
Figure 1.
(A, B) Transmission electron microscopy (TEM) images of the magnetic MIL-101 sorbents, and their corresponding (C) N2 adsorption isotherms and (D) X-ray photoelectron spectroscopy (XPS) spectrum of Fe 2p.
Degradation of perfluorooctanoic acid by inductively heated Fenton-like process over the Fe3O4/MIL-101 composite
Xun Zhu , Chenchen Zhang , Yingying Li , Yin Lu , Na Huang , Dawei Wang

The satisfactory resistance of poly- and perfluoroalkyl substances (PFAS) to heat and oxidation make them useful for various applications in industries and consumer products [1]. However, their wide detection in the environment has raised great concerns on public health and ecological safety [2-4]. As one of the most studied PFAS, perfluorooctanoic acid (PFOA) has been proven to cause highly adverse health effects [5]. However, PFOA cannot be efficiently removed by conventional water treatment processes [6]. Even though adsorption processes have demonstrated promising performance towards PFAS removal [7-9], they require careful disposal of the sorbents and eluents [10]. Several other techniques have thus been proposed, including photolysis [11-13], sonochemical process [6,14], electrochemical process [15,16], and photocatalysis [10,17,18], to decompose PFOA. However, applications of above-mentioned techniques are often limited by their slow kinetics as the concentrations of PFOA in the environment are in low levels (ppb or even lower) and chemical reaction rates are highly dependent on the concentrations of reactants according to Arrhenius equations [2]. It is, therefore, reasonable to expect fast reaction kinetics by developing a system which is able to bring PFOA to the redox active sites.
In this communication, we report a step-wise treatment process for the rapid removal of PFOA from groundwater: The first step is adsorption, and the second step is decomposition. A magnetic MIL-101 (MIL-101 is a class of metal organic framework (MOF)) sorbent adsorbs PFOA at µg/L level within 20 min from synthetic groundwater even in the presence of multiple groundwater components (e.g., ions, anions, and natural organic matter) because of its high surface area and the complexation between Fe3+ within MIL-101 and carboxylate functional group of PFOA. Once PFOA is adsorbed, the sorbent is collected by magnet and transferred to bench reactor for the subsequent decomposition. Because Fe2+/Fe3+ within MIL-101 can catalyze Fenton reactions and produce various reactive oxygen species [19,20], the adsorbed PFOA is decomposed with the addition of H2O2. An inductive-heating effect is introduced to accelerate the decomposition of PFOA. A fast decomposition rate of PFOA is achieved as its concentration near MIL-101 is high.
Fe3O4 nanocrystals were embedded within MIL-101 to make the sorbent magnetic. The as-synthesized Fe3O4 nanocrystals are of spherical morphology and with diameters of ~20 nm (Fig. S1A in Supporting information). After the Fe3O4 nanocrystals are mixed with the precursor of MIL-101 and heated in the microwave reactor, the composites of Fe3O4 nanocrystals and MIL-101 crystals with octahedral morphology are obtained (Figs. 1A and B). As shown in Fig. 1B, Fe3O4 nanocrystals are located either on the surface or inside of the octahedral particles. Several studies have reported the application of magnetic materials/MOFs composites for the removal of contaminants from water [21-25], which often use core/shell structure where MOFs serve as shell. Presumably due to the crystal imperfections of MOFs in those studies, few of them have demonstrated rapid uptake of contaminants. In this study, we achieved producing the magnetic Fe3O4/MIL-101 with completely octahedral morphology (size ranged from 150 nm to 500 nm) [26]. Fe3O4 nanocrystals displayed a typical type-II isotherm, suggesting that they are non-porous, whereas the magnetic Fe3O4/MIL-101 composites display a typical type-I isotherm, indicating that they are microporous [27]. Because of the completely octahedral morphology of MIL-101, the Brunauer-Emmett-Teller (BET) surface area increases significantly from 52.6 m2/g for the pristine Fe3O4 nanocrystals to 960.3 m2/g for the composite (Fig. 1C). The X-ray diffraction (XRD) patterns of the materials also confirmed that the pristine spherical nanocrystals are Fe3O4 and the octahedral crystals are MIL-101 (Fig. S1B in Supporting information). The existence of coordinatively unsaturated sites of MIL-101 were verified by X-ray photoelectron spectroscopy (XPS, full spectrum is shown in Fig. S2 in Supporting information). As shown in Fig. 1D, the spectrum of Fe 2p was deconvoluted into two major peaks for 2p3/2 (710.4 eV) and 2p1/2 (724.1 eV), respectively [28]. Notably, peaks for Fe2+ (710.2 eV) and Fe3+ (712.2 eV) can be deconvoluted from the peak for Fe 2p3/2, verifying the existence of coordinatively unsaturated (Fe2+/Fe3+) sites [19].
To demonstrate the feasibility of the proposed step-wise treatment process, we spiked PFOA into a synthetic groundwater to make PFAS contaminated groundwater with a PFOA concentration of 2 ppb (4.83 nmol/L) [29]. PFOA adsorption over Fe3O4 nanocrystals and activated carbon were also conducted for comparison (Fig. 2A). The Fe3O4 nanocrystals demonstrated little adsorption towards PFOA within 20 min, possibly because of its low surface area (BET surface area is 52.6 m2/g). It was noted that activated carbon and magnetic Fe3O4/MIL-101 showed significantly different adsorption performance despite their similar BET surface areas (~800 m2/g for activated carbon and ~960 m2/g for magnetic MIL-101). Specifically, magnetic Fe3O4/MIL-101 completely removed PFOA in 20 min, whereas activated carbon removed only ~30% of PFOA during the same period. Adsorption isotherms of magnetic Fe3O4/MIL-101 were studied to plot its adsorption capacity (Fig. S3 in Supporting information). It was found that the calculated Langmuir adsorption capacity of magnetic Fe3O4/MIL-101 was among the highest ones as compared to some recently developed adsorbents for PFOA adsorption, even though it was obtained by using synthetic groundwater (possible competitive adsorption from other water components) while distilled water was used in most of other studies (Table S1 in Supporting information).
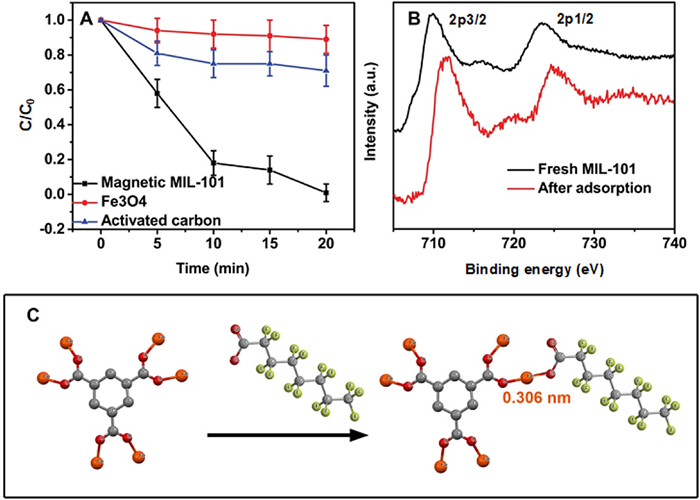
It has been recognized that the PFOA adsorption over activated carbon is mainly ascribed to physical adsorption, e.g., electrostatic forces and hydrophobic interactions [29,30]. Apparently, large surface area of activated carbon also contributed to the adsorption capacity. However, the case for magnetic Fe3O4/MIL-101 could be different as Fe3+ has been proposed to be able to form complex with carboxylate group [13,31,32]. We noted that the Fe 2p3/2 peak of MIL-101 shifted from 709.9 eV to higher binding energy (711.6 eV) after PFOA adsorption as compared to the pristine one (Fig. 2B). This shift is consistent with a previous study that the formation of Fe3+/carboxylate complex increases the binding energy of Fe 2p [33]. We presume that the complexation of carboxylate group with Fe3+ from Fe3O4 nanocrystals is less likely compared to that from MIL-101 because (1) Fe3+ within Fe3O4 formed covalent bond with oxygen, which is less accessible to carboxylate group and (2) Fe3O4 nanocrystals have a much lower surface area compared to MIL-101. The complexation between Fe3+ and carboxylate group was further strengthened by the relatively high bonding energy between them according to density functional theory (DFT) simulation: the bonding energy between the Fe3+ of MIL-101 and carboxylate group of PFOA is 120.5 kcal/mol and the bonding distance is 0.306 nm (Fig. 2C, simulation details see Supporting information) [34]. As a result, the PFOA adsorption over magnetic Fe3O4/MIL-101 can be ascribed to the complexation between PFOA and the Fe3+ within MIL-101. Meanwhile, because the porous structure and high surface area of MIL-101, a variety of Fe3+ are exposed to PFOA, benefitting the complexation and thus adsorption.
Followed by the adsorption step, magnetic Fe3O4/MIL-101 along with the adsorbed PFOA were separated from the treated groundwater using magnet (Fig. 3A) and transferred into distilled water to decompose PFOA. H2O2 was added to the distilled water solution of magnetic Fe3O4/MIL-101/PFOA to initiate the Fenton reactions. As mentioned above, MIL-101 with coordinatively unsaturated (Fe2+/Fe3+) sites was selected in this study because of its potential role as modified Fenton's reagent (Eqs. 1–5) [20]:
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
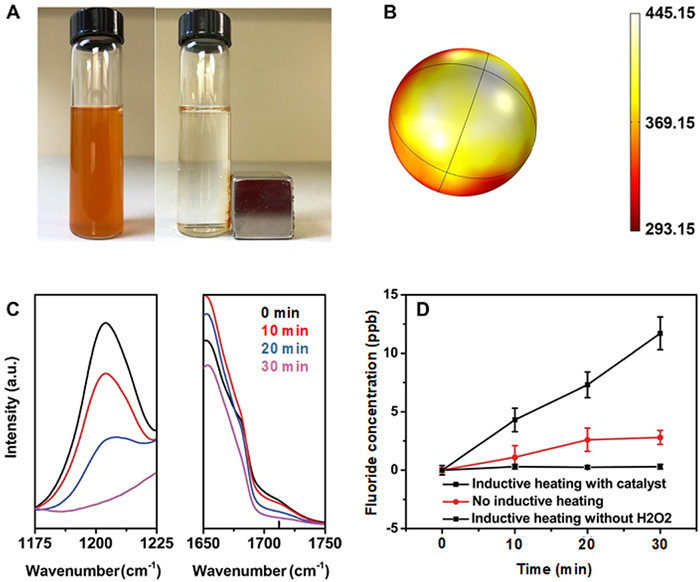
To boost the reaction rate, we introduced the inductive heating for the first time into the Fenton reactions by applying the alternative magnetic field on the reaction medium (details see Experimental section in Supporting information). The principle of inductive heating is based on the heating properties of magnetic nanoparticles upon exposure to a constantly changing magnetic field [35]. Recently, this concept has been becoming an alternative option to introduce thermal energy to a reaction system and been used in the chemical synthesis [36-38]. Since the thermal energy is directly produced by the magnetic nanoparticles, it is possible to heat the catalysts to a temperature higher than the boiling point of the reaction medium (e.g., water for the Fenton reactions). In this way, we can expect a fast reaction rate which is otherwise difficult to achieve by using conventional heating method. We simulated the temperature gradient near a Fe3O4 nanoparticle (diameter = 20 nm) and found that the temperature of the particle was 445.15 K (Fig. 3B) under the experimental conditions (simulation details see Supporting information). According to Arrhenius equation, the increase of temperature can result into an exponential increase of reaction rate. Since Fe3O4 nanocrystals are located either inside or on the surface of MIL-101, it can accelerate the relevant reactions significantly which happen nearby. It should be noted that MIL-101 is one of the most thermally stable MOFs, which can maintain its stability up to 543 K [39].
To monitor the decomposition of PFOA on the surface of magnetic Fe3O4/MIL-101, we used FTIR (Fourier transform infrared spectrometer) to measure the intensities of characteristic peaks of the functional groups in PFOA. As shown in the full FTIR spectrum of pure PFOA chemical, it has two characteristic peaks at around 1200 and 1700 cm−1, corresponding to -CF2 and -COO−, respectively (Fig. S4A in Supporting information) [40]. These two peaks were also observed for the magnetic MIL-101 after PFOA adsorption (Fig. S4B in Supporting information). The zoomed-in spectra of -CF2 and -COO− (Fig. 3C) clearly showed that their FTIR peak intensities decreased over time when the magnetic MIL-101 along with the adsorbed PFOA were subject to the inductive heating-assisted Fenton process. Initially, the PFOA adsorbed MIL-101 showed an intense peak for -CF2 at 1207 cm−1. After 30-min inductive heating-assisted Fenton process, this characteristic peak totally disappeared. Similar phenomenon was also observed for the -COO− (1712 cm−1). The FTIR results suggest that the adsorbed PFOA by magnetic MIL-101 is possibly removed after 30-min inductive heating-assisted Fenton process. This conclusion was further strengthened by the fluoride liberation results (Fig. 3D). We found that the concentration of fluoride in the solution increased over time and ~85% of fluorine recovered as fluoride in the solution (calculation details see Supporting information), confirming that the decrease of FTIR peaks was mainly because of the degradation of PFOA, not desorption. Meanwhile, we only observed a negligible increase of PFOA concentration during the process, further confirming the PFOA degradation during the Fenton process (Fig. S5 in Supporting information). This low amount of PFOA in the solution also indicated that most of PFOA was decomposed on the surface of magnetic Fe3O4/MIL-101, even though the desorption of PFOA may happen due to the increased temperature of magnetic Fe3O4/MIL-101 particles during the inductive heating process. On the contrary, without assistance of inductive heating, the Fenton reactions with MIL-101 as agent showed slow fluoride liberation kinetics (Fig. 3D), highlighting the advantage of inductive heating effect. According to the chemisorption measurements, the active site (Fe2+/Fe3+) density of Fe3O4 nanocrystals is 1.3 nmol/g while that for magnetic Fe3O4/MIL-101 is much higher (2.1 µmol/g), probably because of the high surface area and porous structure of MOF. We thus presume that the magnetic Fe3O4/MIL-101 has more active sites than conventional heterogeneous Fenton catalysts do though few studies have reported the active sites number of catalysts for Fenton reactions. Another control experiment which was conducted in the absence of H2O2 (but with MIL-101 and inductive heating) only showed a negligible production of fluoride, suggesting that pyrolysis and thermally generated reactive oxygen species (ROS) play a minor role in PFOA degradation in our case.
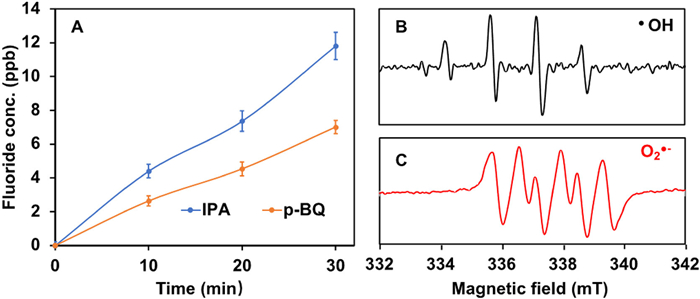
Conclusions from previous studies provide an immediate insight as hydroxyl radicals (•OH) produced by Fenton reactions is not active toward PFOA, and thus the other ROS including superoxide radical anion (O2•−) and hydroperoxide anion (HO2−) may contribute most significantly to the rapid decomposition of PFOA [20]. Indeed, the scavenging experiments showed that when isopropanol alcohol (IPA) was added as the scavenging agent of •OH, the fluoride liberation was not affected (Fig. 4A) as compared to the case in the absence of any scavengers, although the electron paramagnetic resonance (EPR) results confirmed the presence of •OH (Fig. 4B) [41-43]. However, when p-benzoquinone (p-BQ) was added as the scavenger of O2•−, the fluoride liberation was significantly inhibited, suggesting the dominating role of O2•− in the PFOA degradation. The presence of O2•− was further verified by EPR (Fig. 4C) [41]. We also noted that in the absence of the alternative magnetic field, EPR tests only confirmed the production of •OH not O2•−, indicating that the alternative magnetic field changed the production pathways of ROS. A thoroughly mechanistic study on how these different reactive oxygen species evolved will be the focus of future study.
As clearly shown in the full FTIR spectra (Fig. S4B in Supporting information), the peak intensity of magnetic Fe3O4/MIL-101 decreased over time during the inductive heating-assisted Fenton process, indicating a possible destruction of the MIL-101 crystallinity. This is reasonable as the reaction process is highly intensive and MIL-101 is made of organic compounds, which can also possibly be decomposed by the active species produced by the Fenton process. This decomposition of MIL-101 was verified by total organic carbon measurement, which increased from 0.09 mg/L to 3.69 mg/L after the one cycle. TEM image of the used magnetic Fe3O4/MIL-101 showed rougher surface as compared to the pristine one, also implying that its structural integrity has been undermined (Fig. S6 in Supporting information). However, the PFOA adsorption kinetic rates over magnetic Fe3O4/MIL-101 did not show any significant decrease over the next four consecutive runs, probably because the amount of PFOA in the synthetic groundwater was negligible compared to the adsorption capacity of MIL-101 (Fig. S7A in Supporting information). The inductive heating-assisted Fenton process was also conducted between each adsorption cycle, but the fluoride liberation kinetic rates decreased over runs (Fig. S7B in Supporting information). Therein, the destruction of MIL-101 may decrease the available amount of Fe3+/Fe2+ sites serving as Fenton agents. It is reasonable that when stronger magnetic field is applied in pilot-scale and even full-scale applications, the destruction of MIL-101 will be faster. Despite the unsatisfactory stability during the inductive heating Fenton process, magnetic Fe3O4/MIL-101 along with the proposed adsorption and subsequent decomposition strategy is still promising for practical applications as the magnetic MIL-101 can be considered as a consumable "Fenton agent". Meanwhile, by modifying magnetic Fe3O4/MIL-101 with specific functional groups, it could achieve the fast adsorption of other PFACs [44-46], and thus enable their possibly subsequent decomposition. Even though the test with field groundwater is necessary to fully demonstrate the promising features of this technology in the future, we can still envision a scenario that contaminants in the environment can be moved and thus treated under industrial conditions. This process may help address the concerns on byproducts formation in the environment during the advanced oxidation/reduction processes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors would like to thank the financial support from the National Key Research and Development Program of China (No. 2022YFC3205300) and National Natural Science Foundation of China (Nos. 52100178 and 52370072).
Supplementary material associated with this article can be found, in the online version, at doi:
J.P. Giesy, K. Kannan, Environ. Sci. Technol. 36 (2002) 146A–152A. doi: 10.1021/es022253t
X.C. Hu, D.Q. Andrews, A.B. Lindstrom, et al., Environ. Sci. Technol. Lett. 3 (2016) 344–350. doi: 10.1021/acs.estlett.6b00260
M. Houde, A.O. De Silva, D.C.G. Muir, R.J. Letcher, Environ. Sci. Technol. 45 (2011) 7962–7973. doi: 10.1021/es104326w
M. Murakami, K. Kuroda, N. Sato, et al., Environ. Sci. Technol. 43 (2009) 3480–3486. doi: 10.1021/es803556w
R.C. Lewis, L.E. Johns, J.D. Meeker, Int. J. Environ. Res. Health 12 (2015) 6098–6114. doi: 10.3390/ijerph120606098
C.D. Vecitis, H. Park, J. Cheng, B.T. Mader, M.R. Hoffmann, Front. Environ. Sci. Eng. 3 (2009) 129–151. doi: 10.1007/s11783-009-0022-7
A. Alsbaiee, B.J. Smith, L. Xiao, et al., Nature 529 (2015) 190.
L. Xiao, Y. Ling, A. Alsbaiee, et al., J. Am. Chem. Soc. 139 (2017) 7689–7692. doi: 10.1021/jacs.7b02381
Y. Ling, M.J. Klemes, S. Steinschneider, W.R. Dichtel, D.E. Helbling, Water Res. 154 (2019) 217–226. doi: 10.1016/j.watres.2019.02.012
D. Wang, A.L. Junker, M. Sillanpää, Y. Jiang, Z. Wei, Engineering 23 (2023) 19–23. doi: 10.1016/j.eng.2022.08.005
Y. Gu, W. Dong, C. Luo, T. Liu, Environ. Sci. Technol. 50 (2016) 10554–10561. doi: 10.1021/acs.est.6b03261
L. Jin, P. Zhang, T. Shao, S. Zhao, J. Hazard. Mater. 271 (2014) 9–15. doi: 10.1016/j.jhazmat.2014.01.061
D. Liu, Z. Xiu, F. Liu, et al., J. Hazard. Mater. 262 (2013) 456–463. doi: 10.1016/j.jhazmat.2013.09.001
J. Cheng, C.D. Vecitis, H. Park, B.T. Mader, M.R. Hoffmann, Environ. Sci. Technol. 44 (2010) 445–450. doi: 10.1021/es902651g
H. Lin, J. Niu, J. Xu, et al., Environ. Sci. Technol. 47 (2013) 13039–13046. doi: 10.1021/es4034414
J. Niu, H. Lin, C. Gong, X. Sun, Environ. Sci. Technol. 47 (2013) 14341–14349. doi: 10.1021/es402987t
D. Huang, L. Yin, J. Niu, Environ. Sci. Technol. 50 (2016) 5857–5863. doi: 10.1021/acs.est.6b00652
X. Li, P. Zhang, L. Jin, et al., Environ. Sci. Technol. 46 (2012) 5528–5534. doi: 10.1021/es204279u
P. Zhou, W. Ren, G. Nie, et al., Angew. Chem. Int. Ed. 59 (2020) 16517–16526. doi: 10.1002/anie.202007046
S.M. Mitchell, M. Ahmad, A.L. Teel, R.J. Watts, Environ. Sci. Technol. Lett. 1 (2014) 117–121. doi: 10.1021/ez4000862
Q. Shi, M. Cheng, Y. Liu, et al., Chem. Rev. 499 (2024) 215500.
C. Guo, M. Cheng, G. Zhang, et al., Environ. Sci. Nano 10 (2023) 1528–1552. doi: 10.1039/D3EN00007A
H. Peng, W. Xiong, Z. Yang, et al., Chem. Eng. J. 457 (2023) 141317. doi: 10.1016/j.cej.2023.141317
C. Tang, M. Cheng, C. Lai, et al., Chem. Eng. 11 (2023) 110395.
Y.F. Huang, Y.Q. Wang, Q.S. Zhao, Y. Li, J.M. Zhang, RSC Adv. 4 (2014) 47921–47924. doi: 10.1039/C4RA05515B
M. Zhao, K. Yuan, Y. Wang, et al., Nature 539 (2016) 76–80. doi: 10.1038/nature19763
S. Lowell, J.E. Shields, M.A. Thomas, M. Thommes, Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Springer, Netherlands, 2004.
S. Vadahanambi, S.H. Lee, W.J. Kim, I.K. Oh, Environ. Sci. Technol. 47 (2013) 10510–10517.
X. Xiao, B.A. Ulrich, B. Chen, C.P. Higgins, Environ. Sci. Technol. 51 (2017) 6342–6351. doi: 10.1021/acs.est.7b00970
Q. Yu, R. Zhang, S. Deng, J. Huang, G. Yu, Water Res. 43 (2009) 1150–1158. doi: 10.1016/j.watres.2008.12.001
B.C. Faust, R.G. Zepp, Environ. Sci. Technol. 27 (1993) 2517–2522. doi: 10.1021/es00048a032
Y. Wang, P. Zhang, G. Pan, H. Chen, J. Hazard, Mater. 160 (2008) 181–186.
R.D. Feltham, P. Brant, J. Am. Chem. Soc. 104 (1982) 641–645. doi: 10.1021/ja00367a001
A. López-Cruz, G.E. López, Mol. Phys. 107 (2009) 1799–1804. doi: 10.1080/00268970903078567
S. Ceylan, C. Friese, C. Lammel, K. Mazac, A. Kirschning, Angew. Chem. Int. Ed. 47 (2008) 8950–8953. doi: 10.1002/anie.200801474
J. Hartwig, S. Ceylan, L. Kupracz, L. Coutable, A. Kirschning, Angew. Chem. Int. Ed. 52 (2013) 9813–9817. doi: 10.1002/anie.201302239
H.M. Torres Galvis, J.H. Bitter, C.B. Khare, et al., Science 335 (2012) 835– 838. doi: 10.1126/science.1215614
A. Meffre, B. Mehdaoui, V. Connord, et al., Nano Lett. 15 (2015) 3241–3248. doi: 10.1021/acs.nanolett.5b00446
P. Horcajada, S. Surble, C. Serre, et al., Chem. Commun. 27 (2007) 2820– 2822.
X. Gao, J. Chorover, Environ. Chem. 9 (2012) 148–157. doi: 10.1071/EN11119
Y. Jiang, P. Wang, T. Chen, et al., Appl. Catal. B: Environ. 343 (2024) 123468. doi: 10.1016/j.apcatb.2023.123468
L. Jin, S. You, N. Ren, B. Ding, Y. Liu, Environ. Sci. Technol. 56 (2022) 11750–11759. doi: 10.1021/acs.est.2c03904
Y. Liu, F. Li, Q. Xia, et al., Nanaoscale 10 (2018) 4771–4778. doi: 10.1039/C7NR09435C
M.J. Klemes Ling, L. Xiao, et al., Environ. Sci. Technol. 51 (2017) 7590–7598. doi: 10.1021/acs.est.7b00906
K. Liu, S. Zhang, X. Hu, et al., Environ. Sci. Technol. 49 (2015) 8657–8665. doi: 10.1021/acs.est.5b00802
M. Ateia, M.F. Attia, A. Maroli, et al., Environ. Sci. Technol. Lett. 5 (2018) 764–769. doi: 10.1021/acs.estlett.8b00556

Figure 1 (A, B) Transmission electron microscopy (TEM) images of the magnetic MIL-101 sorbents, and their corresponding (C) N2 adsorption isotherms and (D) X-ray photoelectron spectroscopy (XPS) spectrum of Fe 2p.

Figure 2 (A) Adsorption kinetics of three adsorbents used in this study. (B) Fe 2p XPS spectra of fresh magnetic MIL-101 and the one after PFOA adsorption. (C) Proposed adsorption mechanism through the complexation between Fe and -COO−.

Figure 3 (A) Magnetic separation of PFOA adsorbed MIL-101 from synthetic groundwater, the separation time was around 15 min. (B) Temperature gradient near a Fe3O4 nanoparticle (Unit of temperature: K, inductive heating source: 300 W and 60 Hz). (C) Time-dependent FTIR spectra of the asymmetric stretching band for -CF2 (left, located at ~1207 cm−1) and for -COO− (right, located at ~1712 cm−1) of the MIL-101 during the inductive heating Fenton reactions and (D) fluoride liberation during the process.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: