Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
b.
School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
Received Date:
27 November 2023 Accepted Date:
05 March 2024 Revised Date:
29 February 2024 Available Online:
15 August 2024
Abstract:
Organic thermoelectric (OTE) materials and devices have garnered significant attention in the past decade for flexible and wearable electronics. Due to the numerous combinations of different backbones, side chains, and functional groups for polymer molecules, further efficient developments of high performance OTEs rely on reverse and rational molecular design as well as fundamental understanding to the structure-property relationship, which both require precise theoretical input. Recently, many theoretical efforts and progresses have been made to predict TE properties and develop high performance OTE materials. Here, we present first the general methods and principles for OTE theoretical calculations. Subsequently, the latest theoretical advances regarding the effects of molecular design, chemical doping, ambipolar charge transport etc., to TE conversion are carefully reviewed. These theoretical advances not only significantly deepen the fundamental understanding of OTEs, but also provide precise guidance to the molecular design of OTE materials. Finally, we propose several perspectives for future theoretical investigations of OTEs.
With two-thirds of total energy consumption being lost in the form of waste heat, thermoelectricity has become a compelling solution for the key issues of energy and environmental protection, as it could realize the direct conversion between heat and electricity or vice versa in a clean manner [1,2]. The performance of thermoelectric (TE) materials is evaluated by the dimensionless figure of merit ZT = S2σT/κ, where S is Seebeck coefficient, σ is electrical conductivity, and κ is the thermal conductivity [3]. Physically the ZT value represents the TE energy conversion efficiency [4,5]. In the past decade, organic thermoelectrics (OTEs) have attracted great attention due to their unique advantages in TE conversion such as ultralow intrinsic thermal conductivity and excellent room temperature performance. Moreover, from the perspective of functional TE devices, their intrinsic ultrahigh flexibility, light-weight, and non-toxic are particular desired to power the rapid developing flexible, wearable, and implantable micro-electronics for artificial intelligence, health monitoring, Internet of things (IoTs), and other emerging fields, by utilizing environmental and even body heat at room temperature in a versatile way [6-10].
Significant experimental as well as theoretical progresses have been made in the field of OTEs very recently [11-13]. For example, by selenium substitution, Ding et al. reported a high performance diketopyrrolopyrrole (DPP) derivative PDPPSe-12 with enhanced intermolecular interaction, which resulting in a maximum power factor and ZT up to 364 µW m−1 K−2 and 0.25, respectively [14]. In 2022, Wang et al. further boosted the TE performance of a p-type DPP-based polymer (DPP-BTz) via molecular packing orientation engineering, which ZT value remarkably reaches 0.40 [15]. For the n-type OTE material, in 2023 Han et al. developed a PSpF-doped PDPIN blended all-polymer system with a record-high maximum ZT of 0.53 by using polymer dopant, which simultaneously exhibits high power factor and ultralow thermal conductivity [16]. Also in 2023, Yu et al. reported a series of thiophene-based TBDOPV copolymer (TBDOPV-T) with excellent bipolar TE performance [17]. Through modulating the energetic disorder by reducing the driving force of alkyl side chains, the N-DMBI–doped n-type TBDOPV-T-518 reveals a high power factor more than 200 µW m−1 K−2 while the p-type FeCl3-dopeTBDOPV-T-518 shows the high conductivity and power factor over 400 S/cm and 100 µW m−1 K−2, respectively. On the theoretical side, Ma et al. developed an elegant semi-empirical relation between the Seebeck coefficients and electrical conductivity, which could phenomenologically illustrate the charge transport picture in organic polymers [18]. Later in 2022, Ge et al. by combining the hopping transport theory described by Fermi's golden law with the bandlike transport described by the Boltzmann transport equations, proposed a unified model for the general TE conversion process in polymeric semiconductors [19].
Even so, OTE materials are still far away from practical applications in terms of performance. There are several major challenges for further developments of OTE materials and devices. One is how to break the trade-off relation among the Seebeck coefficients, electrical conductivity, and thermal conductivity. Although by developing high mobility organic semiconductors, density of states engineering, and fine controlling of polymer microstructures, the trade-off relation between electrical conductivity and Seebeck coefficients have been partially overcome and both n-type and p-type organic semiconductors could exhibit conductivity near or over 1000 S/cm while simultaneous exhibit a promise Seebeck coefficient, the thermal conductivity has become a new limitation. Because, high electrical conductivity will lead to a pronounced electronic contribution to the thermal conductivity while high mobility organic semiconductors often have relatively good crystallinity which also lead to relatively high lattice thermal conductivity. Another challenge would be the stability in the doped state, which is essential for practical OTE applications. Due to the nature of chemical doping, most doped organic semiconductors would experience relatively fast de-doping process in ambient environment. The mechanism of de-doping and effects of counterions remains to be elusive. Finally, a central obstacle for further developments of OTE materials is the lack of fundamental understanding to the structural-property relationship as well as their puzzling charge transport mechanism. Although different charge transport mechanisms range from localized (hopping), de-localized (band-like), transient localization, and several semi-empirical models have been proposed, no one has been fully verified by experiments. These limitations urgently require systematic theoretical efforts to overcome. Because, from the view point of molecular and dopant design, current developments of high performance OTE systems are more relies on an empirical or semi-empirical way, while the various effects of different functional groups, conjugated cores, side chains, solutions, and packing orderings to the macroscopic TE performance remain elusive [20-22]. Although experimentally they can be summarized by extensive investigations with novel characterization techniques on different length scales, precise theoretical calculations would be a more efficient and scientific route. Since it not only performs high-capacity predictions of various TE properties including Seebeck coefficients, conductivity, and thermal conductivity, but also could advance the understanding to the charge and phonon transport on the molecular level. In addition to the intrinsic structural effects of polymers, external effects such as doping and various physical field-modulations have become key factors to improve the OTE performance, which also greatly require theoretical guidance [23,24]. For example, theoretical predictions of the optimized doping level of polymers for best TE performance would be highly desired before experimental efforts are devoted. Therefore, as illustrated in Fig. 1, it is crucial to establish a systematic bidirectional relationship between the basic the molecular design as well as external influences such as chemical doping with practical TE properties and performance, via theoretical investigations of the TE conversion process and predictions of charge and phonon transport properties in OTE materials [25].
Figure 1
Figure 1.
Illustration of the relationship between molecular design of OTE materials, electronic structure and thermoelectric properties. Start with basic molecular structure and packing, by calculating the electronic structure, their TE properties and performances can be predicted. (a) The conjugated core structure of organic semiconductors facilitates charge transport. Side chains play a role in solution processing and can impact the generation of charge carriers and the assembly of molecules in the solid state. (b) Doping adjusts the HOMO and LUMO energy levels of organic thermoelectric materials by altering charge distribution and electron affinity. This modulation influences the charge transport properties and thermoelectric performance of the material. Such control can enhance the conductivity and thermoelectric efficiency, making it more suitable for thermoelectric conversion applications. (c) The molecular packing in organic thermoelectric materials exhibits both face-on and edge-on configurations, where molecules can stack either with their faces oriented toward each other or with their edges in alignment. The molecular orientations also could significantly affect the TE performance. (d) The electronic energy levels are determined by the highest occupied molecular orbital (HOMO), representing ionization energy, and the lowest unoccupied molecular orbital (LUMO), representing electron affinity. These levels can be adjusted through molecular design. Energetic disorder may lead to a distribution of states around these levels in perfectly ordered materials. Schematic diagram of the energy band structure of organic thermoelectric materials. (e) Schematic representation of the phonon spectrum of organic thermoelectric materials. (f) Schematic diagram illustrating the variation of thermoelectric parameters with carrier concentration. With increasing carrier concentration, electrical conductivity, thermal conductivity, Seebeck coefficient, and power factor exhibit different trends. The ZT value (thermoelectric figure of merit), as a comprehensive parameter, the optimal ZT value often corresponds to a specific carrier concentration, providing crucial insights for the design optimization of thermoelectric materials.
So far, many reviews have focused on the experimental side, including both p-type [26,27] and n-type materials [28,29], chemical doping [30,31], and device applications [32,33]. However, with the rapid development of OTEs, it is necessary to review the basic theoretical methods for OTE polymeric materials as well as the latest theoretical advances. Here, we introduce the primary theoretical principles and procedures for computing various TE properties. In the next, recent theoretical progresses on the OTE properties as well as single-molecular TEs are reviewed. Finally, perspectives toward OTE theoretical calculation prospects are highlighted such as establishing OTE database, which could guide future experiments and boost the OTE performance.
These theoretical advancements play a pivotal role in the field of organic thermoelectric (OTE) materials, contributing to a better understanding, design, and optimization of such materials, thereby driving the development of flexible and wearable electronic products. Firstly, through theoretical models, we can delve into the electronic structure and transport properties of OTE materials, providing profound insights into their intrinsic working principles. This aids in predicting and interpreting experimental observations, laying the theoretical foundation for improving material performance.
Secondly, these theoretical advancements offer critical insights guiding the design of OTE materials. By simulating and calculating the impact of different molecular structures, doping methods, and interface effects on material performance, we can strategically design materials to optimize their thermoelectric properties. This theoretical support for innovative material design holds the potential to propel breakthroughs in the performance of organic thermoelectric materials.
Ultimately, these theoretical advancements are crucial for advancing the development of flexible and wearable electronic products. With a deeper understanding and more effective design, OTE materials can better adapt to flexible and bendable electronic devices, enhancing their performance and stability in practical applications. This opens up new possibilities for innovation in fields such as smart textiles and wearable sensors, positioning organic thermoelectric materials to play a more critical role in the realm of electronic products. Therefore, these theoretical advancements have a significant and positive impact on driving the development of flexible and wearable electronic products.
Organic thermoelectrics pose challenges due to their relatively low thermoelectric conversion efficiency, complex structure, and difficulty in preparation. There may also be problems with stability, sustainability and scalability. Moreover, for the doping and transport processes in semiconductor polymers, it is challenging in experiments to pinpoint the doping sites. The theoretically determined doping sites are difficult to incorporate into models, considering the interactions between different phases. Constructing models for polymer systems is challenging, and the debate between band-like models and hopping models adds complexity. The definitive determination of which model accurately represents polymers remains unsettled.
2.
Methods and key parameters
The film of organic polymers is composed by both crystalline and amorphous regions. For the amorphous regions, there are no available unified physical models which have demonstrated to be successful to describe the charge transport inside. So far, the theoretical calculations of organic polymers are mostly focused on the crystalline regions. Fig. 2 illustrated the common calculation procedures. Usually, theoretical calculations begin with constructing the structural models for the crystalline regions and identifying their Brillouin zones, based on their molecular structures and the experimentally measured molecular distances of different orientations by GIWAXS or other structural characterization techniques, using packages such as VASP or Materials Studio.
Figure 2
Figure 2.
(a) Modelling of organic crystal region and the Brillouin zone of organic polymers. (b) Theoretical methods of calculation: molecular dynamics, ab initio DFT, and Boltzmann transport theory. (c) Performance predictions of OTE materials versus carrier concentration and temperature.
Various calculation methods including molecular dynamics (MD), density functional theory (DFT), and Boltzmann transport theory are applied to the as-constructed models of organic polymers [34]. In particular, MD can directly calculate the motion of atomic nuclei based on the classical Newton's laws under the force field generated by all other particles and charges. The resulting system physical properties are derived by statistical averaging of each particle. The DFT is a quantum mechanical computational method, which has been demonstrated to be an important approach to calculate the electronic properties of realistic multi-electron systems. Once the electronic structure of OTE materials is achieved either classically or quantum-mechanically, the semiclassical Boltzmann transport equations will be employed to describe their charge transport processes under external physical fields including electric field, magnetic field, and thermal or chemical potential gradients [35,36]. By solving the Boltzmann transport equations, specific charge transport properties can be directly obtained, as it takes into account both the material electronic information and charge scattering mechanisms, allowing for a prediction of the TE properties of organic polymers including Seebeck coefficients, electrical conductivity, and thermal conductivity versus various physical parameters such as carrier density and temperature [37]
2.1
Seebeck coefficient
The Seebeck coefficient of a material is defined as the magnitude of the generated electrical field divided by the applied temperature gradient. Theoretically, it is be described by Boltzmann transport equation as follows (Eqs. 1 and 2),
(1)
(2)
where kBis the Boltzmann constant, e is the elementary charge, T is temperature, σ is the conductivity, EF is the Fermi level, and σ(E) is the density of states and Fermi distribution involved energy dependent conductivity [38,39]. As illustrated in Eq. 1, it is obvious that the magnitude of the Seebeck coefficient is highly dependent on the environmental temperature, the Fermi level position, and the distribution of density of states.
For metals or degenerate semiconductors with parabolic band, the Seebeck coefficients can be simplified by assuming the charge mobility is energetically independent around the Fermi level as shown in Eq. 3.
(3)
This equation clearly indicates the Seebeck coefficients is inversely correlated with carrier concentration while proportional to the temperature, which is qualitatively consistent with the typical doping effect and thermal activation picture of most organic semiconductors [38].
In addition to T and charge carrier concentration n, the effective mass m* is another important physical parameter which is closely related to the TE conversion as it is positively proportional to the Seebeck coefficients while reversely correlated with charge carrier mobility. In general, the effective mass m* can be obtained from the curvature of the top of the valance band or the bottom of conduction band for holes or electrons, respectively, as follows (Eq. 4),
(4)
where k is parallel to its spatial vector, E is the energy of the band, and ℏ is the reduced Planck constant [40,41]. Therefore, once the electronic information including band structure and energy dependent density of states plot are achieved, the m* can be obtained. The isotropic material Seebeck coefficients can then be straightforwardly calculated and plotted with temperature or carrier concentration. In particular, the negative calculated Seebeck coefficients would indicate an electron dominating n-type charge transport, while positive values indicate p-type hole dominating charge transport.
By applying the presented method, Shi et al. have predicted the Seebeck coefficients of C8-BTBT based on molecular structure and electronic structure information [42]. Fig. 3a shows the optimized crystal structure of C8-BTBT in the ab and ac plane. Based on this, its band structures and DOS were achieved by performing the first-principle DFT calculations. As shown in Fig. 3b, the C8-BTBT is calculated to be p-type with a direct band-gap of 2.06 eV, since its Fermi level is obviously closer to its HOMO level than its LUMO. Subsequently, by applying the Boltzmann transport theory [43], the Seebeck coefficient of C8-BTBT is achieved and further plotted versus the charge density in Fig. 3c, which is isotropic and similar for electrons and holes along both a and b axis.
Figure 3
Figure 3.
(a) Structure of C8-BTBT in the ab and ac plane. (b) Band structures and DOS of C8-BTBT, the reciprocal coordinates of high-symmetry points are Γ=(0, 0, 0), Y = (0, 0.5, 0), X = (0.5, 0, 0), and Z = (0, 0, 0.5). The red curves indicate the valence band maximum and conduction band minimum. (c) The calculated Seebeck coefficient (S), electrical conductivity (σ), along the a-direction (blue lines) and the b-direction (red lines) for C8-BTBT, as a function of carrier concentration at 298 K. (d) TE power factor (S2σ), and electronic thermal conductivity change with the carrier concentration. Copied with permission [42]. Copyright 2014, American Chemical Society.
The electrical conductivity σ = neµ is another important parameter for OTEs, in which high charge carrier mobility µ holds the promise to beak the trade-off relation between Seebeck coefficients and electrical conductivity with a relatively small n. The key in calculating the electrical conductivity lies in the determination of the charge carrier drift mobility, which is defined as average carrier drift velocity produced by unit electric field. Physically, it represents the magnitude of carrier's ability to conduct the electricity, which can be expressed as follows (Eqs. 5–7),
(5)
(6)
(7)
where m* is the effective mass in the transport direction, T is the temperature, kB is Boltzmann constant, e is the electron charge. ħ is the approximate Planck constant, E represents the deformation potential constant along the transport direction for holes at the top of the valence band (VBM) or electrons clustered at the bottom of the conduction band (CBM) as determined by the equation E = ΔE/(Δl/l0), where ΔE is the change in energy of the CBM or the VBM under compressive or tensile strains, l is the lattice constant in the transport direction, and Δl is the amount of deformation of l0. md is the average effective mass of the carriers. C is the modulus of elasticity of a uniformly deformed crystal [42,44,45].
Since the band structure is typically non-symmetric along different path between the high symmetry points of the Brillouin zone, the resulting charge effective mass and mobility are often anisotropic depending on the crystalline direction and carrier type [46]. For polymers, such anisotropy is particularly strong, as the intrachain charge transport along the backbone direction is expected to be dramatically different from the interchain transport along the π-π direction. For example, Northrup et al. predicted the intrachain hole effective mass of 0.10m0 is much less than corresponding interchain value of 1.61m0, where m0 is the electron mass, suggesting significantly higher mobility along the backbone direction than the π-π direction [45]. In Fig. 3c, Shi et al. found the hole mobility along both a and b directions of 180 and 165 cm2 V-1 s-1 are much larger than the corresponding electron mobility of 13.4 and 28.1 cm2 V-1 s-1 along the same axes for p-type C8-BTBT, respectively [42]. The resulting electrical conductivity and TE power factors are then plotted versus the carrier concentration, as shown in Figs. 3c and d. In contrast to the isotropic Seebeck coefficient of C8-BTBT for electrons and holes, their values of conductivity are different and anisotropic.
Xi et al. have made significant advancements in the development of first-principles methods such as Bardeen and Shockley's deformation potential theory for predicting intrinsic charge mobility in carbon-based and organic nanomaterials [47]. Notably, calculations of packed organic crystals reveal that the intrinsic charge carrier mobilities for graphene sheets and naphthalene are approximately 3 × 105 and 60 cm2 V−1 s−1, respectively, at room temperature. These results are in reasonable agreement with experimental studies. Furthermore, there are novel theoretical studies indicate very high charge carrier mobilities for a class of recently discovered diacene-fused thienothiophenes reaching 100 cm2 V−1 s−1. These findings contribute to the understanding and advances of organic materials for the design and optimization of future high mobility OTE materials.
2.3
Marcus theory and Holstein-Peierls description
What we mentioned above are both based on the band-like model and the deformational potential approximation, which are usually applicable to metallic or semi-metallic organic semiconductors which often exhibit relatively good crystallinity and enable a coherent charge transport, as represented by PBTTT. These semi-metallic semiconductors normally have a very high doping-level as well as electrical conductivity. On the other hand, the hopping model and the polaron model are considered to be applicable to intrinsic or relatively low doping-level organic semiconductors. Under the hopping mechanism, the calculations of charge mobility rely on Marcus theory, considering barriers and reaction coordinates during electron transfer between molecules. These two theoretical approaches contribute to a comprehensive understanding of the mechanisms and properties of charge transport in organic thermoelectric materials.
When charge carriers migrate from one position to another, the mobility is primarily determined by transfer integrals. This formula is applicable only to intermolecular charge carrier migration since the coupling between molecules is weak [48,49]. For the calculation of interchain charge carrier mobility, a hopping model is employed due to weaker interchain couplings. The calculation methods of mobility are shown below ((8), (9), (10), (11), (12)) [50]:
(8)
where D is the diffusion coefficient, kB is the Boltzmann constant, T is the temperature [51].
(9)
here d is the intermolecular spacing and the mobility is related to D by the Einstein relation.
(10)
(11)
(12)
The charge transfer rate and interchain charge carrier mobility in a molecular system are primarily determined by two crucial factors: the reorganization energy (λ) and the transfer integral (V) between adjacent molecules. The total reorganization energy comprises internal reorganization energy (λint) and external reorganization energy (λext). However, for simplicity and due to its negligible contribution, only λint is considered, as evaluating λext, induced by the polarization of surrounding molecules, is challenging and its impact is minimal [52-55]. These values are calculated at the B3LYP/6–31G(d, p) level using Gaussian 09 code. The hole transfer integral (Vh/e), describing the strength of electronic coupling between two stacked chains, is determined by the band-fitting method based on Koopmans' theorem. Here, EH/L+1 and EH−1/L represent the energy of the HOMO/LUMO+1 and HOMO-1/LUMO orbitals, respectively [56]. These energies are obtained from the closed-shell geometry of two stacked chains in a neutral state.
In a broad sense, a polaron can be conceptualized as a quasi-particle formed by a charge entity along with its surrounding polarization cloud. The Holstein–Peierls model offers a more comprehensive framework for elucidating charge transport in molecular crystals.
(13)
Within this model, Rm denotes the Cartesian coordinate of lattice site m in the α-th direction, while Γ serves as a phenomenological parameter addressing inhomogeneous line broadening. The parameter G represents the effective electron–phonon coupling constant, calculated as . The phonon occupation number N is expressed as . The incoherent scattering factor is defined as Φλ(t)=(1+Nλ)e−iωλt+Nλeiωλt. Eq. 13 is quite versatile, incorporating both hopping and band mechanisms. When g ≫ 1, the high-temperature approximation is applicable, leading Eq. 13 to converge to the classical Marcus theory. Conversely, g ≪ 1, Eq. 13 transforms into the band description [57,58].
2.4
Thermal conductivity
The thermal conductivity is composed by electronic thermal conductivity κe, and lattice thermal conductivity κL (Eqs. 14 and 15).
(14)
(15)
For κe, it can be calculated from electrical conductivity and temperature in a straightforward way based on the Lorentz constant L [59]. For the lattice contribution of thermal conductivity κL, it is typically achieved by solving Boltzmann transport equations using non-equilibrium molecular dynamics (NEMD), within the single-mode relaxation-time approximation. Software packages such as LAMMPS are usually applied, which could simulate energy transport in a non-equilibrium state and calculate the displacement and velocity of each atom. Specifically, after building the crystalline models, simulated temperature gradients will be applied, and κL can be obtained from the resulting time dependent temperature distribution and heat flow behavior across the crystals.
Much progresses have been made in recent years in calculating thermal conductivity [12]. As shown in Fig. 4a, Shi et al. calculated the thermal conductivity of C8-BTBT along three different crystal axes using NEMD. For both a, b directions, same κL of 0.18 W m-1 K-1 are obtained due to its crystal symmetry. The resulting ultralow thermal conductivity of C8-BTBT facilitates a high ZT value reaching 0.7. Similarly, for C10-BTBT and C12-BTBT, their κL are calculated to be 0.18/0.18 W m-1 K-1 and 0.25/0.22 W m−1 K−1 for a/b direction, respectively, which are also extremely low [42].
Figure 4
Figure 4.
(a) Calculated lattice thermal conductivity of Cn-BTBTs along the a- and b-axes at room temperature and experimental results for pentacene, TPD, and Alq3. Copied with permission [85]. Copyright 2014, American Chemical Society. (b) Calculated dimensionless thermoelectric (ZT) versus the mobility for C8-BTBT, pentacene, rubrene, and TiOPc. Copied with permission [42]. Copyright 2019, Wiley Publishing Group.
With the predictions of Seebeck coefficient, charge mobility, and thermal conductivity, the material ZT values versus carrier density or temperature can be theoretically calculated by (Eq. 16) [42]:
(16)
where kB is Boltzmann constant, e is the elementary charge, T is the temperature, σ is the electrical conductivity, Np is the hole concentration, Neff is the effective density of states. Simultaneously, the thermoelectric power factor PF = S2σ, which is another important parameter to evaluate the TE performance of materials, can also be predicted. Usually, larger PF indicates higher energy output.
Therefore, from Eq. 16, what ultimately determine the OTE material ZT values are the charge mobility, lattice thermal conductivity, as well as the distribution of density of states which can be evaluated by the degree of energetic disorder. As shown in Fig. 4b, even though the effect of energetic disorder was not studied, Shi et al. calculated the ZT values of different molecules versus charge mobility and lattice thermal conductivity, in which higher charge mobility with lower lattice thermal conductivity will generally produce higher ZT values [42].
Some of the molecular structures discussed in this review are presented in Fig. 5, which contains conjugated conducting polymers such as PEDOT: TOS and poly[Ni-ett], organic semiconductors like DPP derivatives, and crystalline small molecules such as rubrene and C8-BTBT. The calculated performance of these various OTE materials is further compared with the corresponding experimentally measured values. Meanwhile, the theoretically predicted ZT values and power factors of representative high performance OTE materials are summarized and displayed in Fig. 6.
Figure 5
Figure 5.
Molecular structures of mentioned organic thermoelectric materials.
By comparison, insights into the underlying difference between the theoretical calculations and experimental values can be obtained. Specially, the theoretically calculated ZT values range from 0.01 to 20 while the experimentally measured values are much lower ranging from 0.011 to 2. The highly conducting quasi one-dimensional (Q1D) organic crystals attract a special attention [60], as they are predicted to exhibit ZT values around 20 [61]. The ZT values of molecular nanowires were also predicted to be extremely high reaching over 15 [33,62]. On the other hand, the power factors of various p- and n-type OTE materials range from 0.00051 µW m-1 K-2 to 2790 µW m-1 K-2. It should be note that the single-molecular TE devices exhibit unparalleled theoretically predicted as well as experimental performance, as compared to other bulk OTE materials, due to various quantum effects. For example, the constructive quantum interference (CQI)) could enhance the transmission probability of electrons which in turn improves the electrical transport capacity in single molecule devices.
Moreover, it can be found that in general the theoretically computed values for thermoelectric figure of merits and power factors tend to be higher than the corresponding experimental values of OTE materials. This disparity can be attributed to several factors. For example, theoretical calculations only account for single crystalline regions while realistic OTE materials could be polycrystalline, paracrystalline, or partially amorphous. The non-crystalline regions normally would hinder the effective charge transport as well as the TE conversion process. Other factors that influence the realistic performance of OTE materials such as grain boundaries, defects and impurities, coulomb interaction, and charge scatterings are often simplified with idealized assumptions in theoretical models.
3.
Recent advances in OTE calculation
In this section, recent theoretical advances in the field of OTE materials and devices are comprehensively reviewed, including the effects of doping and temperature on TE properties, ambipolar TE conversion, structural and disorder effects, and single-molecular TE properties.
3.1
Doping effect
Doping has been extensively utilized to regulate the concentration of charge carriers as well as stimulate novel physical properties in inorganic semiconductors [63,64]. However, when it comes to conjugated polymers based OTE materials, achieving effective chemical doping faces challenges due to the inherent trade-off between doping level and charge carrier mobility. This issue arises because dopants tend to distribute randomly within the polymer, leading to significant structural and energetic disorder, which cannot be probed directly by most experimental techniques. Therefore, as discussed above, theoretical investigations of microscopic doping mechanism, the aggregate structure of doped polymers, and the best doping condition for the highest TE performance are essential to guide further experimental optimizations and to assist the design of high-performance OTE materials and dopants. Specifically, DFT calculations could predict the ideal amount, doping site, and charge transfer quantities of different target dopants in polymer thin film structures, via calculating the electronic structure variations before and after the doping process as well as the performance under different doping conditions.
For p-type doping, using DFT methods, Di Nuzzo et al. employed vibrational spectrum simulations to investigate the effect of dopant positioning along the copolymer backbone to the charge carrier transport properties. They revealed that efficient charge transfer exclusively takes place when the dopant is in proximity to the donor moiety. Interestingly, the results also demonstrate that the donor-acceptor configuration hampers effective doping, as the acceptor moiety remains inactive for p-type doping. When F4-TCNQ is placed on the donor site of PCPDTBT, efficient charge transfer occurs, while on the acceptor site, it does not [65]. Li et al. utilized density-functional theory (DFT) and the Boltzmann transport equations to investigate the electronic transport properties of polypyrrole (PPy). Additionally, molecular dynamics simulations were employed to analyze its thermal transport properties. Notably, the crystalline phase exhibited remarkably high electrical conductivity, whereas the amorphous phase demonstrated a high Seebeck coefficient and low thermal conductivity.
Furthermore, the ZT value is higher in the crystalline phase along the chain direction compared to the amorphous phase. These findings provide valuable insights for enhancing the ZT value of PPy and other conducting polymers, potentially enabling more energy-efficient thermoelectric applications [66]. Moreover, the doping can also be affected by the shape of polymer phase. For example, Wu et al. theoretically investigated the TE properties of p-toluenesulfonic acid (p-TSA) doped PANI and found that its nanowire form exhibits a higher Seebeck coefficient and a lower thermal conductivity than corresponding form of nanorod with the same doping level [67].
As compared with tremendous progress in the p-doping of organic polymers, there are only limited theoretical investigations of n-doping reported [68]. Liu et al., by coupling the density functional theory with the Boltzmann transport equations, investigated the effects of potassium doping on n-type poly(nickel-ethylenetetrathiolate). The formation of an electron polaron band (EPB) is revealed by calculations, which is distinct from the conduction band (CB) and predicted to play a dominant role in electron transport. At low doping concentrations, as the temperature increases, both the EPB and the upper CB contribute to electron transport. This results in a significantly enhanced Seebeck coefficient and power factor. At higher temperatures, the CB begins to dominate electron transport, leading to a plateau and subsequent decrease in the Seebeck coefficient. This "exotic" nonmonotonic temperature effect, previously observed in experiments but unexplained, can be primarily attributed to the polaron effect. Importantly, the presence of a doping-induced polaron band can be harnessed to enhance the Seebeck coefficient thereby endowing the organic coordination polymer with unique characteristics as an n-type thermoelectric material.
The thermoelectric properties are greatly influenced by the polaron band narrowing and charge localization effects. Fig. 7a shows the band structure and PDOS of K1Ni5, CB, VB and EPB by Liu et al. In Fig. 7b, schematic representations of the two-band transport model for K-doped poly[Ni-ett] are depicted. Notably, the Fermi level is positioned within the half-filled EPB. As temperature rises, electrons in the EPB can undergo thermal activation, transitioning to the CB due to a small energy gap between these bands. The two-band transport model has effectively elucidated the temperature dependence of thermoelectric properties in lightly doped polymers. It has also emphasized the significant role played by the polaron-induced charge localization effect in enhancing the Seebeck coefficient of K1Nin, making it a promising candidate for thermoelectric applications. Fig. 7c plots the electrical conductivity, the Seebeck coefficient, and the power factor of K1Ninversus carrier density. The maximum power factor occurs at a doping level of n = 14, as the doping-induced polaron band lean to an enhancement of the Seebeck coefficients (Eq. 17).
(17)
Figure 7
Figure 7.
(a) The calculated band structure and PDOS of K1Ni20.The electron polaron bandwidths are highlighted in pink. (b) Illustration of the band structure of two-band transport model for poly[Kx(Ni-ett)n]. (c) Conductivity, Seebeck coefficient and mobility as a function of carrier number for poly[Kx(Ni-ett)n]. Copied with permission [68]. Copyright 2019, American Chemical Society. (d) Orthorhombic and triclinic phases of rubrene molecule. Copied with permission [69]. Copyright 2016, The Royal Society of Chemistry. (e) Seebeck coefficient versus hole density nh for pristine orthorhombic rubrene crystals (without counter ions) at different temperatures. (f) Seebeck coefficient versus hole density nh for modulation-doped orthorhombic rubrene crystals at 300 K, 320 K, 340 K, 360 K. (g) Modulation-doped triclinic rubrene crystals Seebeck coefficient versus hole density nh with a doping concentration of nd = 10% and nd = 16% in different temperature. The pictures in top right corner show the DOS around the Fermi energy. Copied with permission [70]. Copyright 2022, American Association for the Advancement of Science.
Modulation doping represents a novel and unexplored approach for achieving exceptional performance in organic thermoelectric materials. Wang et al. theoretically studied the impact of crystal packing on the excitonic properties of different modulation-doped polymorphs of rubrene (Fig. 7d) [69]. To investigate their TE properties and electronic structure, a theoretical model involving vibronic material parameters for electronic coupling and thermal disorder due to intramolecular electron-phonon interaction was applied, which depends on the electronic band width. Figs. 7e-g show the simulated Seebeck coefficient versus the carrier concentration nh at various temperatures for pristine orthorhombic rubrene crystals, modulation-doped orthorhombic rubrene crystals, and modulation-doped triclinic rubrene crystals [70]. From Fig. 7e, the Seebeck coefficient is decrease with carrier concentration, because the shift in the chemical potential toward the transport level with increasing free carrier density.
Figs. 7f and g show that the optimized doping concentration is 16%. As the carrier density increases, Seebeck coefficient first decreases and then increases. Negative values can be observed at carrier concentrations around 10%. In order to explain such peculiar phenomena, Wang et al. conducted theoretical simulations and attributed these unusual behaviors to the influence of dopant BPAPF. Specially, they were found to be correlated with the Higher Occupied Molecular Orbital (HOMO) of the density of states (DOS). In the case of pristine crystals (Fig. 7e), the Seebeck coefficient diminishes as the carrier concentration increases. This behavior arises from the chemical potential shifting towards the transport level with an increasing density of free carriers. Notably, a negative Seebeck coefficient is only anticipated to occur above half-filling of the HOMO-derived states in the pristine rubrene crystals. However, within the plotted density range, this negative Seebeck coefficient is not observed. For modulation-doped systems, the doping concentration of nd is 16% for both the orthorhombic and triclinic systems in Figs. 7f and g. At lower carrier densities, the Seebeck coefficient also decreases with an increasing carrier density, similar to the pristine case. However, in contrast to the pristine case, at higher carrier densities, the Seebeck coefficient becomes negative, and the slope with nh is reversed. This change in behavior signifies a distinct response in the Seebeck coefficient due to the modulation doping process in the orthorhombic and triclinic rubrene crystals.
3.2
Ambipolarity and temperature dependence
From the TE point view, the charge transport polarity of organic polymers is determined by the sign of Seebeck coefficients, in which positive values indicate a p-type charge transport while negative values mean n-type. In addition to the p- and n-type organic semiconductors, ambipolarity, which is the basis for the practical constructions of modern organic electronics such as field-effect transistors and organic solar cells, has been generally observed in polymers and drawn significant interests. Bipolar OTE materials can transport both electrons and holes, which both will diffuse from the hot end to the cold end under a temperature gradient. In particular, depending on the temperature as well as doping condition and method, bipolar OTE materials allow the transition between p-type and n-type or vice versa. For example, as early as 1986, Zhu et al. discovered that the Seebeck coefficient of β(BEDT-TTF)2Br2I2 decreases at beginning and then increases with temperature, as well as the mysterious Seebeck coefficient sign switch phenomenon from negative to positive, indicating the transition of the apparent charge transport polarity from n-type to p-type [70]. Moreover, it is found recently that as the degree of p-doping increases, the Seebeck coefficient will then switch from positive to negative, indicating a p-type to n-type transition [71,72].
On the molecular level, ambipolar polymer is usually composed of a donor-acceptor (D-A) backbone, and intramolecular charge transfer could occur between the donor and the acceptor units [73]. D-A copolymers have gained much attention for their prominent bipolar charge transport properties including conductivities and mobilities, which are originated from their highly dissociated π-conjugated HOMO and LUMO level, strong intermolecular π-π interactions, and low band gap. In particular, the strong intermolecular interactions are the result of charge transfer between donor and acceptor units within the polymer, which leads to the enhanced coplanarity of polymer backbones.
Wang et al. reported that DPPTTT, a D-A polymer doped with FeCl3 solution, exhibiting high TE performance. Compared with the monopole polymer, it shows both high p-type and n-type conductivity [74]. However, for TEs, the simultaneous transport of electrons and holes in D-A bipolar polymers would normally compensate and therefore reduce the Seebeck coefficients. Therefore, to further optimize the TE performance of bipolar polymers, their quantitative bipolar charge transport nature is required to be fundamentally investigated via theoretical methods, since experimentally it is extremely difficult to separate the TE conversion process of electrons and holes and study individual charge transport properties. Theoretically, Wang et al. calculated the effect of different substituents (-NH2, –OCH3, -F, -CF3, and -CN) to DPP-TVT [diketopyrrolopyrrole-(E)-1,2-di(2-thienyl) ethane] [75]. Interestingly, distinct bipolar characters occur with different functional groups and they predicted that the charge transport polarity can be switched conveniently through changing of substituted groups. The density functional theory was applied to simulate the influence of the TVT substituents in DPPTVT copolymers.
Specifically, as shown in Figs. 8a-d, the fluorine substituent can convert the charge transport polarity from purely p-type to ambipolar. Two additional typical electron-donating groups and two electron-withdrawing groups are chosen for comparison. The calculated ratios of hole to electron mobilities of -CN and -NH2 substituted materials are 0.64 and 1.08. Amino and cyanide groups are beneficial to electron and hole transport, and the values are higher than those of fluorine atom substituted DPP-TVT.
Figure 8
Figure 8.
(a) Calculated intrachain ratios of hole to electron mobilities for ambipolar copolymers by introducing -NH2, -OCH, -F, -CF3, and -CN. (b) Calculated hole and electron effective mass. (c) Calculated elastic constants. (d) Hole and electron DP constants for all calculated polymers. Copied with permission [75]. Copyright 2020, American Chemical Society. (e) Schematic illustration of one-band model, two band model and ambipolar behavior. (f) Molecular structure, band structure and DOS of poly(Ni-ttftt). (g) Seebeck coefficients of valanced band and conduction band in n-type doping poly(Ni-ttftt) versus temperature. (h) Seebeck coefficients of valance band and conduction band of p-type doping poly(Ni-ttftt) versus temperature. Copied with permission [76]. Copyright 2021, Chinese Chemical Society.
It is observed that the CF3-substituted DPP-TVT has the largest effective mass of holes and electrons, and the other substituent groups make less change. The elastic constants of the CF3-substituted DPP-TVT are minimal, indicating unfavorable structural planarity. Optimal backbone planarity and rigidity could significantly enhance the efficiency of intra-chain charge transfer, while poor planarity happens due to large substituent like -CF3 group.
As mentioned above, the ambipolar charge transport can be largely modulated by the operating temperature, which diverse dependence can be studied via theoretical calculations to reveal the elusive mechanism of ambipolarity in polymers. For most unipolar polymers, the temperature dependence of TE properties is usually monotonic, either increasing or decreasing with temperature. However, different phenomena would occur for bipolar polymers, exhibiting and non-monotonic behavior, where it initially increases and then decreases. Within the framework of thermal activation, the Seebeck coefficient displays a monotonous temperature dependence in the one-band model for unipolar polymers, but could exhibit a non-monotonic relationship under the two-band transport picture for bipolar polymers with decreased bandgap between conduction band (CB) and valence band (VB), as schematically shown in Fig. 8e [76]. The theoretical investigation of temperature dependence of bipolar charge transport is not straightforward, as in terms of the theoretical basis of modern DFT calculations, the quantum mechanical calculations exclude the temperature. Therefore, the direct computing of the electronic structure at different temperatures is prohibitied [77-79].
However, by combining first-principles calculations with the semiclassical Boltzmann transport equation, the temperature-dependent trends of the Seebeck coefficient can be analyzed. Fig. 8f shows the chemical structure, band structure and DOS of poly(Ni-ttftt)[66]. The p-type and n-type doping effects are further investigated versus temperature. In particular, the poly(Ni-ttftt) Seebeck coefficients were calculated for both valence band and conduction band contributions at varying temperatures, exhibiting a bipolar charge transport behavior in general while different temperature dependence for electrons and holes, as shown in Figs. 8g and h.
3.3
Molecular design and disorder influences
Through the rational design of molecular structures based on theoretical principles, it is possible to achieve high-performance OTE materials [80], [81]. By carefully considering factors such as electronic structure, molecular packing, and charge transport properties, we can tailor the molecular architecture to optimize the thermoelectric performance. This includes selecting suitable electron-donor and acceptor groups, controlling the conjugation length as well as structural and energetic disorder, and enhancing intermolecular interactions. Additionally, strategies such as interface optimization can further enhance the OTE performance.
Yan et al. reported a novel molecular structural design strategy, that the planar backbone, high torsional barrier, and zigzag backbone together exhibit favorable characteristics for doping, as shown in Figs. 9a-c [82]. In fact, the electrical conductivity of the designed polymer exceeded 120 S/cm, highlighting its superior performance. Specifically, they demonstrated that a designed conjugated polymer, featuring a predominant planar backbone conformation, a high torsional barrier at each dihedral angle, and a zigzag backbone curvature, can withstand dopant-induced disorder. The torsional potential energy scans for different combinations confirm that the Pz-2FT has good disorder tolerant ability. From the MD simulation of the polymers in crystalline and disorder regions, P(PzDPP-2FT) and P(PzDPP-4F2T) show narrower torsion-angle distributions and more planar backbone. They expanded the π-π stacking and lamellar distances to simulate the disorder. From crystalline to disorder, the torsion angle distribution width is improved. As the DOS calculated via DFT and comparison of the characteristic depth of trap states, the Eb of P(PzDPP-4F2T) increased slightly from 18 meV to 23 meV and while it of the P(PzDPP-2FT) increased more from 19 meV to 31 meV, also indicating the P(PzDPP-4F2T) has better tolerant ability.
Figure 9
Figure 9.
(a) Molecular dynamics simulation of P(PzDPP-2FT), P(PzDPP-4F2T), P(PzDPP-T) in crystalline and disorder regions. (b) Calculated torsion angle distribution width of P(PzDPP-2FT), P(PzDPP-4F2T, P(PzDPP-T) of φ1-φ7. (c) Calculated DOS distributions of P(PzDPP-2FT), P(PzDPP-4F2T). Copied with permission [82]. Copyright 2021, Springer Nature. (d) Seebeck coefficient versus electrical conductivity with increasing energetic disorder. (e) Comparison of the conductivity as function of Seebeck plot with increasing positional disorder. Copied with permission [83]. Copyright 2019, Springer Nature.
Upadhyaya et al. demonstrated that decreasing energetic disorder and positional correlation while simultaneously increasing positional disorder, can result in higher TE power factors. They employed the Gaussian disorder model (GDM) to represent site energies, and to model the hopping between localized sites the approach of Pauli's master equation is applied. Fig. 9d plots the Seebeck coefficient versus conductivity for increasing disorder, which clearly reveal the strong effect ofdisorder on conductivity [83], indicating reducing the energetic disorder could enhance the TE performance. On the other hand, Fig. 9e shows when the positional disorder increases, the TE power factors also increase. Therefore, for high TE performance copolymers, larger positional disorder while smaller energetic disorder is desired.
Meanwhile, Lu et al. theoretically investigated the control of TE power factor and mobility in a 2D-MOF system via side-chain engineering. Fig. 10a show the structures of Ni3(HATI_CX)2 with different lengths of alkyl chains, such as Ni3(HATI_C1)2, Ni3(HATI_C3)2, and Ni3(HATI_C4)2 [84]. In Fig. 10b, the results of DFT (density functional theory) calculations are presented, showing the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) as well as the optimized structures. These calculations reveal significant changes in the electronic structure and charge transport properties of the material.
Figure 10
Figure 10.
(a) Structures of Ni3(HATI_CX)2 with different lengths of alkyl chains. (b) HOMO and LUMO orbital of researched molecules. The calculated band structures and projected density of states (PDOS) of (c) bulk Ni3(HATI_C1)2, (d) Ni3(HATI_C3)2, (e) Ni3(HATI_C4)2. (f) Calculated electrical conductivity versus constant-relaxation-time and average effective mass of Ni3(HATI_C1)2, Ni3(HATI_C3)2, Ni3(HATI_C4)2 by VASP. Copied with permission [84]. Copyright 2022, Springer Nature.
For the Ni3(HATI_CX)2 monolayers, their band structures are similar, featuring a band gap of 0.57 eV in Figs. 10c-e. However, in the case of the layer-stacked Ni3(HATI_CX)2, the interlayer interactions lead to distinct dispersions in the band structure. The average dispersions for the layer-stacked systems are 0.17 eV, 0.12 eV, and 0.09 eV, respectively, showing variations depending on the specific alkyl chains. These findings suggest that the layer stacking and interlayer interactions play a significant role in shaping the electronic properties and charge transport behaviors of Ni3(HATI_CX)2, providing valuable insights for understanding and engineering the properties of these materials for various applications. In Fig. 10f, the bulk effective masses of Ni3(HATI_C1)2, Ni3(HATI_C3)2, and Ni3(HATI_C4)2 are calculated to be mave* = 1.10m0, mave* = 5.90m0, and mave*=7.10m0. The higher dispersion of the bands resulting in lower effective mass and higher carrier mobility along the interlayer direction compared with the intralayer direction. By precisely adjusting the layer spacing, the band gap has successfully widened, and significantly improved the Seebeck coefficient. The power factor of Ni3(HATI_C3)2 is predicted to be 68 ± 3 nW m−1 K−2. This method paves the way for enhanced thermoelectric performance and potential applications in energy conversion and related fields.
A promising strategy to enhance TE performance is to incorporate long alkyl side chains into conjugated backbones. For instance, in the case of Cn-BTBTs (where n represents the alkyl chain length), introducing longer alkyl chains leads to increased hole mobilities [85]. This effect arises from the closer packing of conjugated backbones, which facilitates more efficient charge transport. Additionally, the long alkyl side chains introduce numerous low-frequency phonon modes, effectively suppressing lattice thermal conductivity to an exceptionally low level. This approach offers the advantages of enhanced charge transport through improved packing of conjugated backbones, and reduced thermal conductivity via the introduction of low-frequency phonon modes.
DFT simulations were employed to investigate the impact of lactone maximization on the conformation of the backbone in oligomers of P-0, P-50, and P-75 by Alsufyani et al. [86]. The results revealed that the presence of double bond connections substantially decreased the twist angle between the building blocks. Furthermore, the dihedral angle decreased as the lactone unit was increased through benzene maximization, as observed from P-0 to P-75. This reduction in torsional angle signifies that increasing the number of lactone units influences the planarization of the backbone, thereby leading to a more rigid molecular structure. The anticipated consequence of this enhanced planarity in P-75 is a greater orbital overlap, extended polaron delocalization, and consequently, an improved charge carrier transport. The increased lactone content in P-75 is therefore expected to promote stronger electronic coupling between the molecular units, facilitating more efficient charge transport in the material.
3.4
Single molecular TE performance
Single-molecule TE device is based on the molecular level TE effect with the merits of quantum size, high efficiency, fast response, high controllability, and low power consumption [87]. Single-molecule devices offer a potential route to sub-10 nm electronics. However, the experimental measurements of single molecular scale TE properties are extremely difficult, in particular due to the challenges in building and calibrating the temperature gradient and thermal transport within the molecular junctions. Therefore, theoretical predictions become the only convenient route to evaluate their potentially remarkable TE performance and investigate various interesting quantum charge, heat, and spin transport phenomena on the single molecular limit.
One of the most successful methods in studying single-molecule charge transport is to build the single-molecule junctions, comprising individual molecules attached to two electrodes [88]. Over the past two decades, significant advancements in both technology and theory have substantially enhanced our understanding of the TE properties within the single-molecule junctions. More importantly, there are several theoretical investigations which predict the ZT values on the single-molecule limit are remarkably high, even exceeding the current experimental limits. For example, Almughathawi et al. studied the DPP molecules and discovered that the conductivity relies on the connectivity of thiophene rings. Specifically, as shown in Fig. 11a, rotating the thiophene rings can get different conductivity and ZT values. They further calculated the room-temperature quantum interference effect and demonstrated the destructive quantum interference could much enhance the Seebeck coefficients [89]. Fig. 11b shows the resulting ZT values of different thiophene-DPP isomers as the function of the Fermi energy at 300 K, from which can be seen the ZT of DPP-3b is the largest, due to the above quantum effects. Fig. 11c shows the ZT values versus temperature, and the TE figure of merits of thiophene-DPP isomers are strikingly up to 800.
Figure 11
Figure 11.
(a) The transmission function log G/G0 as well as ZT values of different single molecules: DPP 1-a, DPP 2-a, DPP 3-a, DPP 1-b, DPP 2-b, and DPP 3-b. (b) Calculated ZT values of different thiophene-DPP isomers as the function of Fermi energy at room temperature. (c) Corresponding calculated ZT values of (b) as a function of temperature. Copied with permission [89]. Copyright 2020, American Chemical Society. (d) The calculated various TE properties of different number of benzene ring units versus the chemical potential. (e) Calculated maximum ZT values as a function of benzene ring unit numbers n. (f) The transmission probability T(E) and ZT of different phonon transmission functions versus the chemical potential. Copied with permission [90]. Copyright 2010, American Chemical Society.
Bergfield et al. suggested that exploiting higher-order interferences can lead to a substantial enhancement in the TE performance of molecular scale junctions. This quantum mechanical approach has the potential to unlock new avenues for improving the efficiency of single-molecule TE devices. In Fig. 11d, molecule of n meta-connected phenyl groups in series is shown. The corresponding ZT values and thermopower are calculated with muti-body theory. With the repeating unit number n increasing, the Seebeck coefficients and ZT values are enhanced. Fig. 11e shows the predicted ZT values as a function of repeated phenyl unit number n and energy µ-µ0. In a junction composed of just four phenyl groups (n = 4), the max {ZTel} reaches a significant value of 4.1 [90,91]. Due to the high-order destructive interferences, the ZT is further greatly enhanced reaching 50 when n = 30. Fig. 11f illustrates the relationship between various phonon transmission functions and the chemical potential, depicting their impact on the transmission probability (T(E)) and the thermoelectric performance parameter ZT.
4.
Summary and perspectives
In summary, the theoretical investigation of OTEs is indeed a new field, which is developing from fundamental molecular and electronic structure calculations to predictions of TE properties [92-94]. Previous theoretical endeavors have already significantly accelerated the development of high-performance OTE materials for the realization of flexible energy-harvesting applications into a foreseeable future. However, the current predictions of TE properties usually require significant computation resources and the results are often not quantitatively consistent with experimental results. Therefore, there is still a long way to go for providing precise, effective, and efficient theoretical guidance for experimental advances. Other than the reviewed progresses, we put forward the following perspectives to boost future directions for OTE theoretical calculations.
First of all, there are still several key factors which could significantly affect the OTE performance yet lack theoretical understanding, such as the film morphology and counterions in the doped state. The film morphology including crystallinity and roughness, which can often be manipulated by fabrication techniques, annealing condition, as well as solvent, could largely affect the charge transport and thermoelectric properties. For example, Xiong et al. demonstrates that the aggregation of P(PzDPP-CT2) in solution is highly sensitive to solvent, temperature, and aging time. By controlling the aggregation dynamics, they achieved precise manipulation of solid-state microstructures and dopant miscibility. This strategic approach resulted in a substantial increase in the conductivity of P(PzDPP-CT2), representing a significant advancement in doping efficiency for conjugated polymers and indicating its potential applicability across diverse polymer systems with analogous aggregation behaviors observed in various polymeric semiconductors [95,96]. Regarding the influence of counterions, in general at relatively low doping level, separated counterions could create steep Coulomb traps which decrease the charge mobility, while higher doping level would lead to overlapping traps, which restrict the delocalization of free charged carriers. In both cases, the thermoelectric properties would be significantly affected. Recently, an anion-exchange method has been proposed to overcome the limitations of counterion effects to achieve efficient doping, by reducing the Coulomb interaction between charge carriers and ionized dopants. Moreover, several recent studies have demonstrated the size and shape of counterions could produce a substantial influence to the doping and thermoelectric properties of polymers. However, the effects of morphology and counterions still lack fundamental understanding, as they have rarely been investigated theoretically. This is primarily because such calculations require modeling the doping or aggregation dynamics of a large and disordered polymer system rather than monomers or oligomers, thus significantly enhance the difficulties and consumptions in computing. In most cases, current theoretical calculations only account for the crystalline regions of conjugated polymers and neglect the morphology effect, while for OTE doping, the counterion effects are often be extremely simplified by replacing the actual dopant molecule with single atom or charged ion and by assuming an ideal doping process. These treatments however certainly deviate from the reality. We anticipate with the rapid development of chip technology and algorithm, such limitations can be overcome in the near future to thoroughly illustrate the various effects which affect the OTE performance.
Furthermore, it is required to establish a theoretical database for diverse OTE materials, including predictions of molecular structure, packing ordering, electronic structure, charge transport properties, and TE performance, in an easily accessible way [97,98]. The establishment of a comprehensive OTE properties database, encompassing diverse molecular structures, formulations, and experimental conditions, stands as a crucial step towards unraveling structure-property correlations and expanding the repertoire of available materials for researchers. Similar to the inorganic crystal structure database, an OTE materials database can offer a platform to enhance the development efficiency of novel OTE materials and devices with improved performance and various functions. This would be achieved through direct searching for desired molecular structures or properties within the database, as well as the development of innovative material design strategies or tools using data mining techniques. Traditionally, experimental synthesis and discovery of new molecules have been the primary sources of input for such databases. However, this approach is no longer sufficient to meet the current requirements of OTE development, particularly in terms of achieving the desired ZT values necessary to power the fast-growing flexible and wearable electronics sector. Therefore, theoretical input is expected to become another important avenue for generating significant amounts of molecular data. This is due to the rapid advancements in various calculation methods, models, and computational technologies in recent years. Once the complete open-source OTE database is established, as the latest computational advance, the data-driven AI and machine learning hold the promise for discovering revolutionized OTE materials and devices, in terms of not only performance and functions, but also its fundamentally elusive nature [99]. By machine learning of large amount of data, artificial intelligence can identify superior OTE materials and provide ideal optimization routes. However, the challenge lies in the development of innovative computational models and algorithms for the complex OTE materials with intricate charge transport mechanisms and interfacial effects.
Rapid screening of advanced OTE materials and precise prediction of their quantitative structure-property relationship are crucial for the development of new organic energy-harvesting materials. This requires striking a balance between calculation accuracy, time, and computational cost. To address this challenge, the development of innovative high-throughput calculation methods has gained significant attention. High-throughput calculations emerge as a powerful tool, enabling the efficient screening of an extensive array of molecules and structures to identify potential high-performance OTE materials. The integration of artificial intelligence and machine learning techniques further accelerates the prediction and optimization of material properties, facilitating the identification of patterns within vast datasets and expediting the material design process. In the realm of inorganic crystals, several high-throughput platforms based on first-principle density functional theory (DFT) calculations have been established. These platforms, available internationally, have played a vital role in various material fields. However, when it comes to polymeric OTE materials and devices, high-throughput computing becomes necessary due to the complexity of their molecular structures. Even a single molecular unit of a polymer can consist of hundreds of atoms, resulting in a massive structural model that requires weeks-long computing time for high accuracy calculations. To tackle this issue, it is feasible to combine existing high-throughput DFT calculation platforms with fast solving of semiclassical Boltzmann transport equations. This approach would enable the development of specific high-throughput computing methods for OTE materials. By integrating the strengths of these two computational techniques, researchers can efficiently screen and predict the thermoelectric properties of organic materials, paving the way for the discovery of novel energy-harvesting materials.
In conclusion, the prospects for future theoretical research on organic thermoelectrics (OTE) hold great promise, with strategies such as database construction, high-throughput computation, and artificial intelligence-assisted computation offering exciting avenues for exploration.
While these methodologies promise to enhance our understanding of organic thermoelectric materials, it is essential to acknowledge and address the challenges and limitations. The intricate balance between accuracy and computational cost in dealing with the complex structures of OTE materials highlights the need for continuous refinement of theoretical methods. Experimental validation remains indispensable to ensure the reliability of theoretical calculations, emphasizing the importance of synthesizing and testing new materials.
Moreover, the challenge of insufficient data for database construction underscores the necessity for collaborative efforts to gather diverse and comprehensive experimental data. Overcoming these challenges requires interdisciplinary collaboration, ongoing research endeavors, and a commitment to refining theoretical and experimental approaches.
As we navigate these challenges and embrace the opportunities presented by advanced theoretical methods, the future of organic thermoelectrics holds the promise of accelerated material discovery, leading to the development of high-performance materials that will undoubtedly contribute to the advancement of the field. Through persistent efforts and collaborative endeavors, the integration of theoretical insights and experimental validation will pave the way for transformative discoveries in the realm of organic thermoelectrics. Currently, there are reports on inorganic thermoelectric materials. However, organic semiconductor materials, due to their relatively lower production costs, flexibility, and lightweight characteristics, are widely utilized in fields such as flexible electronics, organic optoelectronics, and organic field-effect transistors (OFETs). We believe that the application of organic thermoelectric materials will become increasingly widespread [100-106].
The construction of databases plays a pivotal role in accelerating the development of thermoelectric materials. By assembling a comprehensive dataset of experimentally synthesized thermoelectric materials and their properties, machine learning models achieve high predictive accuracy [107]. Additionally, the introduction of material descriptors enhances the extrapolation capabilities of these models. The database, utilizing these descriptors, significantly improves predictive accuracy for unexplored material groups. The creation of searchable, interactive databases for thermoelectric properties facilitates efficient data-driven exploration and visualization, enabling researchers to leverage machine learning for rapid discovery of novel materials [108]. Moreover, such databases can be extended to various functional materials, fostering accelerated innovation in materials science [109]. High-throughput computational methods significantly accelerate the development of thermoelectric materials by efficiently prescreening a large number of candidates. In the study, algorithms based on machine-learning techniques, physical insights, and automatic ab initio calculations were employed to estimate the lattice thermal conductivity (κω) for approximately 79,000 half-Heusler entries. The results revealed a broad range of κω and identified 450 mechanically stable ordered semiconductors. Further screening for thermodynamical stability narrowed down the list to 75 systems, with three semiconductors having κω < 5 W m−1 K−1 recommended for experimental study [110]. This high-throughput approach enables the rapid identification of promising thermoelectric materials, showcasing the potential of computational methods in accelerating materials discovery for energy applications [111-113].
The development of thermoelectric materials is hindered by labor-intensive processes [114]. AI-driven frameworks, utilizing machine learning, expedite material discovery by predicting thermoelectric properties with high efficiency. This work focuses on overcoming traditional methods' limitations, showcasing AI models' accuracy in identifying promising thermoelectric materials [115]. Validated through density-functional theory, the framework achieves over 90% accuracy, enhancing material feature representation and streamlining the discovery process. The approach not only accelerates thermoelectric material design but highlights AI's broader potential in advancing materials science [116,117].
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors acknowledge financial support from the National Natural Science Foundation of China (Nos. 22125504, 22305253, 62205347), the Beijing Natural Science Foundation (No. Z220025) and the K.C. Wong Education Foundation (No. GJTD-2020–02).
[1]
N. Toshima, S. Ichikawa, J. Electron. Mater. 44 (2014) 384–390.
[2]
Q. Zhang, Y. Sun, W. Xu, D. Zhu, Adv. Mater. 26 (2014) 6829–6851. doi: 10.1002/adma.201305371
[3]
E.H. Suh, J.G. Oh, J. Jung, et al., Adv. Energy Mater. 10 (2020) 2002521. doi: 10.1002/aenm.202002521
B. Russ, M.J. Robb, F.G. Brunetti, et al., Adv. Mater. 26 (2014) 3473–3477. doi: 10.1002/adma.201306116
[9]
O. Bubnova, Z.U. Khan, A. Malti, et al., Nat. Mater. 10 (2011) 429–433. doi: 10.1038/nmat3012
[10]
I. Petsagkourakis, K. Tybrandt, X. Crispin, et al., Sci. Technol. Adv. Mater. 19 (2018) 836–862. doi: 10.1080/14686996.2018.1530938
[11]
Y. Ma, Y. Zou, C. Di, D. Zhu, Introduction of organic thermoelectrics, in: D. Zhu (Ed.), Organic Thermoelectrics: From Materials to Devices, WILEY-VCH GmbH, Weinheim, 2022, pp. 3–24.
[12]
J. Chen, D. Wang, Z. Shuai, J. Chem. Theory Comput. Theory 8 (2012) 3338–3347. doi: 10.1021/ct3004436
[13]
Y. Ge, R. Liu, Z. Shuai, Appl. Phys. Lett. 118 (2021) 123301. doi: 10.1063/5.0043863
[14]
J. Ding, Z. Liu, W. Zhao, et al., Angew. Chem. Int. Ed. 58 (2019) 18994–18999. doi: 10.1002/anie.201911058
[15]
D. Wang, J. Ding, X. Dai, et al., Adv. Mater. 35 (2023) e2208215. doi: 10.1002/adma.202208215
[16]
J. Han, Y. Jiang, E. Tiernan, et al., Angew. Chem. Int. Ed. 62 (2023) e202219313. doi: 10.1002/anie.202219313
J. Wu, Y. Sun, W. Xu, Q. Zhang, Synth. Met. 26 (2014) 6829–6851.
[68]
Y. Liu, W. Shi, T. Zhao, D. Wang, Z. Shuai, J. Phys. Chem. Lett. 10 (2019) 2493–2499. doi: 10.1021/acs.jpclett.9b00716
[69]
X. Wang, T. Garcia, S. Monaco, B. Schatschneider, N. Marom, CrystEngComm 18 (2016) 7353–7362. doi: 10.1039/C6CE00873A
[70]
S.J. Wang, M. Panhans, I. Lashkov, et al., Sci. Adv. 8 (2022) eabl9264. doi: 10.1126/sciadv.abl9264
[71]
D.B. Zhu, P. Wang, M.X. Wan, et al., Proceedings of the Yamada Conference XV on Physics and Chemistry of Quasi One-Dimensional Conductors, 1986, pp. 281–284.
[72]
E. Dalas, S. Sakkopoulos, E. Vitoratos, J. Mater. Sci. 29 (1994) 4131–4133. doi: 10.1007/BF00355982
[73]
S. Hwang, W.J. Potscavage, Y.S. Yang, et al., Phys. Chem. Chem. Phys. 18 (2016) 29199–29207. doi: 10.1039/C6CP04572C
E. Yildirim, Ö. C. Yelgel, Using Machine Learning Techniques to Discover Novel Thermoelectric Materials, New Materials and Devices for Thermoelectric Power Generation, IntechOpen, 2023, doi: 10.5772/intechopen.1003210.
[117]
G. Han, Y. Sun, Y. Feng, et al., Adv. Electron. Mater. 9 (2023) 2300042. doi: 10.1002/aelm.202300042
Figure 1
Illustration of the relationship between molecular design of OTE materials, electronic structure and thermoelectric properties. Start with basic molecular structure and packing, by calculating the electronic structure, their TE properties and performances can be predicted. (a) The conjugated core structure of organic semiconductors facilitates charge transport. Side chains play a role in solution processing and can impact the generation of charge carriers and the assembly of molecules in the solid state. (b) Doping adjusts the HOMO and LUMO energy levels of organic thermoelectric materials by altering charge distribution and electron affinity. This modulation influences the charge transport properties and thermoelectric performance of the material. Such control can enhance the conductivity and thermoelectric efficiency, making it more suitable for thermoelectric conversion applications. (c) The molecular packing in organic thermoelectric materials exhibits both face-on and edge-on configurations, where molecules can stack either with their faces oriented toward each other or with their edges in alignment. The molecular orientations also could significantly affect the TE performance. (d) The electronic energy levels are determined by the highest occupied molecular orbital (HOMO), representing ionization energy, and the lowest unoccupied molecular orbital (LUMO), representing electron affinity. These levels can be adjusted through molecular design. Energetic disorder may lead to a distribution of states around these levels in perfectly ordered materials. Schematic diagram of the energy band structure of organic thermoelectric materials. (e) Schematic representation of the phonon spectrum of organic thermoelectric materials. (f) Schematic diagram illustrating the variation of thermoelectric parameters with carrier concentration. With increasing carrier concentration, electrical conductivity, thermal conductivity, Seebeck coefficient, and power factor exhibit different trends. The ZT value (thermoelectric figure of merit), as a comprehensive parameter, the optimal ZT value often corresponds to a specific carrier concentration, providing crucial insights for the design optimization of thermoelectric materials.
Figure 2
(a) Modelling of organic crystal region and the Brillouin zone of organic polymers. (b) Theoretical methods of calculation: molecular dynamics, ab initio DFT, and Boltzmann transport theory. (c) Performance predictions of OTE materials versus carrier concentration and temperature.
Figure 3
(a) Structure of C8-BTBT in the ab and ac plane. (b) Band structures and DOS of C8-BTBT, the reciprocal coordinates of high-symmetry points are Γ=(0, 0, 0), Y = (0, 0.5, 0), X = (0.5, 0, 0), and Z = (0, 0, 0.5). The red curves indicate the valence band maximum and conduction band minimum. (c) The calculated Seebeck coefficient (S), electrical conductivity (σ), along the a-direction (blue lines) and the b-direction (red lines) for C8-BTBT, as a function of carrier concentration at 298 K. (d) TE power factor (S2σ), and electronic thermal conductivity change with the carrier concentration. Copied with permission [42]. Copyright 2014, American Chemical Society.
Figure 4
(a) Calculated lattice thermal conductivity of Cn-BTBTs along the a- and b-axes at room temperature and experimental results for pentacene, TPD, and Alq3. Copied with permission [85]. Copyright 2014, American Chemical Society. (b) Calculated dimensionless thermoelectric (ZT) versus the mobility for C8-BTBT, pentacene, rubrene, and TiOPc. Copied with permission [42]. Copyright 2019, Wiley Publishing Group.
Figure 7
(a) The calculated band structure and PDOS of K1Ni20.The electron polaron bandwidths are highlighted in pink. (b) Illustration of the band structure of two-band transport model for poly[Kx(Ni-ett)n]. (c) Conductivity, Seebeck coefficient and mobility as a function of carrier number for poly[Kx(Ni-ett)n]. Copied with permission [68]. Copyright 2019, American Chemical Society. (d) Orthorhombic and triclinic phases of rubrene molecule. Copied with permission [69]. Copyright 2016, The Royal Society of Chemistry. (e) Seebeck coefficient versus hole density nh for pristine orthorhombic rubrene crystals (without counter ions) at different temperatures. (f) Seebeck coefficient versus hole density nh for modulation-doped orthorhombic rubrene crystals at 300 K, 320 K, 340 K, 360 K. (g) Modulation-doped triclinic rubrene crystals Seebeck coefficient versus hole density nh with a doping concentration of nd = 10% and nd = 16% in different temperature. The pictures in top right corner show the DOS around the Fermi energy. Copied with permission [70]. Copyright 2022, American Association for the Advancement of Science.
Figure 8
(a) Calculated intrachain ratios of hole to electron mobilities for ambipolar copolymers by introducing -NH2, -OCH, -F, -CF3, and -CN. (b) Calculated hole and electron effective mass. (c) Calculated elastic constants. (d) Hole and electron DP constants for all calculated polymers. Copied with permission [75]. Copyright 2020, American Chemical Society. (e) Schematic illustration of one-band model, two band model and ambipolar behavior. (f) Molecular structure, band structure and DOS of poly(Ni-ttftt). (g) Seebeck coefficients of valanced band and conduction band in n-type doping poly(Ni-ttftt) versus temperature. (h) Seebeck coefficients of valance band and conduction band of p-type doping poly(Ni-ttftt) versus temperature. Copied with permission [76]. Copyright 2021, Chinese Chemical Society.
Figure 9
(a) Molecular dynamics simulation of P(PzDPP-2FT), P(PzDPP-4F2T), P(PzDPP-T) in crystalline and disorder regions. (b) Calculated torsion angle distribution width of P(PzDPP-2FT), P(PzDPP-4F2T, P(PzDPP-T) of φ1-φ7. (c) Calculated DOS distributions of P(PzDPP-2FT), P(PzDPP-4F2T). Copied with permission [82]. Copyright 2021, Springer Nature. (d) Seebeck coefficient versus electrical conductivity with increasing energetic disorder. (e) Comparison of the conductivity as function of Seebeck plot with increasing positional disorder. Copied with permission [83]. Copyright 2019, Springer Nature.
Figure 10
(a) Structures of Ni3(HATI_CX)2 with different lengths of alkyl chains. (b) HOMO and LUMO orbital of researched molecules. The calculated band structures and projected density of states (PDOS) of (c) bulk Ni3(HATI_C1)2, (d) Ni3(HATI_C3)2, (e) Ni3(HATI_C4)2. (f) Calculated electrical conductivity versus constant-relaxation-time and average effective mass of Ni3(HATI_C1)2, Ni3(HATI_C3)2, Ni3(HATI_C4)2 by VASP. Copied with permission [84]. Copyright 2022, Springer Nature.
Figure 11
(a) The transmission function log G/G0 as well as ZT values of different single molecules: DPP 1-a, DPP 2-a, DPP 3-a, DPP 1-b, DPP 2-b, and DPP 3-b. (b) Calculated ZT values of different thiophene-DPP isomers as the function of Fermi energy at room temperature. (c) Corresponding calculated ZT values of (b) as a function of temperature. Copied with permission [89]. Copyright 2020, American Chemical Society. (d) The calculated various TE properties of different number of benzene ring units versus the chemical potential. (e) Calculated maximum ZT values as a function of benzene ring unit numbers n. (f) The transmission probability T(E) and ZT of different phonon transmission functions versus the chemical potential. Copied with permission [90]. Copyright 2010, American Chemical Society.

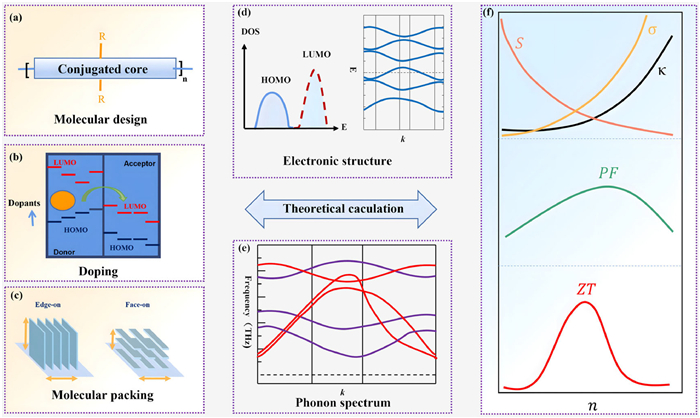
 DownLoad:
DownLoad:
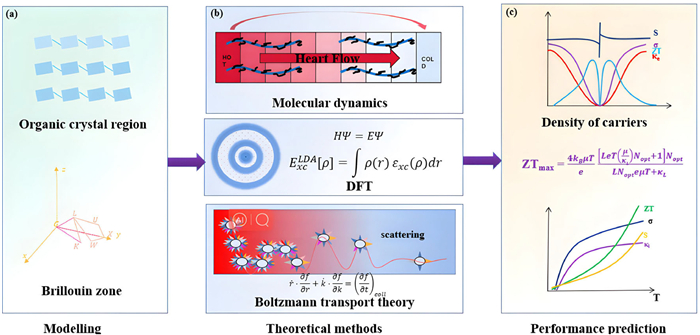
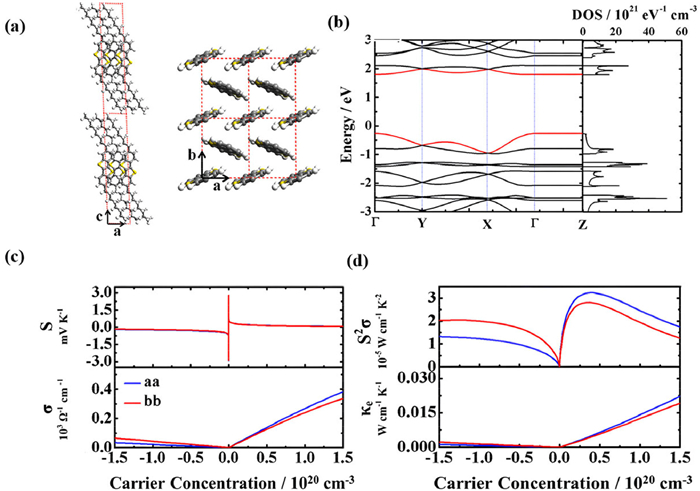


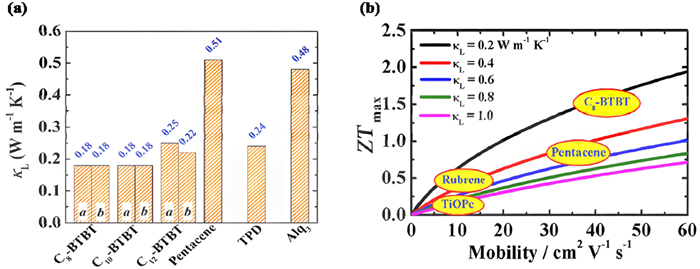
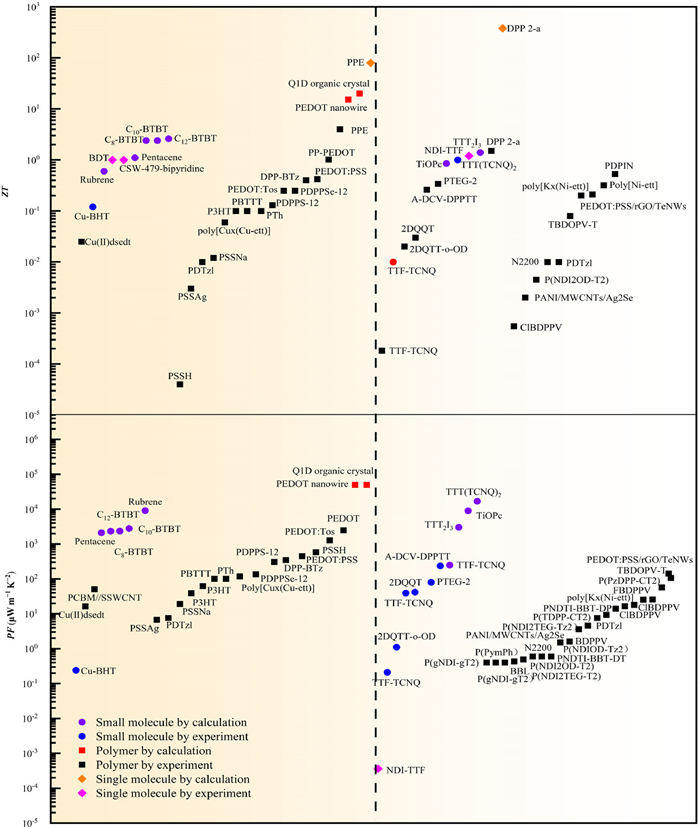
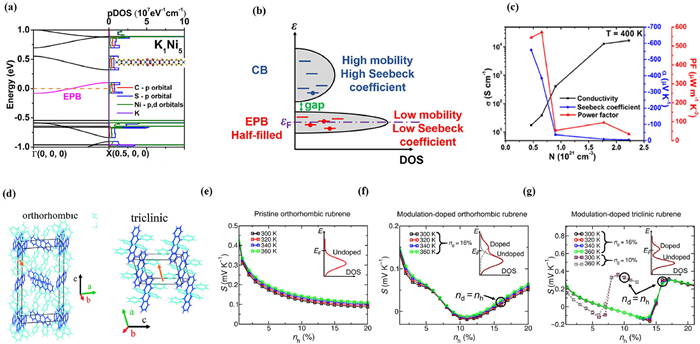
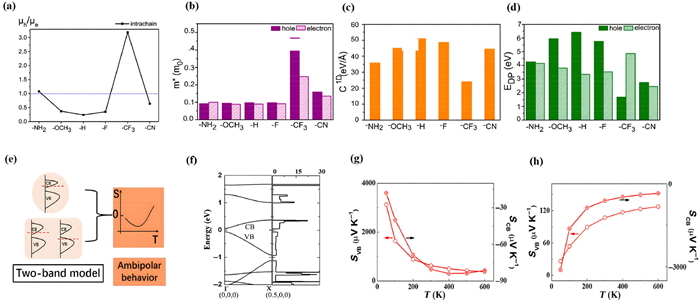
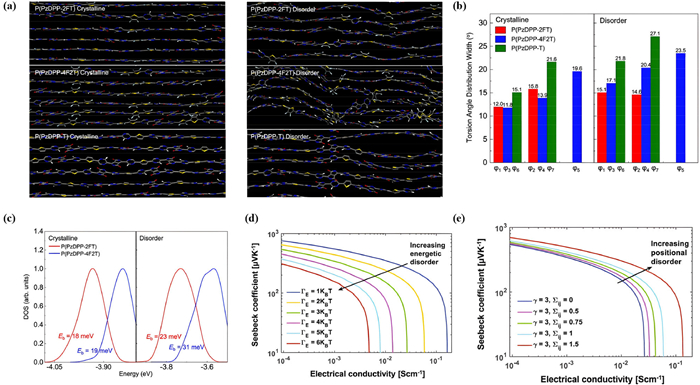
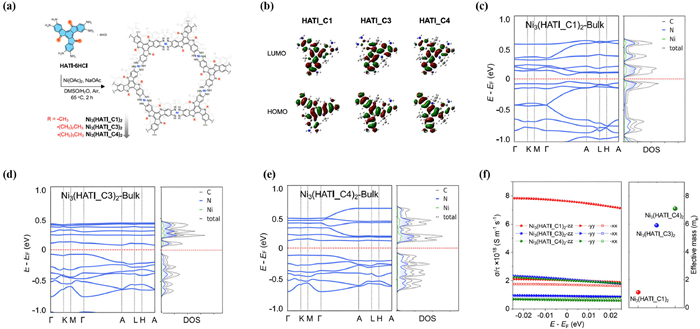
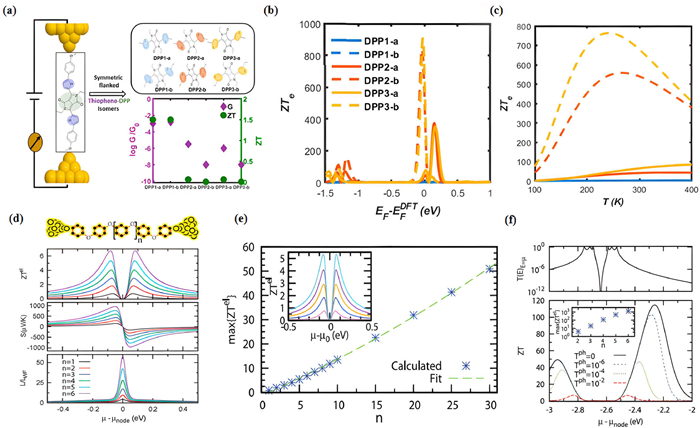

 下载:
下载:












 下载:
下载:
