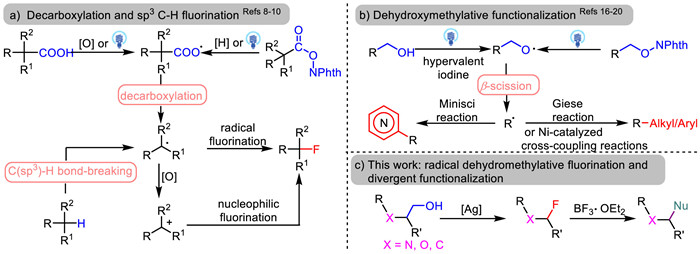
Scheme 1.
Dehydroxymethylative functionalization of primary alkoxy radicals via β-fragmentation.
Radical dehydroxymethylative fluorination of aliphatic primary alcohols and diverse functionalization of α-fluoroimides via BF3·OEt2-catalyzed C‒F bond activation
Peng Wang , Jianjun Wang , Ni Song , Xin Zhou , Ming Li

The incorporation of fluorine atom in an organic molecule has shown potential in regulating the pKa, increasing the lipophilicity, improving metabolic stability and bioavailability, and exerting the conformation control of the molecule [1]. As a consequence, the fluorinated settings increasingly attract attention in pharmaceuticals, agrochemicals, and materials science [2,3]. The development of novel synthetic methods for fluorine atom incorporation has become a hot topic in organic synthesis [4]. Besides conventional nucleophilic and electrophilic fluorination [5,6], radical fluorination reactions have emerged as powerful strategies for efficiently constructing C–F bonds [7]. The recent significant advances include decarboxylative fluorination and C–H fluorination (Scheme 1a) under transition-metal catalysis [8], photocatlysis [9], and electrocatalysis [10]. These transformations involve either a radical reaction mechanism or a radical-polar crossover reaction pathway.
Alkoxy radicals [11] have proven to be versatile reactive intermediates and usually engage in three elementary reactions, namely 1,n-hydrogen atom transfer (1,n-HAT, n = 5, 6, 8, etc.), β-scission, and addition to alkenes [12-14]. It is well-recognized that compared to tertiary and strained cyclic alkoxy radicals, primary alkoxy radicals show the relatively smaller tendency to undergo β-cleavage. However, such processes have been designed and applied in a plethora of organic transformations over the past years [15]. Photocatalyzed generation of primary alkoxy radicals and the following β-fragmentation have been achieved by the Zuo [16], Chen [17], and Martin groups [18] using the corresponding alcohols or N-alkoxyphthalimides as the starting materials (Scheme 1b). These processes have been employed in the formation of various carbon-carbon bonds by means of the Giese reactions and the nickel-catalyzed cross-coupling reactions with alkyl and aryl bromides, providing elegant routes to convert readily available alcohols into high-valued chemicals. The Minisci reactions based on β-fragmentation of primary alkoxy radicals have also been independently documented by Liu, Chen, and their co-workers [19,20]. These reactions used hypervalent iodine reagents as oxidants with resort to visible light irradiation (Scheme 1b). In continuation of our interest in β-scission of alkoxy radicals [21], we herein report radical dehydroxymethylative fluorination of aliphatic alcohols. The reaction accommodates various alcohols and enables access to α-fluoroimides and 1-fluoroalkyl benzoates as well as secondary and tertiary alkyl fluorides. Divergent functionalization of α-fluoroimides is also established through BF3·OEt2-catalyzed C–F bond activation, providing a novel route to amine derivatives.
α-Amino C-centered radicals that are stabilized by the adjacent N-substituents are considered reactive intermediates. These species have been successfully applied to construction of carbon-carbon bonds and carbon-heteroatom bonds [22,23]. Based on this knowledge, we chosen L-N-phthaloyl-leucinol 1a as a model substrate to determine the optimal reaction parameters for dehydroxymethylative fluorination of aliphatic primary alcohols. Since Ag(Ⅱ) species are recognized to be capable of converting alcohols into the corresponding alkoxyl radicals by single electron transfer [24], and the ensuing β-scission is highly expedited by hydrogen bond-donating solvents [25], we thereby conducted silver salt-promoted oxidative radical dehydroxymethylation of 1a using Selectfluor as fluorine source in aqueous acetone. After scrutinizing various reaction parameters, including composition of solvent, reaction concentration, reaction temperature, type and equivalent of silver salts and fluorinating source, and additives HF·H2O or HF·pyridine (Table S1 in Supporting information), we found that treatment of alcohol 1a with 0.5 equiv. of Ag2CO3 and 5.0 equiv. of Selectfluor in acetone/H2O (v/v = 4:1) delivered monofluoride 2a and difluoride 2a’ in 89% overall yield and at the highest ratio of 2a/2a’ = 4:1 after stirring for 30 min at 28 ℃ (Scheme 2). Based on these observations, the control experiments, and the precedents in the literature [16-21], a radical reaction mechanism involving the formation of intermediate alkoxy radical and the following β-fragmentation was proposed (Scheme S1 in Supporting information).
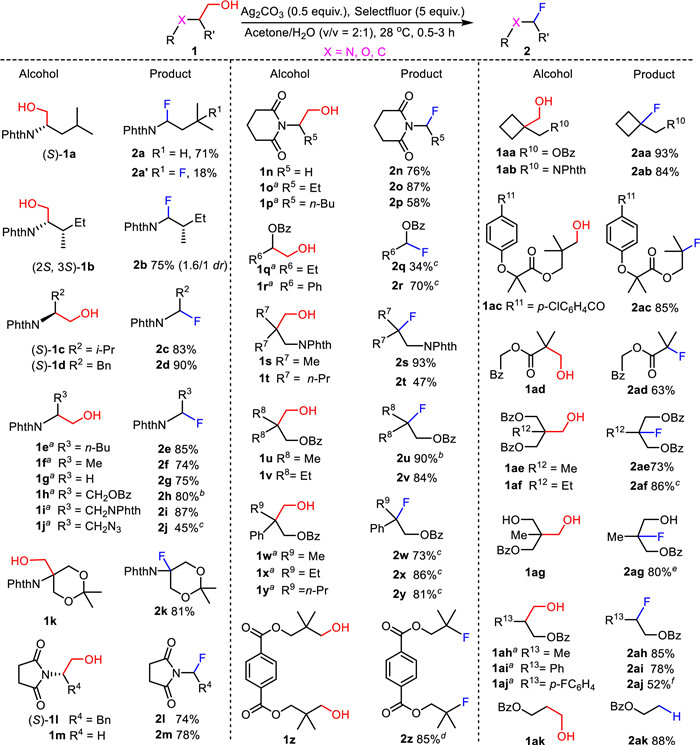
With the optimized condition established, we then examined the reaction scope and limitations with respect to structurally diverse primary alcohols (Scheme 2). A variety of N-phthaloyl-β-amino alcohols were first examined, and it was found that alcohols 1b–1g were amenable to dehydroxymethylative fluorination, affording the desired α-fluoroimides 2b–2g in 74%–90% yields. These transformations deserve further comments. Distinct from leucinol derivative 1a, dehydroxymethylative fluorination of N-phthaloyl-l-isoleucinol 1b and 2-phthalimido-hexanol 1e produced α-fluoroimides 2b and 2e in 75% and 85% yields as the sole product, leaving the secondary C‒H moiety at the δ position untouched. We ascribe these differences to the lower bond dissociation energy of the δ tertiary C‒H bond in 1a than that of δ the secondary C‒H bonds in 2b and 2e [26]. Ready access to 2c and 2d demonstrates the tolerance of γ tertiary C‒H and benzyl C‒H next to hydroxy group to the radical dehydroxymethylative fluorination. N-Phthaloyl-glycinol 1g was uneventfully converted into phthalimidomethyl fluoride 2g in 75% yield. This result highlights that phthalimidomethyl radical is easily generated and enables fluorine abstraction from Selectfluor although it is a primary alkyl radical.
Taking serinol derivatives 1h–1j as examples, we evaluated the influence of the functional group on the side chain to dehydroxymethylative fluorination. 3-O-Benzoyl-2-N-phthaloyl serinol 1h and its 3-deoxy-3-phthalimido congener 1i underwent smoothly dehydroxymethylative fluorination to afford fluorides 2h and 2i in 80% and 87% yields, respectively. 3-Azido-3-deoxy-N-phthaloyl serinol 1j, however, furnished the expected fluoride 2j in much lower yield of 45%. This observation might be attributed to the stronger electron-withdrawing nature of azido group than benzoyloxy and phthalimido groups [27]. This property of azido group disfavors the formation of α-phthalimido alkyl radicals. It is worth noting that the high-yielding transformation of alcohols 1h into 2h was carried out on one mmol scale, demonstrating the feasibility of scale-up for dehydroxymethylative fluorination.
N-Phthaloyl-tris(hydroxymethyl)aminomethane derivative 1k was subjected to dehydroxymethylative fluorination, supplying fluoride 2k in 81% yield with the quaternary C‒F bond formation. It should be noted that the acetonide group to protect 1,3-diol is compatible with the reaction conditions although the reaction medium is found to be acidic [28]. Besides N-phthaloyl-β-amino alcohols, N-succinyl-β-amino alcohols 1l and 1m as well as N-glutaryl congeners 1n–1p are all competent substrates, enabling the production of fluorides 2l–2p in 58%–87% yields.
Glycosyl fluorides and reverse glycosyl fluorides are typical representatives of α-fluoroethers [21,29]. These architectures have been fruitfully applied in carbohydrate chemistry as glycosylating agents. Inspired by these advances, we attempted to make 1-fluoroalkyl carboxylates through dehydroxymethylative fluorination. Pleasingly, β-benzoyloxy-propanol 1q and β-benzoyloxy-β-phenyl ethanol 1r participated in dehydroxymethylative fluorination reaction, and yielded 1-fluoroalkyl benzoates 2q and 2r in 34% and 70% yields. The higher yield for 2r compared with that for 2q might result from the stabilizing effect of phenyl on the adjacent C-centered radicals.
Next, we moved our focus to dehydroxymethylative fluorination of β,β-disubstituted 1,3-diols and derivatives. To our delight, primary alcohols 1s–1y were uneventfully converted into fluorides 2s–2y in the yields ranging from 47% to 93%. Compound 1z bearing two primary hydroxy groups smoothly underwent dehydroxymethylative fluorination to give rise to difluorides 2z in 85% yield. The suitability of the present method for scale-up reaction was again demonstrated by 90% conversion of 1u into 2u. Both 1,1-cyclobutanedimethanol monobenzoate 1aa and its phthalimido analogue 1ab were exposed to dehydroxymethylative fluorination, efficiently giving rise to fluorides 2aa and 2ab in 84% and 93% yields. Fenofibrate is an anti-hyperlipidemic drug with a wide range of uses in clinic [30]. Recently, this compound was found to reveal potential in reducing the risk of SARS-CoV-2 infection [31]. Notably, subjection of monoester 1ac, obtained by reacting β,β-dimethyl 1,3-diol with fenofibric acid, to dehydroxymethylative fluorination reaction afforded 85% yield of 2ac as a structurally intriguing fluorinated congener of fenofibrate. α-Fluoro pivalate 2ad was successfully made by treating 1ad under the established conditions. This result demonstrates that a tertiary C-centered radical next to an ester group is an enabling species for fluorine abstraction from Selectfluor.
1,1,1-Tris(hydroxymethyl)ethane dibenzoates 1ae and its propane congener 1af as well as 1,1,1-tris(hydroxymethyl)ethane monobenzoate 1ag were suitable for dehydroxymethylative fluorination, uneventfully furnishing fluorides 2ae, 2af, and 2ag in satisfactory yields. Radical decarboxylative fluorination of α,α-disubstituted malonic acids has proven to be an efficient approach to gem–difluoro constructs [32]. However, we failed to convert diol 1ag into the corresponding gem–difluoropropyl benzoate by concomitant dehydroxymethylative fluorination of the two hydroxymethyl units. These results could be rationalized by the observations that radical decarboxylation is easier and faster than the corresponding radical dehydroxymethylation [33]. Since 2-alkyl 1,3-diols serve as readily modifiable building blocks in synthetic chemistry [34,35], and 2-alkyl 1,3-diol-based moieties are frequently found in bioactive compounds [36,37], ready access to 2ae–2ag lays a solid foundation for preparing 2-fluoro-2-methyl/ethyl 1,3-diol-based derivatives like fluorinated drug candidates [38,39] and functionalized polymer [40,41].
Dehydroxymethylative fluorination of 2-methyl and phenyl propane-1,3-diol derivatives 1ah and 1ai worked well, giving fluorides 2ah and 2ai in 85% and 78% yields. However, treatment of 2-para-fluorophenyl-propane-1,3-diol monobenzoate 1aj in aqueous acetone (v/v = 6:1) afforded fluoride 2aj in 52% yield along with the formation of benzoyloxymethyl aryl ketone S21 (see Supporting information for details) in 25% yield.
Treatment of propane-1,3-diol monobenzoate 1ak with the current conditions for dehydroxymethylative fluorination supplied ethyl benzoate 2ak in 88% yield without the desired fluoride detected. This result implies that 2-benzoyloxyethyl radical, generated by β-fragmentation of 3-benzoyloxypropoxy radical, prefers to abstract hydrogen atom from the medium rather than abstracts fluorination from Selectfluor.
Silver-catalyzed decarboxylative fluorination has proven to be a powerful method to make aliphatic fluorides [28]. For comparison purpose, a competition experiment was set up using each about 0.1 mmol of N-phthaloyl L-phenylalanine S22 and 1e as the substrates (see Supporting information for details) under the established conditions. The transformation gave 2e in 49% (based on the quantities of the used S22 and 1e) along with 56% of 1e recovered (based on the quantities of the used 1e). These results showed a possibility of selective decarboxylative fluorination over the dehydroxymethylative fluorination due to the faster rate the former reaction than the latter. These findings are consistent with the observations in the literature [42].
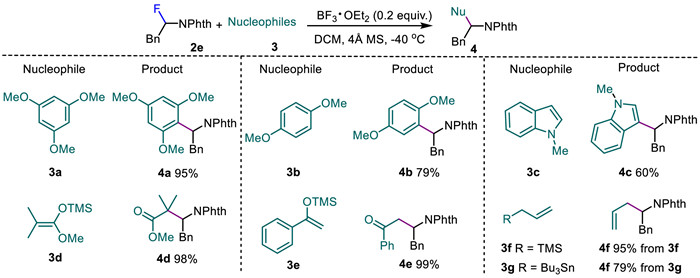
N,N-Aminals, N,O-acetals, α-chloroaminals, and α-aminosulfides are popular equivalents of N-iminium ions which are an class of important reactive species and play significant roles in organic synthesis [43-46]. With the various α-fluoroimides in hand, we explored the feasibility of these compounds as the masked surrogates of iminium ions through C‒F bond activation under the catalysis of Lewis acid (Scheme 3) [47]. It was found that 0.2 equiv. of BF3·OEt2 was capable of catalyzing the Friedel-Crafts alkylation of electron-rich aromatics 3a-3c with α-fluoroimide 2e through C‒F bond activation. The reactions afforded α-aryl imides 4a-4c in 60%–95% yields. With enol silyl ethers as nucleophiles, 2e was converted into β-imido ester 4d and β-imido ketone 4e in excellent yields. Furthermore, the couplings of 2e with allyltrimethylsilane 3f and allyltributyltin 3g furnished homoallylic imide 4f in 95% and 79% yields, respectively.
Encouraged by these results, we moved to examine the scope of α-fluoroimides as coupling partners using 1,3,5-trimethyloxy benzene 3a, enol silyl ether 3d, and allylic trimethylsilane 3f as nucleophiles. As listed in Scheme 4, all BF3·OEt2-catalyzed transformations performed well, giving the corresponding products 4g–4t in 48%–96% yields. 1,2-Amino alcohols are privileged building blocks prevalent in pharmaceuticals, agrochemicals, natural products, and catalyst architectures [48]. Efficient access to 4h in 95% yield shows the potential of 2h as an electrophile in introducing an β-amino alcohol unit. It should be mentioned that the reaction of α-fluorophthalimide 2a with allyltrimethylsilane 3f afforded homoallylimide 4p and enamine 4p’ (see Supporting information for details) in 66% and 13%, respectively. The formation of 4p’ highlights the involvement of iminium ion intermediates during the reaction. In addition, α-fluoroimide 2b possessing a chiral side chain did not show apparently diastereoselective bias, affording 4m as a mixture (dr = 1/1). Access to benzyl imide 4g and homoallyl imide 4q indicates that phthalimidomethyl fluoride 2g is alternative aminomethylating agent, but it required to be activated at a temperature up to 0 ℃ for the successful couplings. Activation of α-fluorosuccinimide 2l and α-fluoroglutarimide 2n–2p entailed more forcing conditions, and imides 4i, 4j, 4n, 4o, and 4r–4t were obtained in good yields at 0 ℃ in the presence of 1.2 equiv. of BF3·OEt2.
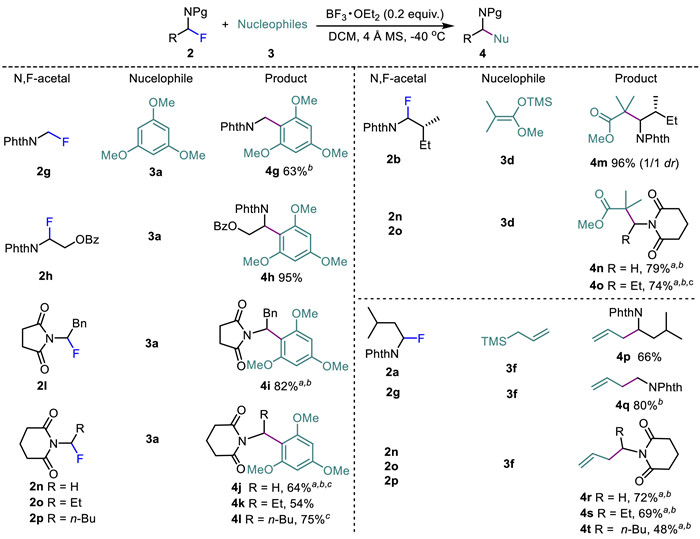
Notice that imido moieties are not only the precursors for amines but also versatile building blocks to construct structurally complex and biologically intriguing natural products and pharmaceuticals which contain nitrogen atom [49]. For instance, partial reduction of phthalimidyl, succinimidyl, and glutarimidyl groups with NaBH4 have the potential to be transformed into the corresponding acylaminoacetals which are a class of acyliminium ions precursors of synthetic importance [50]. Consequently, ready accessibility of imides 4a–4t offers promising platform for synthesis of biologically active amines.
In summary, a novel and efficient protocol has been established for radical dehydroxymethylative fluorination of aliphatic primary alcohols with Ag2CO3 as promoter and Selectfluor as fluorine source and oxidant. The reaction proceeded under mild reaction conditions, and accommodated a wide range of substrates, efficiently providing various alkyl fluorides of importance. Preliminary mechanistic studies suggest that a β-cleavage of primary alkoxy radicals might be involved. Diverse functionalization of α-fluoroimides was also achieved by BF3·OEt2-catalyzed C‒F bond activation. The present work opens up a door for installation of fluorine atom in organic molecules with primary alcohols as the starting materials and adds another tool to the arsenal for the synthesis of structurally diversified amines of biological relevance. With α-fluoroimides as electrophiles, enantioselective construction of carbon-carbon bonds is underway in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are grateful for financial support from the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. 2022QNLM030003–2), the National Natural Science Foundation of China (No. 21977088), and the National Natural Science Foundation of China-Shandong Joint Fund (No. U1906213).
Supplementary material associated with this article can be found, in the online version, at doi:
W.K. Hagmann, J. Med. Chem. 51 (2008) 4359–4369. doi: 10.1021/jm800219f
K. Müller, C. Faeh, F. Diederich, Science 317 (2007) 1881–1886. doi: 10.1126/science.1131943
Q. Wang, H. Song, Q. Wang, Chin. Chem. Lett. 33 (2022) 626–642. doi: 10.1016/j.cclet.2021.07.064
H. Yan, C. Zhu, Sci. China Chem. 60 (2017) 214–222. doi: 10.1007/s11426-016-0399-5
J. Wu, Tetrahedron Lett. 55 (2014) 4289–4294. doi: 10.1016/j.tetlet.2014.06.006
K. Yamamoto, J. Li, J.A.O. Garber, et al., Nature 554 (2018) 511–514. doi: 10.1038/nature25749
M. Rueda-Becerril, C. Sazepin, J.C.T. Leung, et al., J. Am. Chem. Soc. 134 (2012) 4026–4029. doi: 10.1021/ja211679v
H. Chen, Y. Liu, X. Liao, Synthesis 53 (2021) 1–29. doi: 10.1055/s-0040-1707273
M. Rueda-Becerril, O. Mahé, M. Drouin, et al., J. Am. Chem. Soc. 136 (2014) 2637–2641. doi: 10.1021/ja412083f
M.C. Leech, D. Nagornîi, J.M. Walsh, et al., Org. Lett. 25 (2023) 1353–1358. doi: 10.1021/acs.orglett.2c04305
L. Chang, Q. An, L. Duan, et al., Chem. Rev. 122 (2022) 2429–2486. doi: 10.1021/acs.chemrev.1c00256
M. Salamone, M. Bietti, Acc. Chem. Res. 48 (2015) 2895–2903. doi: 10.1021/acs.accounts.5b00348
M. Murakami, N. Ishida, Chem. Lett. 46 (2017) 1692–1700. doi: 10.1246/cl.170834
E. Tsui, A.J. Metrano, Y. Tsuchiya, et al., Angew. Chem. Int. Ed. 59 (2020) 11845–11849. doi: 10.1002/anie.202003959
A. Boto, D. Hernández, R. Hernández, et al., J. Org. Chem. 68 (2003) 5310–5319. doi: 10.1021/jo034442f
Y. Chen, X. Wang, X. He, et al., J. Am. Chem. Soc. 143 (2021) 4896–4902. doi: 10.1021/jacs.1c00618
J. Zhang, Y. Li, R. Xu, et al., Angew. Chem. Int. Ed. 56 (2017) 12619–12623. doi: 10.1002/anie.201707171
F. Cong, X. Lv, C. Day, et al., J. Am. Chem. Soc. 142 (2020) 20594–20599. doi: 10.1021/jacs.0c11172
Y. Wang, L. Yang, S. Liu, et al., Adv. Synth. Catal. 361 (2019) 4568–4574. doi: 10.1002/adsc.201900975
X. Hu, G. Li, G. He, et al., Org. Chem. Front. 6 (2019) 3205–3209. doi: 10.1039/c9qo00786e
X. Zhou, H. Ding, P. Chen, et al., Angew. Chem. Int. Ed. 59 (2020) 4138–4144. doi: 10.1002/anie.201914557
M.L. Coote, C.Y. Lin, A.L.J. Beckwith, et al., Phys. Chem. Chem. Phys. 12 (2010) 9597–9610. doi: 10.1039/c003880f
T. Shao, X. Ban, Z. Jiang, Chem. Rec. 23 (2023) e202300122. doi: 10.1002/tcr.202300122
A. Clerici, F. Minisci, K. Ogawa, Tetrahedron Lett. 19 (1978) 1149–1152. doi: 10.1016/S0040-4039(01)94484-3
G. Litwinienko, A.L.J. Beckwith, K.U. Ingold, Chem. Soc. Rev. 40 (2011) 2157–2163. doi: 10.1039/c1cs15007c
Y. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007.
C.H. Hsu, S.C. Hung, C.Y. Wu, et al., Angew. Chem. Int. Ed. 50 (2011) 11872–11923. doi: 10.1002/anie.201100125
F. Yin, Z. Wang, Z. Li, et al., J. Am. Chem. Soc. 134 (2012) 10401–10404. doi: 10.1021/ja3048255
A. Lepak, A. Gutmann, B. Nidetzky, ACS Catal. 8 (2018) 9148–9153. doi: 10.1021/acscatal.8b02685
G. Prasad, P. Govardhan, G. Deepika, et al., Infammopharmacology 26 (2018) 973–981. doi: 10.1007/s10787-017-0428-y
S. Alkhayyat, H. Alkuraishy, A. AlGareeb, et al., Inflamm. Res. 71 (2022) 1159–1167. doi: 10.1007/s00011-022-01615-w
Z. Wang, C.Y. Guo, C. Yang, et al., J. Am. Chem. Soc. 141 (2019) 5617–5622. doi: 10.1021/jacs.9b00681
C. Fittschen, H. Hippler, B. Viskolcz, Phys. Chem. Chem. Phys. 2 (2000) 1677–1683. doi: 10.1039/b000009o
D.S. Reddy, N. Shibata, J. Nagai, et al., Angew. Chem. Int. Ed. 47 (2008) 164–168. doi: 10.1002/anie.200704093
L. Banfi, G. Guanti, Synthesis 11 (1993) 1029–1056.
D. Herkommer, B. Schmalzbarer, D. Menche, Nat. Prod. Rep. 31 (2014) 456–467. doi: 10.1039/C3NP70093C
C. Hertweck, Angew. Chem. Int. Ed. 48 (2009) 4688–4716. doi: 10.1002/anie.200806121
J. Liang, J.R. Zbieg, R.A. Blake, et al., J. Med. Chem. 64 (2021) 11841–11856. doi: 10.1021/acs.jmedchem.1c00847
G. Haufe, A. Burchardt, Eur. J. Org. Chem. (2001) 4501–4507.
D.S. Honeycutt, W.F. Charbonneau, A.J. North, et al., Polymer 242 (2022) 124584. doi: 10.1016/j.polymer.2022.124584
H. Peng, Polymer Rev. 59 (2019) 739–757. doi: 10.1080/15583724.2019.1636390
H. Togo, M. Kaqtohgi, Synlett 2001 (2001) 565–581. doi: 10.1055/s-2001-13349
C. Simon, J. Peyronel, J. Rodriguez, Org. Lett. 3 (2001) 2145–2148. doi: 10.1021/ol010056d
Y. Huang, C. Cai, X. Yang, et al., ACS Catal. 6 (2016) 5747–5763. doi: 10.1021/acscatal.6b01725
A. Kataja, G. Masson, Tetrahedron 70 (2014) 8783–8815. doi: 10.1016/j.tet.2014.06.101
E. Marcantoni, A. Palmieri, M. Petrin, Org. Chem. Front. 6 (2019) 2142–2182. doi: 10.1039/c9qo00196d
Q. Long, J. Gao, N. Yan, et al., Org. Chem. Front. 8 (2021) 3332–3341. doi: 10.1039/d1qo00211b
D.J. Ager, I. Prakash, D.R. Schaad, Chem. Rev. 96 (1996) 835–876. doi: 10.1021/cr9500038
P. Huang, Z. Guo, Y. Ruan, Org. Lett. 8 (2006) 1435–1438. doi: 10.1021/ol0602203
P. Wu, T. Nielsen, Chem. Rev. 117 (2017) 7811–7856. doi: 10.1021/acs.chemrev.6b00806

Scheme 1 Dehydroxymethylative functionalization of primary alkoxy radicals via β-fragmentation.

Scheme 2 Scope of dehydorxymethylative fluorination. a Racemic substrate was used. b 1 mmol scale; c Acetone/H2O (v/v = 4:1); d Ag2CO3 (1.0 equiv.), Selectfluor (10.0 equiv.); e Selectfluor Ⅱ as fluorinating reagent; fAcetone/H2O (2.1 mL, v/v = 6:1).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:


 下载:
下载: