for beam-sensitive crystals.
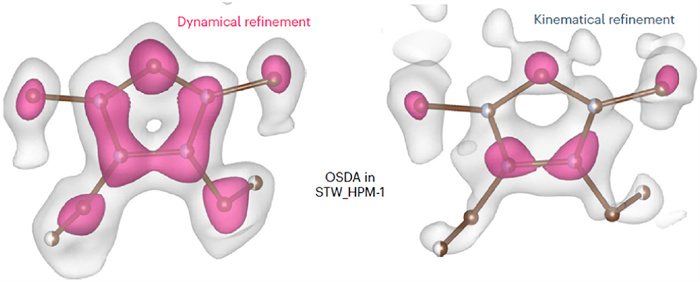
The 3D ED methods have also been reported for determining the absolute configuration of chiral zeolites. In 2021, Palatinus et al. used the method of continuous RED to determine the absolute configuration of HPM-1 pure silica STW-type zeolite crystals mentioned in Section 2.2 [28]. Through dynamical refinement, the wRall values of a pair of enantiomeric structures converged to 0.306 (for P6122) and 0.234 (for P6522), respectively. The significantly lower R-factor of the latter one determined its true absolute configuration, which was in accordance with that measured by the single-crystal XRD result of the same crystal in advance, demonstrating the effectiveness of the dynamical effect of electron diffraction in determining the absolute configuration of chiral zeolites. However, the limitation of this report is that the samples they tested were zeolite materials containing OSDA molecules (2-ethyl-1,3,4-trimethylimidazolium salt, 13 in Fig. 1), and the differences in dynamical refinement involved the contribution of specific chiral conformations of OSDA in confined cavities (Fig. 9). There is still no relevant report on whether the remaining chiral channel of zeolitic materials upon removal of OSDAs can still exhibit such a large R-factor difference to determine their absolute configuration.
To achieve enantioselective applications is the ultimate goal of studying chiral zeolitic materials. Compared to other chiral reagents such as organic catalysts or transition metal complexes, zeolitic molecular sieves showcase several advantages, including structural stability, easy recycling and regeneration, the capability of solid-phase loading and easy set-up of continuous processes, tolerance to varied temperatures, pressure and pH ranges, almost non-toxic and pollution-free at low cost, etc. [120,121]. Chiral zeolites take advantage of their channel shapes to match the reaction substrate, producing shape-selective activity without the need for specific anchoring group interactions. This complements traditional chiral catalytic modes. However, since there are limited reports on the synthesis of enantiomerically enriched zeolitic materials, research on the adsorption and catalysis of chiral zeolites was limited to only a few cases.
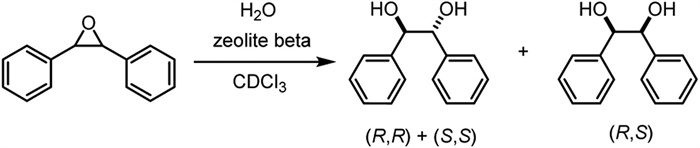
As early as 1992, Davis et al. reported an enantioselective catalysis using their synthesized zeolite beta [86], which achieved asymmetric hydrolytic ring-opening of 1,2-diphenylepoxyethane (Scheme 1) and claimed to acquire an ee value of 5%.
In 2017, Davis et al. conducted simultaneous studies on enantioselective adsorption and catalytic reactions after achieving the first chiral STW zeolite synthesis with enantiomeric enrichment [23]. The selected chiral adsorbent for adsorption research is 2-butanol, a small organic compound with a single chiral center. The results show that chiral STW zeolite has a certain degree of enantioselective adsorption function.
For catalytic reaction research, the asymmetric ring opening reaction of simple primary alcohols on epoxy compounds was conducted [14,23]. (R)-, (S)- and racemic STW zeolites with aluminum loading about Si/Al = 100:1 were tested for catalytic reactions. Methanol, ethanol, n-propanol and n-butanol were used as nucleophilic reagents respectively to the ring-opening reaction of 1,2-epoxybutane, 1,2-epoxyhexane and 1,2-epoxyoctane (Scheme 2). The optimal ee value obtained was reported to be 22.9% under the reaction between n-butanol and 1,2-epoxyoctane. Moreover, comparing different reaction substrates reveals the phenomenon that a longer carbon chain leads to better enantioselectivity, which could possibly be explained as the result of the product molecule's matching toward the chiral pore of STW (Fig. 10). Considering the relatively small pore size, catalytic reactions appropriate for STW may also be limited.

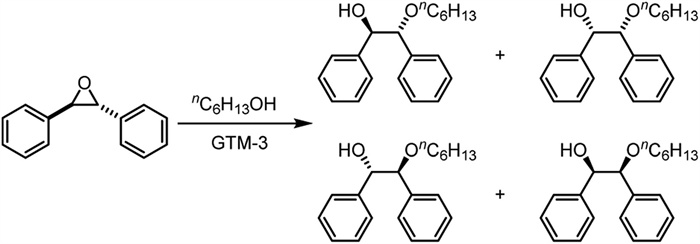
In 2022, Gómez-Hortigüela et al. reported the successful synthesis of mesoporous GTM-3, and their key experimental evidence to verify its enantiomeric enrichment was the catalytic activity of enantioselectivity [24]. They imitated the example of Davis et al. in 1992 and used n-hexanol instead of water as a nucleophilic reagent to the asymmetric ring-opening reaction of trans-1,2-diphenylepoxyethane (Scheme 3), achieving a maximum ee value of 30%, which also represents the best results in the field of chiral zeolite catalysis at present.
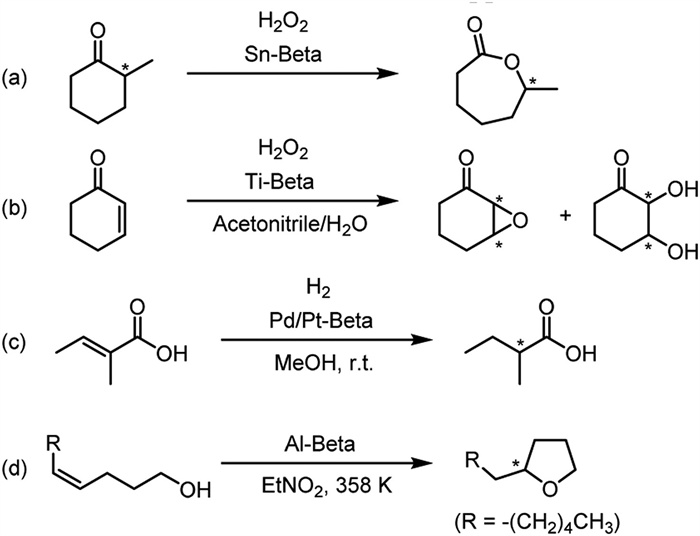
Since it is currently only chiral zeolites with STW and -ITV topologies were reported possible to prepare enantiomeric-enriched samples to be used in asymmetric catalysis, researchers are eagerly looking forward to that of *BEA as well. This is because there are already many reports on the employment of zeolite beta for producing potentially chiral compounds but temporarily only exist in the racemic form. Once enantiomeric-enriched *BEA emerges, there is great potential for its capability of desymmetrization or kinetic resolution in such reactions. For example, in 2001, Corma et al. studied the application of tin-doped zeolite beta to catalyze Baeyer–Villiger oxidation reaction of cyclohexanone to caprolactone [122]. Catalysis was also performed on the reaction of chiral substrates (in the form of racemic mixtures) such as α-methylcyclohexanone and bicyclo[3.2.0]hept-2-en-6-one, which yielded corresponding ring-expanded chiral lactones (Scheme 4a). In addition, Tatsumi et al. reported that titanium-doped zeolite beta can be used to catalyze the epoxidation of cyclohex-2-en-1-one with hydrogen peroxide, resulting in racemic chiral epoxides or vicinal diols (Scheme 4b) [123]. In 2003, Xia et al. reported the hydrogenation reaction using palladium or platinum-doped zeolite beta as catalyst to reduce prochiral tiglic acid to α-methylbutyric acid (Scheme 4c) [124]. In 2010, Čejka et al. reported another kind of intramolecular cyclization in which zeolite beta activates an olefin to form a ring-closed α-substituted tetrahydrofuran (Scheme 4d) [125]. These products are all important chiral precursors of natural products such as perfumes, medicine and pesticides but so far only racemic productions could have been achieved in these reactions. It is, therefore, greatly expected to testify how much enantiomeric excess it can bring about in reactions catalyzed by enantiomeric-enriched *BEA, and it is believed that there will be much more occasions for other enantioselective applications.
For over two centuries, zeolitic molecular sieves have been a field of intense research and development, while chiral zeolites have already been studied for nearly 40 years with particularly significant progress in various aspects over the past decade. Researchers have comprehensively covered the overall fields including computational prediction and rational design, chiral OSDA-directed hydrothermal synthesis, structure analysis with absolute configuration determination, and enantioselective adsorption and catalysis. Among them, many important achievements have come from the introduction of interdisciplinary applications and the advancement of instrumental characterization methods. For example, new algorithms such as machine learning and neural networks helped with the computational design of chiral OSDAs; electron microscopy crystallography has revolutionarily solved the problems of zeolitic structural analysis and absolute configuration determination, etc.
However, even with these advancements, there are still many challenging issues that need to be resolved. For instance, one of the most daunting situations is that we are still far from truly rational design and precise synthesis towards zeolitic materials instead of relying on high-throughput trial-and-error screening for the optimization of synthetic conditions. Secondly, scoring of OSDA virtual screening always focuses on the thermodynamics of binding energy, but in actual zeolitic systems this physical quantity is hardly observable, making it difficult to judge or calibrate calculated value with practical measurements. Additionally, characterizing the enantiomeric excess of the bulk phase of solid porous materials in place of the simple counting method is also undetermined. Furthermore, the types of catalytic reactions and substrate scope are still extremely limited; to catch up with the enantiomeric excess values to the level of classical organic or transition metal catalysis in organic reactions is also of great expectation. It is because of these challenges that the field of chiral zeolites continues to present exciting possibilities for further meaningful advancements.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support from Institute of Chemistry, Chinese Academy of Sciences is gratefully acknowledged. We thank Prof. Qi-Yu Zheng from Institute of Chemistry, Chinese Academy of Sciences for helpful discussion.
C. Martinez, A. Corma, Coord. Chem. Rev. 255 (2011) 1558–1580. doi: 10.1016/j.ccr.2011.03.014
J.Y. Li, A. Corma, J.H. Yu, Chem. Soc. Rev. 44 (2015) 7112–7127. doi: 10.1039/C5CS00023H
M. Dusselier, M.E. Davis, Chem. Rev. 118 (2018) 5265–5329. doi: 10.1021/acs.chemrev.7b00738
E. Pérez-Botella, S. Valencia, F. Rey, Chem. Rev. 122 (2022) 17647–17695. doi: 10.1021/acs.chemrev.2c00140
B. Yue, S.S. Liu, Y.C. Chai, et al., J. Energy Chem. 71 (2022) 288–303. doi: 10.1016/j.jechem.2022.03.035
B.M. Weckhuysen, J.H. Yu, Chem. Soc. Rev. 44 (2015) 7022–7024. doi: 10.1039/C5CS90100F
Y.C. Chai, W.L. Dai, G.J. Wu, N.J. Guan, L.D. Li, Acc. Chem. Res. 54 (2021) 2894–2904. doi: 10.1021/acs.accounts.1c00274
R.R. Xu, W.Q. Pang, J.H. Yu, Q.S. Huo, J.S. Chen, Chemistry of Zeolites and Related Porous Materials: Synthesis and Structure, John Wiley & Sons (Asia), Singapore, 2007.
J.M. Newsam, Science 231 (1986) 1093–1099. doi: 10.1126/science.231.4742.1093
C. Dryzun, Y. Mastai, A. Shvalb, D. Avnir, J. Mater. Chem. 19 (2009) 2062–2069. doi: 10.1039/b817497k
M.E. Davis, Nature 382 (1996) 583–585. doi: 10.1038/382583a0
M.E. Davis, Top. Catal. 25 (2003) 3–7. doi: 10.1023/B:TOCA.0000003093.74240.23
J.H. Yu, R.R. Xu, J. Mater. Chem. 18 (2008) 4021–4030. doi: 10.1039/b804136a
M.E. Davis, ACS Catal. 8 (2018) 10082–10088. doi: 10.1021/acscatal.8b03080
L. Gómez-Hortigüela, B. Bernardo-Maestro, Chiral organic structure-directing agents, Insights Into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials L. Gómez-Hortigüela (Ed. ), Springer International Publishing AG, Cham, 2017, pp. 201–244.
Y. Wang, J.H. Yu, Y. Li, Z. Shi, R.R. Xu, Chem. Eur. J. 9 (2003) 5048–5055. doi: 10.1002/chem.200305040
J.M. Newsam, M.M.J. Treacy, W.T. Koetsier, C.B. De Gruyter, Proc. R. Soc. Lond, A 420 (1988) 375–405.
J.B. Higgins, R.B. LaPierre, J.L. Schlenker, et al., Zeolites 8 (1988) 446–452. doi: 10.1016/S0144-2449(88)80219-7
X.D. Zou, T. Conradsson, M. Klingstedt, M.S. Dadachov, M.A. Okeeffe, Nature 437 (2005) 716–719. doi: 10.1038/nature04097
L.Q. Tang, L. Shi, C. Bonneau, et al., Nat. Mater. 7 (2008) 381–385. doi: 10.1038/nmat2169
A. Rojas, M.A. Camblor, Angew. Chem. Int. Ed. 51 (2012) 3854–3856. doi: 10.1002/anie.201108753
J.L. Sun, C. Bonneau, Á. Cantín, et al., Nature 458 (2009) 1154–1157. doi: 10.1038/nature07957
S.K. Brand, J.E. Schmidt, M.W. Deem, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 5101–5106. doi: 10.1073/pnas.1704638114
R. de la Serna, D. Nieto, R. Sainz, et al., J. Am. Chem. Soc. 144 (2022) 8249–8256. doi: 10.1021/jacs.2c01874
L. Gómez-Hortigüela, M. Á. Camblor, Introduction to the zeolite structure-directing phenomenon by organic species: general aspects, in: L. Gómez-Hortigüela (Ed. ), Insights Into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, Springer International Publishing AG, Cham, 2017, pp. 1–41.
Y.H. Ma, P. Oleynikov, O. Terasaki, Nat. Mater. 16 (2017) 755–759. doi: 10.1038/nmat4890
Z.Y. Dong, Y.H. Ma, Nat. Commun. 11 (2020) 1588. doi: 10.1038/s41467-020-15388-5
P.B. Klar, Y. Krysiak, H.Y. Xu, et al., Nat. Chem. 15 (2023) 848–855. doi: 10.1038/s41557-023-01186-1
R. de la Serna, J. Pérez-Pariente, L. Gómez-Hortigüela, Catal. Today 426 (2024) 114389. doi: 10.1016/j.cattod.2023.114389
G.P. Moss, Pure Appl. Chem. 68 (1996) 2193–2222. doi: 10.1351/pac199668122193
H. Klapper, T. Hahn, Point-group symmetry and physical properties of crystals, International Union of Crystallography, International Tables For Crystallography, 2006, pp. 804–808.
U. Müller, Symmetry Relationships between Crystal Structures: Applications of Crystallographic Group Theory in Crystal Chemistry, Oxford University Press, Oxford, 2013.
Structure Commission of the International Zeolite Association, Database of Zeolite Structures, 2017.
R.C. Rouse, D.R. Peacor, Am. Mineral. 71 (1986) 1494–1501.
L.B. McCusker, Ch. Baerlocher, E. Jahn, M. Bülow, Zeolites 11 (1991) 308–313. doi: 10.1016/0144-2449(91)80292-8
N. Rajić, N.Z. Logar, V. Kaučič, Zeolites 15 (1995) 672–678. doi: 10.1016/0144-2449(95)00083-I
C.H. Lin, S.L. Wang, Chem. Mater. 14 (2002) 96–102. doi: 10.1021/cm010421v
L.B. McCusker, R.W. Grosse-Kunstleve, C. Baerlocher, M. Yoshikawa, M.E. Davis, Microporous Mater. 6 (1996) 295–309.
P.Y. Feng, X.H. Bu, S.H. Tolbert, G.D. Stucky, J. Am. Chem. Soc. 119 (1997) 2497–2504. doi: 10.1021/ja9634841
A.K. Cheetham, H. Fjellvåg, T.E. Gier, et al., Stud. Surf. Sci. Catal. 135 (2001) 158.
X.W. Song, Y. Li, L. Gan, et al., Angew. Chem. Int. Ed. 48 (2009) 314–317. doi: 10.1002/anie.200803578
R.W. Broach, R.M. Kirchner, Microporous Mesoporous Mater. 143 (2011) 398–400. doi: 10.1016/j.micromeso.2011.03.025
E. Pidcock, Chem. Commun. 27 (2005) 3457–2359.
Á. Valentín-Pérez, P. Rosa, E.A. Hillard, M. Giorgi, Chirality 34 (2021) 163–181.
C.S. Song, Shape-Selective Catalysis, in: Proceedings of the ACS Symposium Series, American Chemical Society, Washington, DC, 1999.
S.M. Csicsery, Zeolites 4 (1984) 202–213. doi: 10.1016/0144-2449(84)90024-1
D.R. Corbin, N. Herron, J. Mol. Catal. 86 (1994) 343–369. doi: 10.1016/0304-5102(93)E0178-J
B. Smit, T.L.M. Maesen, Chem. Rev. 108 (2008) 4125–4184. doi: 10.1021/cr8002642
J. Huang, Y.J. Jiang, V.R.R. Marthala, M. Hunger, J. Am. Chem. Soc. 130 (2008) 12642–12644. doi: 10.1021/ja8042849
C.P. Li, P. Ferri, C. Paris, et al., J. Am. Chem. Soc. 143 (2021) 10718–10726. doi: 10.1021/jacs.1c04818
A. Corma, Chem. Rev. 97 (1997) 2373–2419. doi: 10.1021/cr960406n
S.A. Che, Z. Liu, T. Ohsuna, et al., Nature 429 (2004) 281–284. doi: 10.1038/nature02529
L. Han, S.A. Che, Chem. Soc. Rev. 42 (2013) 3740–3752. doi: 10.1039/C2CS35297D
M.S. Cui, W. Zhang, L.Y. Xie, L. Chen, L. Xu, Molecules 25 (2020) 3899. doi: 10.3390/molecules25173899
J.A. Kelly, M. Giese, K.E. Shopsowitz, W.Y. Hamad, M.J. Maclachlan, Acc. Chem. Res. 47 (2014) 1088–1096. doi: 10.1021/ar400243m
J.C. Fan, N.A. Kotov, Adv. Mater. 32 (2020) 1906738. doi: 10.1002/adma.201906738
Z.S. Han, W. Shi, P. Cheng, Chin. Chem. Lett. 29 (2018) 819–822. doi: 10.1016/j.cclet.2017.09.050
W. Gong, Z.J. Chen, J.Q. Dong, Y. Liu, Y. Cui, Chem. Rev. 122 (2022) 9078–9144. doi: 10.1021/acs.chemrev.1c00740
M.X. Ma, J.H. Chen, H.Y. Liu, et al., Nanoscale 14 (2022) 13405–13427. doi: 10.1039/d2nr01772e
G.F. Liu, J.H. Sheng, Y.L. Zhao, Sci. China Chem. 60 (2017) 1015–1022. doi: 10.1007/s11426-017-9070-1
X. Han, C. Yuan, B. Hou, et al., Chem. Soc. Rev. 49 (2020) 6248–6272. doi: 10.1039/d0cs00009d
H.C. Ma, J. Zou, X.T. Li, G.J. Chen, Y.B. Dong, Chem. Eur. J. 26 (2020) 13754–13770. doi: 10.1002/chem.202001006
Y.L. Yan, X.L. Li, G. Chen, et al., Chin. Chem. Lett. 32 (2021) 107–112. doi: 10.1016/j.cclet.2020.11.063
B. Tang, W. Wang, H.P. Hou, et al., Chin. Chem. Lett. 33 (2022) 898–902. doi: 10.1016/j.cclet.2021.06.089
M.Q. Tong, D.L. Zhang, W.B. Fan, et al., Sci. Rep. 5 (2015) 11521. doi: 10.1038/srep11521
T.T. Lu, W.F. Yan, R.R. Xu, Inorg. Chem. Front. 6 (2019) 1938–1951. doi: 10.1039/c9qi00574a
J.E. Naber, K.P. de Jong, W.H.J. Stork, H.P.C.E. Kuipers, M.F.M. Post, Stud. Surf. Sci. Catal. 84 (1994) 2197–2219.
J. Shi, Y.D. Wang, W.M. Yang, Y. Tang, Z.K. Xie, Chem. Soc. Rev. 44 (2015) 8877–8903. doi: 10.1039/C5CS00626K
A.B. Halgeri, J. Das, Appl. Catal. A 181 (1999) 347–354. doi: 10.1016/S0926-860X(98)00395-0
J.C. Jansen, E.J. Creyghton, S.L. Njo, H. van Koningsveld, H. van Bekkum, Catal. Today 2 (1997) 205–212.
T.D. Baerdemaeker, B. Yilmaz, U. Müller, et al., J. Catal. 308 (2013) 73–81. doi: 10.1016/j.jcat.2013.05.025
Y.Y. Yue, H.Y. Liu, Y.N. Zhou, Z.S. Bai, X.J. Bao, Appl. Clay Sci. 126 (2016) 1–6. doi: 10.1016/j.clay.2016.02.024
G.Q. Zhang, B.C. Wang, W.P. Zhang, M.R. Li, Z.J. Tian, Dalton Trans. 45 (2016) 6634–6640. doi: 10.1039/C6DT00424E
T.T. Lu, L.K. Zhu, X.H. Wang, et al., Inorg. Chem. Front. 5 (2018) 1640–1645. doi: 10.1039/c8qi00265g
T.T. Lu, L.K. Zhu, X.H. Wang, et al., Inorg. Chem. Front. 5 (2018) 805.
J.E. Schmidt, M.W. Deem, M.E. Davis, Angew. Chem. Int. Ed. 53 (2014) 8372–8374. doi: 10.1002/anie.201404076
P. Lu, L. Gómez-Hortigüela, L. Xu, M.A. Camblor, J. Mater. Chem. A 6 (2018) 1485–1495. doi: 10.1039/c7ta10002g
Y. Shinno, K. Iyoki, K. Ohara, et al., Angew. Chem. Int. Ed. 132 (2020) 20274–20278. doi: 10.1002/ange.202008233
F. Jiao, J. Zhang, X.S. Cai, et al., Chem. Commun. 59 (2023) 1649–1652. doi: 10.1039/d2cc05850b
J.H. Kang, L.B. McCusker, M.W. Deem, C. Baerlocher, M.E. Davis, Chem. Mater. 33 (2021) 1752–1759. doi: 10.1021/acs.chemmater.0c04573
K. Qian, J.Y. Li, J.X. Jiang, et al., Microporous Mesoporous Mater. 164 (2012) 88–92. doi: 10.1016/j.micromeso.2012.06.036
F.J. Chen, Z.H. Gao, L.L. Liang, J. Zhang, H.B. Du, CrystEngComm 18 (2016) 2735–2741. doi: 10.1039/C5CE02312B
C.Q. Zhang, X. Li, X.Z. Li, et al., Chem. Commun. 55 (2019) 2753–2756. doi: 10.1039/c8cc08196d
M. Moliner, F. Rey, A. Corma, Angew. Chem. Int. Ed. 52 (2013) 13880–13889. doi: 10.1002/anie.201304713
J.D. Rimer, Nat. Catal. 1 (2018) 488–489. doi: 10.1038/s41929-018-0114-5
M.E. Davis, R.F. Lobo, Chem. Mater. 4 (1992) 756–768. doi: 10.1021/cm00022a005
R.F. Lobo, M.E. Davis, Microporous Mater. 3 (1994) 1-2.
P. Wagner, M. Yoshikawa, M. Lovallo, et al., Chem. Commun. (1997) 2179–2180.
R.F. Lobo, M.E. Davis, J. Am. Chem. Soc. 117 (1995) 3766–3779. doi: 10.1021/ja00118a013
Y. Kubota, M.M. Helmkamp, S.I. Zones, M.E. Davis, Microporous Mater. 6 (1996) 213-229.
Y. Li, J.H. Yu, Z.P. Wang, et al., Chem. Mater. 17 (2005) 4399–4405. doi: 10.1021/cm050536p
A. Turrina, P.A. Cox, Molecular modelling of structure direction phenomena, Insights Into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials L. Gómez-Hortigüela, Springer International Publishing AG, Cham, 2017, pp. 75–102.
M. Moliner, P. Serna, Á. Cantín, et al., J. Phys. Chem. C 112 (2008) 19547–19554. doi: 10.1021/jp805400u
F. Daeyaert, M.W. Deem, J. Mater. Chem. A 7 (2019) 9854–9866. doi: 10.1039/c8ta11913a
F. Daeyaert, F.D. Ye, M.W. Deem, Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 3413–3418. doi: 10.1073/pnas.1818763116
D. Schwalbe-Koda, R. Gómez-Bombarelli, J. Chem. Phys. 154 (2021) 174109. doi: 10.1063/5.0044927
C. Shi, L. Li, L.X. Yang, Y. Li, Chin. Chem. Lett. 31 (2020) 1951–1955. doi: 10.1016/j.cclet.2020.01.016
S. León, G. Sastre, J. Phys. Chem. Lett. 11 (2020) 6164–6167. doi: 10.1021/acs.jpclett.0c01734
M. Roslova, V.J. Cybulskis, M.E. Davis, et al., Angew. Chem. Int. Ed. 61 (2022) e202115087. doi: 10.1002/anie.202115087
Z. Jensen, S. Kwon, D. Schwalbe-Koda, et al., ACS Cent. Sci. 7 (2021) 858–867. doi: 10.1021/acscentsci.1c00024
L.K. Xu, X. Peng, Z.H. Xi, Z.Q. Yuan, W.M. Zhong, Chem. Eng. Sci. 282 (2023) 119188. doi: 10.1016/j.ces.2023.119188
U. Kolb, T. Gorelik, C. Kübel, M.T. Otten, D. Hubert, Ultramicroscopy 107 (2007) 507–513. doi: 10.1016/j.ultramic.2006.10.007
P. Boullay, L. Palatinus, N. Barrier, Inorg. Chem. 52 (2013) 6127–6135. doi: 10.1021/ic400529s
D.L. Zhang, P. Oleynikov, S. Hovmöller, X.D. Zou, Z. Kristallogr. 225 (2010) 94–102. doi: 10.1524/zkri.2010.1202
W. Wan, J.L. Sun, J. Su, S. Hovmöller, X.D. Zou, J. Appl. Cryst. 46 (2013) 1863–1873. doi: 10.1107/S0021889813027714
B.L. Nannenga, D. Shi, A.G.W. Leslie, T. Gonen, Nat. Methods 11 (2014) 927–930. doi: 10.1038/nmeth.3043
Y.F. Yun, X.D. Zou, S. Hovmöller, W. Wan, IUCrJ 2 (2015) 267–282. doi: 10.1107/S2052252514028188
Z.H. Huang, T. Willhammar, X.D. Zou, Chem. Sci. 12 (2021) 1206–1219. doi: 10.1039/d0sc05731b
M. Ge, X.D. Zou, Z.H. Huang, Crystals 11 (2021) 263. doi: 10.3390/cryst11030263
T.M. Yang, T. Willhammar, H.Y. Xu, X.D. Zou, Z.H. Huang, Nat. Protoc. 17 (2022) 2389–2413. doi: 10.1038/s41596-022-00720-8
C. Gao, J. Li, S. Yin, et al., Angew. Chem. Int. Ed. 58 (2019) 9770–9775. doi: 10.1002/anie.201905591
T. Sun, W. Lei, Y.H. Ma, Y.B. Zhang, Chin. J. Chem. 38 (2020) 1153–1166. doi: 10.1002/cjoc.202000120
J. Li, J.L. Sun, Acc. Chem. Res. 50 (2017) 2737–2745. doi: 10.1021/acs.accounts.7b00366
R. Uyeda, Acta. Cryst. A 24 (1968) 175–181.
P. Oleynikov, S. Hovmöller, X.D. Zou, Ultramicroscopy 107 (2007) 523–533. doi: 10.1016/j.ultramic.2006.04.032
S. Miyake, R. Uyeda, Acta. Cryst. 8 (1955) 335–342. doi: 10.1107/S0365110X55001023
J. Jansen, H.W. Zandbergen, Ultramicroscopy 90 (2002) 291–300. doi: 10.1016/S0304-3991(01)00136-X
P. Brázda, L. Palatinus, M. Babor, Science 364 (2019) 667–669. doi: 10.1126/science.aaw2560
L.J. Wang, J.Y. Li, Y. Ling, et al., CCS Chem. 5 (2023) 2466–2472. doi: 10.31635/ccschem.023.202303084
A. Corma, Curr. Opin. Solid State Mater. Sci. 2 (1997) 63–75. doi: 10.1016/S1359-0286(97)80107-6
G. Perot, M. Guisnet, J. Mol. Catal. 61 (1990) 173–196. doi: 10.1016/0304-5102(90)85154-A
A. Corma, L.T. Nemeth, M. Renz, S. Valencia, Nature 412 (2001) 423–425. doi: 10.1038/35086546
M. Sasidharan, P. Wu, T. Tatsumi, J. Catal. 205 (2002) 332–338. doi: 10.1006/jcat.2001.3440
Q.H. Xia, S.C. Shen, J. Song, S. Kawi, K. Hidajat, J. Catal. 219 (2003) 74–84. doi: 10.1016/S0021-9517(03)00154-4
E. Pérez-Mayoral, I. Matos, I. Fonseca, J. Čejka, Chem. Eur. J. 16 (2010) 12079–12082. doi: 10.1002/chem.201001593

Figure 2 Zeolite beta polymorph A with its 41 helical channel structure. Copied with permission [14]. Copyright 2018, American Chemical Society.

Figure 3 The helical channels in SU-32 with the space group P6122. Copied with permission [20]. Copyright 2008, Springer Nature Limited.

Figure 4 Synthesis of enantiomeric-enriched STW using chiral OSDAs. Copied with permission [23]. Copyright 2017, National Academy of Sciences.

Figure 5 The mesoporous channel and gyroidal surfaces in ITQ-37 with the space group P6122. Copied with permission [22]. Copyright 2009, Macmillan Publishers Limited.

Figure 6 (a) Generation of the chiral channel in the space group P6122. (b) Comparison of framework energy vs. framework density. Copied with permission [91]. Copyright 2005, American Chemical Society.

Figure 7 Illustration of a six-fold rotation of STW frameworks with different chirality. Copied with permission [23]. Copyright 2017, National Academy of Sciences.

Figure 8 Illustration of different orientations in a tilt series with different chirality. Copied with permission [27]. Copyright 2020, Authors.

Figure 9 Histogram of difference electron static potential maps of kinematical (grey) and dynamical (pink) refinements. Copied with permission [27]. Copyright 2023, Authors.

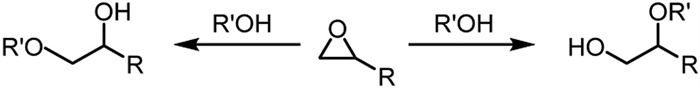
Scheme 1 Ring-opening reaction studies of stilbene oxide substrates with water catalyzed by zeolite beta.

Scheme 2 Ring-opening reaction studies of 1,2-epoxyalkane substrates with alkyl alcohols catalyzed by zeolite STW.

Figure 10 Comparison of the helical pore of STW, the OSDA used in its synthesis and the product of catalytic ring-opening reaction. Copied with permission [14]. Copyright 2018, American Chemical Society.

Scheme 3 Ring-opening reaction studies of trans-stilbene oxide substrates with n-hexanol catalyzed by zeolite GTM-3.

Scheme 4 Previously reported examples on the catalytic reaction of zeolite beta involving chiral substrates or products. Asterisks mark the chiral centers.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
 下载:
下载: