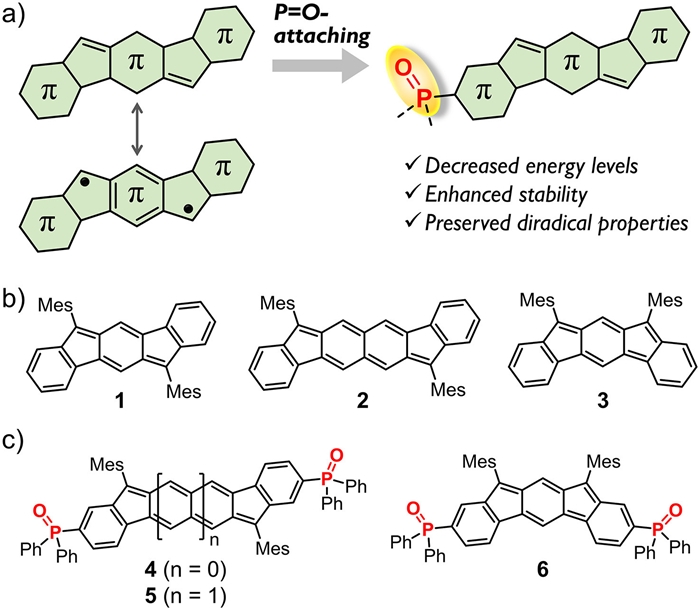
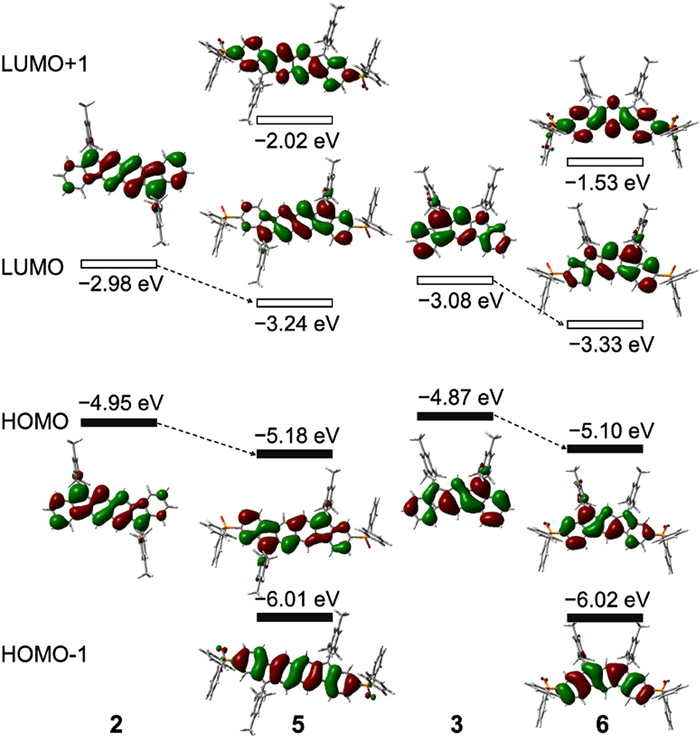
Figure 1.
(a) Schematic illustration of the P=O-attaching strategy toward diradical PHs. (b) Examples of previously reported diindeno-based PHs (1–3). (c) Chemical structures of P=O-attached PHs (4–6).
Enhancing stability of diradical polycyclic hydrocarbons via P=O-attaching
Jingyuan Yang , Xinyu Tian , Liuzhong Yuan , Yu Liu , Yue Wang , Chuandong Dou

Much attention has been paid to diradical polycyclic hydrocarbons (PHs), because they have unique open-shell electronic states and are potential functional materials in organic electronics and spintronics, as well as energy and biological fields [1–5]. However, owing to high reactivity of unpaired electrons toward oxygen, moisture or other electrophiles, such open-shell organic diradicaloids are usually less stable than closed-shell systems, limiting their practical applications (Fig. 1a) [6–8]. To stabilize these reactive radicals, incorporation of the bulky groups (such as mesityl and 2,6-dichlorophenyl) at the radical site has been widely used to achieve kinetical blocking stabilization [9,10]. Extension of π-conjugated frameworks may promote the radicals to delocalize over the whole π-skeletons for thermodynamic stabilization [11,12]. Following these two approaches, a variety of diradical PH systems with diverse topological structures and interesting properties have been developed, such as indenofluorenes (Fig. 1b), bisphenalenyls, anthenes and zethrenes, as well as quinodimethane-embedded macrocycles [13–26]. Despite of these great advances, it is still needed to develop new strategy for stabilizing radicals and enhancing stability of organic diradicaloids, which are crucial for radical chemistry and materials.
Incorporation of heteroatoms onto PHs have developed as an effective approach to modulate their electronic structures and diradical properties [27–36]. For example, the S atom may endow diradical fluorenofluorene derivatives with moderately strong diradical character (y) but large singlet−triplet energy gap (ΔES−T) and excellent stability, which are rarely realized for all-carbon diradicaloids [37]. Introduction of the N atom into indenofluorene derivatives can efficiently tune the spin density and thus afford a stable triplet diradical PH [38]. Our study focused on doping the B atom into PHs to construct B-doped conjugated π-systems [39–43]. We found that the as-developed B-containing organic diradicaloids have good stability and Lewis acidity, further leading to supramolecular diradicaloids via Lewis acid-base coordination. In this context, we envisioned that other heteroatom, such as P, featuring unusual electronic effects has the potential to greatly modulate physical properties of diradical PHs and thereby to expand organic diradicaloids, but the research has very rarely been explored [44,45].
In this study, we report P=O-attaching of diradical PHs as a new strategy to enhance their stability while maintaining diradical properties. We designed and synthesized three P=O-attached PHs (4–6) that have the π-skeletons of indeno[1,2-b]fluorene (1), fluoreno[3,2-b]fluorene (2) and indeno[2,1-b]fluorene (3), respectively (Fig. 1c) [13–15]. The detailed theoretical and experimental studies demonstrate that 5 and 6 have the large diradical characters and open-shell singlet diradical nature, whereas 4 is a closed-shell molecule. Notably, in comparison to the all-carbon analogues, 5 and 6 exhibit much lower LUMO/HOMO energy levels but preserved diradical properties. More importantly, both of them exhibit remarkably high stabilities at ambient condition. The essence for stabilization is attributed to the electron-withdrawing ability of the P=O group, which can decrease the oxidation activity and thereby enhance stability of the quinoidal π-skeletons. This P=O-attaching strategy may be applicable to other diradical PH systems, and thus will lead to the generation of stable P=O-based organic diradicaloids.
The synthesis of the P=O-attached PHs 4–6 is shown in Scheme 1. Starting from boronate-ester-functionalized triphenylphosphine oxide 7, we synthesized 4, 5 and 6 in four steps based on the Suzuki–Miyaura cross-coupling, nucleophilic addition, intramolecular Friedel–Crafts alkylation and oxidative dehydrogenation reactions. The final oxidative dehydrogenation reaction was performed on the dihydro-precursors 9/11/13 using t-BuOK in commercial CH2Cl2 under air, and this reaction condition was rarely employed in the synthesis of the all-carbon indenofluorenes. The desired compounds 4, 5 and 6 were stable enough to be purified under ambient conditions, affording the pink, blue and green solids, respectively. In their high-resolution mass spectrometry (HRMS) spectra (Fig. S1 in Supporting information), one intense signal with isotopic distributions is fully consistent with the simulated data, thus determining the formation of 4, 5 and 6.
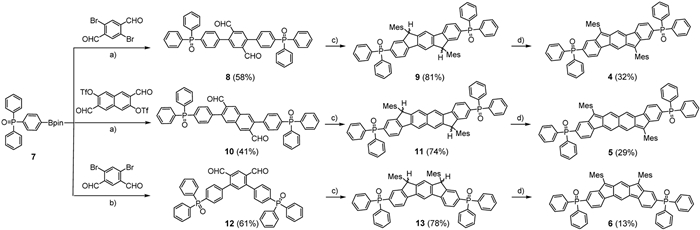
The single crystals of 4 and 5 were successfully obtained by the diffusion of hexane (for 4) and heptane (for 5) into their CH2Cl2 solutions, so that their chemical structures were further determined by X-ray diffraction (XRD) analysis. Both of them have the centrosymmetric structures containing the indeno[1,2-b]fluorene and fluoreno[3,2-b]fluorene skeletons and two terminal P=O groups (Fig. 2 and Fig. S2 in Supporting information), respectively. For the indenofluorene derivatives, the bond a length from the apical methine carbon to the central hexagon is commonly used to estimate their diradical structures. The bond a length (1.381 Å) for 4 is comparable to that (1.381 Å) of 1, indicating their closed-shell electronic structures. For 5, the length of bond a is 1.398 Å, which is longer that (1.391 Å) of indeno[2,1-a]fluorene but shorter than that (1.406 Å) of diindeno[b, i]anthracene [9]. This intermediate band a length in 5 indicates the contribution of the open-shell singlet diradical form to the ground electronic state. The single crystal of 6 was not obtained for bond length analysis.
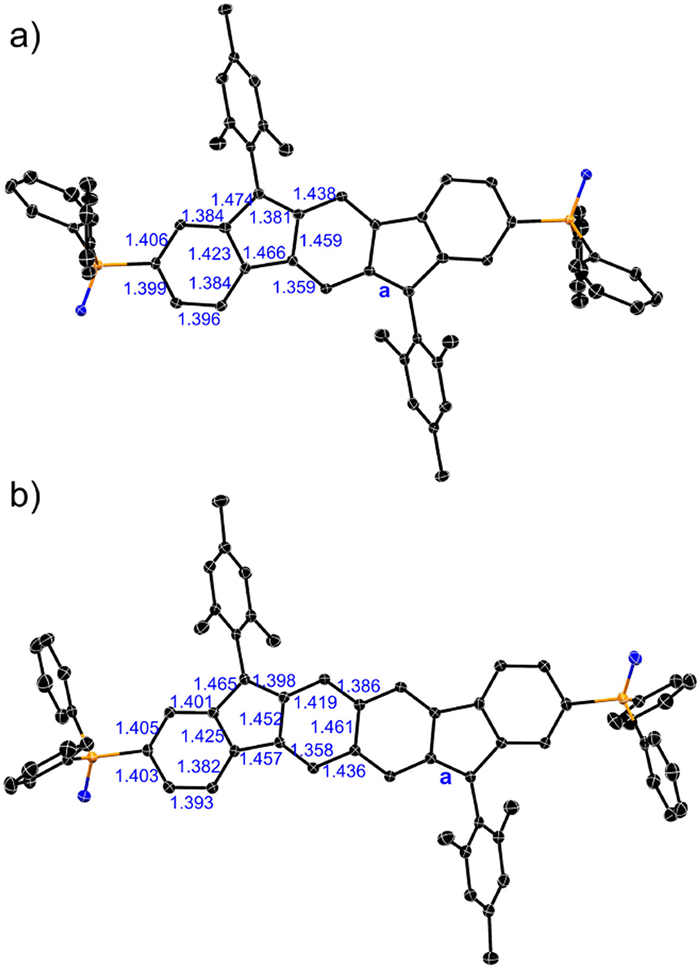
To study the open-shell electronic states, we conducted variable-temperature electron paramagnetic resonance (EPR) measurements on the solid powders of 5 and 6. The unresolved EPR signals with g-factors of 2.0028 and 2.0030 are observed for 5 at 353 K and 6 at 393 K, respectively. Upon increasing the temperature, the signal intensity gradually increases (Fig. 3), thus suggesting more significant contribution of thermally accessible triplet state at high temperature. Fitting I·T versus T to the Bleaney–Bowers equation leads to the singlet–triplet energy gap of ‒7.03 kcal/mol for 5 and ‒3.66 kcal/mol for 6. These spectral changes and relatively small ΔES–T values thus prove the open-shell singlet diradical ground states of 5 and 6 (Fig. S3 in Supporting information).
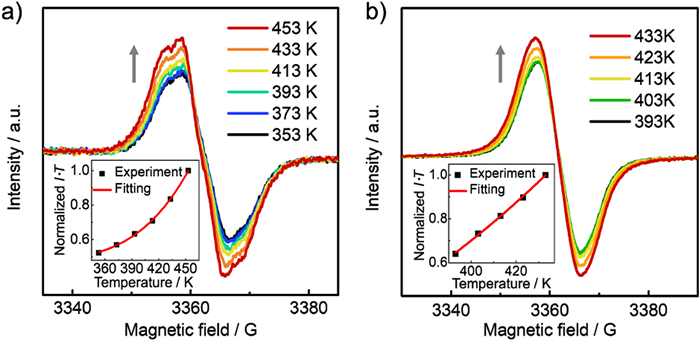
More evidence for the singlet diradical ground states of 5 and 6 was brought to light by detailed theoretical calculations. According to the density functional theory (DFT) calculations (UB3LYP/6-311 G(d) level), the diradical character values of 5 and 6 are determined to be 0.524 and 0.688 (Fig. S4 in Supporting information), respectively, based on occupation numbers of the spin-unrestricted Hartree–Fock natural orbitals. The theoretical ΔES‒T is calculated to be ‒8.98 kcal/mol for 5 and ‒2.22 kcal/mol for 6, which are in consistence with the experimental results. These data unambigiously prove the contributions of their open-shell diradical states. For the all-carbon analogues, 2 and 3 have the y values of 0.523 and 0.582, respectively. Thus, 2 and 5 have the almost identical diradical character, whereas 6 has the larger open-shell extent than 3. This comparison reveals that P=O-attaching may perserve and even enlarge the open-shell singlet diradical characteristics.
In Figs. 4a and b, the spin-density distribution maps (calculated at UB3LYP/6–311 G(d) level) of 5 and 6 are fully delocalized on the π-skeletons, and spin-distribution is not extended to the P=O unit. The highest spin population is located on the apical sp2 carbons in rings B, along with the spin-density value of 0.288 for 5 and 0.475 for 6. According to the spin-density distributions, the resonance structures of 5 and 6 can be illustrated in the closed-shell and open-shell forms (Fig. 4c and Fig. S5 in Supporting information). To reveal the electronic effects of P=O-attaching, nucleus-independent chemical shift (NICS) calculation at the GIAO-UB3LYP/6–311+G(d) level was carried out. As shown in Fig. 4d, 2 and 5 have almost the same NICS(1)ZZ values for their corresponding rings, thus in consistence with their identical diradical character. From 3 to 6, the NICS(1)ZZ values for rings B/C are decreased to a certain extent, indicating P=O-attaching may reduce local antiaromaticity in 6 (Fig. 4e). The reduced antiaromaticity provides additional driving force for recovery of the Clar sextet, further leading to the enhanced open-shell diradical extent of 6.
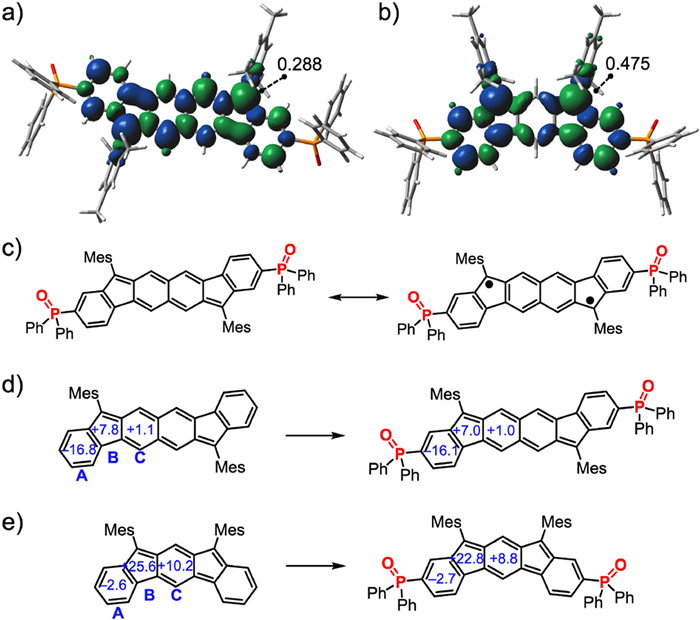
The electrochemical properties of 4, 5 and 6 were investigated using cyclic voltammetry (Fig. 5). Two quasi-reversible reductions are observed for 4 and 5 with half-wave reduction potentials (E1/2red) at −1.40/−1.95 V and −1.28/−1.74 V, respectively, whereas 6 has three quasi-reversible reductions with E1/2red of −0.94/−1.37/−1.77 V. All of them have two quasi-reversible oxidation progresses with half-wave oxidation potentials (E1/2ox) at +0.87/+1.27 eV for 4, +0.47/+1.01 eV for 5, and +0.31/+0.94 eV for 6. Their HOMO and LUMO energy levels were estimated to be −5.67/−3.40 eV for 4, −5.27/−3.52 eV for 5 and −5.11/−3.86 eV for 6, based on the first reversible oxidation and reduction potentials. From the all-carbon analogue to the P=O-attached molecule, the HOMO and LUMO energy levels are decreased by 0.2−0.6 eV, revealing that the electron-withdrawing P=O group significantly stabilize these PHs in term of the oxidation activity [13–15].
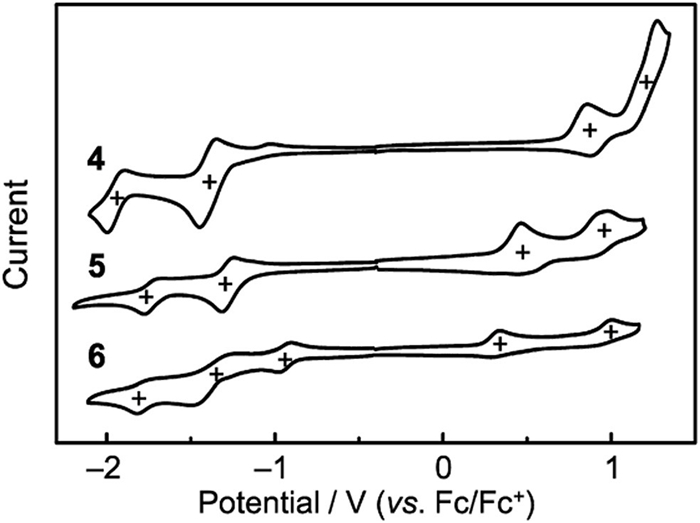
The UV−vis−NIR absorption spectra of 4, 5 and 6 in toluene are shown in Fig. 6. Their solutions exhibit the pink, blue and green colors, respectively. The absorption bands of 4 and 5 are located in the visible region with the main absorption peaks (λabs) around 523/334 nm for 4 and 609/356 nm for 5, along with a low-energy shoulder at 662 nm for 5. For 6, the absorption spectrum exhibits two strong absorption bands at 654/433 nm and a weak absorption band at 1000−1750 nm. The shoulder peak of 5 and the near-infrared absorption band of 6 may be ascribed to the diradical contribution to the ground state, which has been reported for typical indenofluorene system. The absorption bands of these P=O-attached molecules are moderately red-shifted in comparison to that of all-carbon analogues. In addition, their absorption peaks are bule-shifted comparing with our reported B-containing organic diradicaloids, respectively, which are ascribed to the remarkable contribution of the B and O/N atoms [39,40]. These P=O-containing PHs do not emit fluorescence in solutions, owing to their antiaromatic substructure and/or open-shell nature. We then performed the absorption spectral measurements on their solutions under ambient conditions for 20 days to reveal their stabilities (Figs. 6b and c, Fig. S6 in Supporting information). Almost no spectral changes are observed for their time-dependent absorption spectra, thus indicative of their excellent ambient stabilities. It was reported that 3 gradually decomposed in solution. Therefore, P=O-attaching of these PHs plays an important role in enhancing their stabilities.
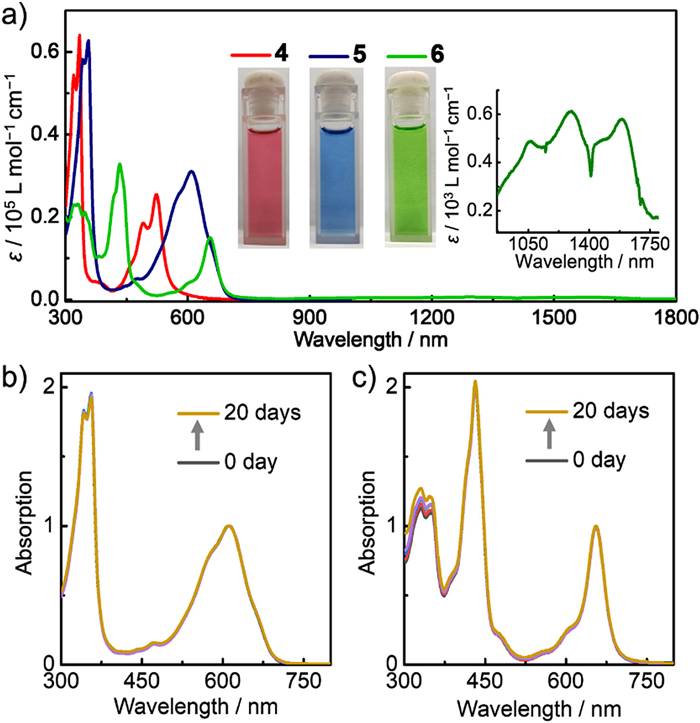
Finally, we would like to clarify their absorption properties and electronic effects of P=O-attaching using theoretical calculations. According to the time-dependent DFT (TD-DFT) calculations, the intense absorption band at 523 nm for 4 and at 609 nm for 5 is assignable to the HOMO → LUMO and HOMO−1 → LUMO+1 transitions (Fig. 7, Figs. S9 and S10 in Supporting information). For 6, the absorption band at 654 nm is assigned to the HOMO−1 → LUMO, HOMO → LUMO and HOMO → LUMO+1 transitions (Fig. S11 in Supporting information). These electronic transitions all involve the HOMO, HOMO−1, LUMO and LUMO+1, and notably, their LUMO+1 are delocalized over the π-skeletons and futher extending to the P=O groups, thus revealing the contributions of the P=O groups to absorption properties. On the other hand, these P=O-attached molecules display the similar HOMO and LUMO distributions to their all-carbon analogues. From the all-carbon analogues to the P=O-attached molecules, the HOMO and LUMO energies are decreased by over 0.2 eV (Fig. 7 and Fig. S7 in Supporting information). It is demonstrated that the electron-withdrawing P=O group can reduce the oxidation activity of these PHs [46], further proved by their electrostatic potential diagrams (Fig. S8 in Supporting information). These theoretical data along with the experimental results indicate that the P=O-attaching strategy is very desirable for the development of stable organic diradicaloids, which are of importance for radical chemistry and materials.
In conclusion, we report the design and synthesis of three P=O-attached PHs (4–6) that have the indeno[1,2-b]fluorene, fluoreno[3,2-b]fluorene and indeno[2,1-b]fluorene π-skeletons, respectively. While 4 is a closed-shell molecule, 5 and 6 have the large diradical characters and open-shell singlet diradical nature, as proved by the theoretical and experimental studies. The attached electron-withdrawing P=O groups endow 5 and 6 with much lower LUMO/HOMO energy levels but preserved magnetic activities and physical properties, such as thermally accessible triplet species and multi-redox ability, in comparison to their all-carbon analogues. Moreover, the P=O groups decrease their oxidation activity and thereby result in their remarkably high stabilities at ambient condition. Thus, this P=O-attaching strategy will be applicable to other diradical PH systems and may lead to the generation of stable organic diradicaloids for radical chemistry and materials. The construction of P=O-containing organic diradicaloids is in progress in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by Jilin Scientific and Technological Development Program (Nos. 20220101054JC and 20220201085GX) and National Natural Science Foundation of China (Nos. 22175074 and 52373182).
Supplementary material associated with this article can be found, in the online version, at doi:
J.J. Dressler, M.M. Haley, J. Phy. Org. Chem. 33 (2020) e4114. doi: 10.1002/poc.4114
T. Kubo, Chem. Rec. 15 (2015) 218–232. doi: 10.1002/tcr.201402065
T.Y. Gopalakrishna, W. Zeng, X. Lu, et al., Chem. Commun. 54 (2018) 2186–2199. doi: 10.1039/C7CC09949E
Z. Cui, A. Abdurahman, X. Ai, et al., CCS Chem. 2 (2020) 1129–1145. doi: 10.31635/ccschem.020.202000210
X. Hu, W. Wang, D. Wang, et al., J. Mater. Chem. C 6 (2018) 11232–11242. doi: 10.1039/C8TC04484H
A. Konishi, Y. Hirao, K. Matsumoto, et al., J. Am. Chem. Soc. 135 (2013) 1430–1437. doi: 10.1021/ja309599m
M. Chen, Y. Duan, X. Liu, et al., CCS Chem. 6 (2024) 353–364. doi: 10.31635/ccschem.023.202302848
P. Hu, S. Lee, T.S. Herng, et al., J. Am. Chem. Soc. 138 (2016) 1065–1077. doi: 10.1021/jacs.5b12532
G.E. Rudebusch, J.L. Zafra, K. Jorner, et al., Nat. Chem. 8 (2016) 753–759. doi: 10.1038/nchem.2518
Y. Ni, T.Y. Gopalakrishna, H. Phan, et al., Angew. Chem. Int. Ed. 57 (2018) 9697–9701. doi: 10.1002/anie.201804276
J. Liu, S. Mishra, C.A. Pignedoli, et al., J. Am. Chem. Soc. 141 (2019) 12011–12020. doi: 10.1021/jacs.9b04718
Z.X. Chen, Y. Li, F. Huang, Chem 7 (2021) 288–332. doi: 10.1016/j.chempr.2020.09.024
D.T. Chase, A.G. Fix, S.J. Kang, et al., J. Am. Chem. Soc. 134 (2012) 10349–10352. doi: 10.1021/ja303402p
J.E. Barker, C.K. Frederickson, M.H. Jones, et al., Org. Lett. 19 (2017) 5312–5315. doi: 10.1021/acs.orglett.7b02605
A. Shimizu, R. Kishi, M. Nakano, et al., Angew. Chem. Int. Ed. 52 (2013) 6076–6079. doi: 10.1002/anie.201302091
K. Sahara, M. Abe, H. Zipse, et al., J. Am. Chem. Soc. 142 (2020) 5408–5418. doi: 10.1021/jacs.0c01003
C.K. Frederickson, B.D. Rose, M.M. Haley, Acc. Chem. Res. 50 (2017) 977–987. doi: 10.1021/acs.accounts.7b00004
H. Han, D. Zhang, Z. Zhu, et al., J. Am. Chem. Soc. 143 (2021) 17690–17700. doi: 10.1021/jacs.1c08262
A. Konishi, Y. Hirao, M. Nakano, et al., J. Am. Chem. Soc. 132 (2010) 11021–11023. doi: 10.1021/ja1049737
J. Ma, J. Liu, M. Baumgarten, et al., Angew. Chem. Int. Ed. 56 (2017) 3280–3284. doi: 10.1002/anie.201611689
T. Shen, Y. Zou, X. Hou, et al., Angew. Chem. Int. Ed. 62 (2023) e202311928. doi: 10.1002/anie.202311928
Y. Li, W.K. Heng, B.S. Lee, et al., J. Am. Chem. Soc. 134 (2012) 14913–14922. doi: 10.1021/ja304618v
J. Guo, Z. Li, J. Zhang, et al., CCS Chem. 4 (2022) 95–103. doi: 10.31635/ccschem.021.202000658
A. Shimizu, M. Uruichi, K. Yakushi, et al., Angew. Chem. Int. Ed. 48 (2009) 5482–5486. doi: 10.1002/anie.200901382
W. Zeng, H. Phan, T.S. Herng, et al., Chem 2 (2017) 81–92. doi: 10.1016/j.chempr.2016.12.001
R. Lu, S. Wu, L. Yang, et al., Angew. Chem. Int. Ed. 58 (2019) 7600–7605. doi: 10.1002/anie.201902028
X. Shi, C. Chi, Top. Curr. Chem. 375 (2017) 68. doi: 10.1007/s41061-017-0154-3
M. Hirai, N. Tanaka, M. Sakai, et al., Chem. Rev. 119 (2019) 8291–8331. doi: 10.1021/acs.chemrev.8b00637
Z. Feng, S. Tang, Y. Su, et al., Chem. Soc. Rev. 51 (2022) 5930–5973. doi: 10.1039/D2CS00288D
J.E. Barker, J.J. Dressler, A. Cárdenas Valdivia, et al., J. Am. Chem. Soc. 142 (2020) 1548–1555. doi: 10.1021/jacs.9b11898
X. Zhang, J. Fang, C. Cai, et al., Chin. Chem. Lett. 32 (2021) 1280–1292. doi: 10.1016/j.cclet.2020.09.058
L. Yuan, J. Guo, Y. Yang, et al., CCS Chem. 5 (2023) 876–884. doi: 10.31635/ccschem.022.202101738
J. Guo, Z. Li, T. Zhang, et al., Chin. Chem. Lett. 35 (2024) 109337. doi: 10.1016/j.cclet.2023.109337
L. Ma, S. Wang, Y. Li, et al., CCS Chem. 4 (2022) 3669–3676. doi: 10.31635/ccschem.022.202201954
L. Mao, M. Zhou, X. Shi, et al., Chin. Chem. Lett. 32 (2021) 3331–3341. doi: 10.1016/j.cclet.2021.05.004
Y. Liu, L. Yuan, J. Guo, et al., Angew. Chem. Int. Ed. 62 (2023) e202306911. doi: 10.1002/anie.202306911
J.J. Dressler, M. Teraoka, G.L. Espejo, et al., Nat. Chem. 10 (2018) 1134–1140. doi: 10.1038/s41557-018-0133-5
Z. Wang, Y. Dai, L. Ding, et al., Angew. Chem. Int. Ed. 60 (2021) 4594–4598. doi: 10.1002/anie.202012989
J. Guo, Y. Yang, C. Dou, et al., J. Am. Chem. Soc. 143 (2021) 18272–18279. doi: 10.1021/jacs.1c08486
J. Guo, Z. Li, X. Tian, et al., Angew. Chem. Int. Ed. 62 (2023) e202217470. doi: 10.1002/anie.202217470
Z. Li, T. Sun, J. Guo, et al., Org. Chem. Front. 10 (2023) 4289–4297. doi: 10.1039/D3QO00900A
L. Yuan, J. Yang, S. Qi, et al., Angew. Chem. Int. Ed. 62 (2023) e202314982. doi: 10.1002/anie.202314982
J. Guo, X. Tian, Y. Wang, et al., Chem. Res. Chin. Univ. 39 (2023) 161–169. doi: 10.1007/s40242-023-2363-3
T. Baumgartner, Acc. Chem. Res. 47 (2014) 1613–1622. doi: 10.1021/ar500084b
D. Käch, A.C. Gasser, L. Wettstein, et al., Angew. Chem. Int. Ed. 62 (2023) e202304600. doi: 10.1002/anie.202304600
M.P. Duffy, W. Delaunay, P.A. Bouit, et al., Chem. Soc. Rev. 45 (2016) 5296–5310. doi: 10.1039/C6CS00257A

Figure 1 (a) Schematic illustration of the P=O-attaching strategy toward diradical PHs. (b) Examples of previously reported diindeno-based PHs (1–3). (c) Chemical structures of P=O-attached PHs (4–6).

Scheme 1 Synthesis of 4, 5 and 6. Conditions: (a) Pd(PPh3)4, K2CO3, THF: H2O (5:1), 75 ℃. (b) Pd(PPh3)4, K2CO3, toluene: EtOH: H2O (5:1:1), 70 ℃. (c) (i) mesitylmagnesium bromide, THF, 25 ℃; (ii) BF3·Et2O, CH2Cl2, 25 ℃. (d) t-BuOK, CH2Cl2, 25 ℃.

Figure 2 Single-crystal structures of (a) 4 and (b) 5 along with selected bond lengths (50% probability for thermal ellipsoids). Hydrogen atoms are omitted for clarity.

Figure 3 Variable-temperature EPR spectra of (a) 5 and (b) 6 at the solid state. Insets are the experimental I∙T‒T plots (black) and Bleaney−Bowers fitting lines (red).

Figure 4 Calculated spin density distribution maps of (a) 5 and (b) 6 along with the spin-density values for the specific carbon atoms. (c) Resonance structures in the closed-shell and open-shell forms of 5. Calculated NICS(1)ZZ values for (d) 2 and 5 and (e) 3 and 6.

Figure 5 Cyclic voltammograms of 4, 5 and 6 in CH2Cl2 (1.0 mmol/L). Fc/Fc+ = ferrocene/ferrocenium.

Figure 6 (a) Absorption spectra of 4, 5 and 6 in toluene. Insets are the photographs for their solutions under daylight and enlarged absorption spectrum of 6. Time-dependent absorption spectra of (b) 5 and (c) 6 in toluene under ambient conditions.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: