Hunan Engineering Technology Research Center for Comprehensive Development and Utilization of Biomass Resources, College of Chemistry and Bioengineering, Hunan University of Science and Engineering, Yongzhou 425199, China
b.
Shanghai Frontiers Science Center of TCM Chemical Biology, Institute of Interdisciplinary Integrative Medicine Research and Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China
c.
Zhejiang Cancer Hospital, Hangzhou Institute of Medicine (HIM), Chinese Academy of Sciences, Hangzhou 310022, China
d.
State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Institute of Medicinal Plant Development, Chinese Academy of Medical Science and Peking Union Medical College, Beijing 100700, China
Received Date:
15 January 2024 Accepted Date:
29 February 2024 Revised Date:
27 February 2024 Available Online:
15 January 2025
Abstract:
Despite ongoing advancements in cancer treatment, the emergence of primary and acquired resistance poses a significant challenge for both traditional chemotherapy and immune checkpoint blockade therapies. The demand for targeted therapeutics for multidrug-resistant cancer is more important than ever. Peptides, as emerging alternatives to current anticancer drugs, offer exquisite versatility in facilitating the design of novel oncology drugs, with the core superiorities of good biocompatibility and a low tendency to induce drug resistance. This review comprehensively introduces the pharmacological mechanisms of peptide-based drugs and strategies for overcoming multidrug resistance (MDR) in cancers, including inducing cell membrane lysis, targeting organelles, activating anticancer immune responses, enhancing drug uptake, targeting ATP-binding cassette (ABC) transporters, and targeting B-cell lymphoma-2 (BCL-2) family proteins. Additionally, the current clinical applications of representative peptides in combating MDR cancers and their potential directions for medicinal chemistry research have been thoroughly discussed. This review offers essential insights into the novel treatment approaches for MDR cancers and highlights the trends and perspectives in this field.
Cancer remains an arduous disease that poses challenges in terms of treatment and cure, contributing significantly to global morbidity and mortality [1,2]. Chemotherapy typically serves as a first-line clinical intervention for cancer patients, but its efficacy is limited by severe side effects and inherent or acquired drug resistance [3]. Despite the groundbreaking impact of immune checkpoint blockades (ICBs) on cancer therapy, their response rate is also limited due to drug resistance issues [4]. Multidrug resistance (MDR) is the primary cause of malignancy development, spread, and clinical chemotherapy failure [5-7]. The mechanisms underlying MDR are extraordinarily complex, including but not limited to the following: (1) increased expression of multidrug resistant efflux transporters like P-glycoprotein (P-gp/ABCB1), breast cancer resistance protein (BCRP/ABCG2), and MDR protein 1 (MRP1/ABCC1) [8-12] (2) the deregulation of apoptosis or programmed cell death, including imbalances in B-cell lymphoma-2 (BCL-2) family levels, p53 inactivation, and overexpression of inhibitor of apoptosis proteins (IAPs) [8,13,14]; (3) intratumor heterogeneity, a significant driver of tumor-acquired resistance [15,16]; (4) the tumor microenvironment (TME) that protects tumor cells from the effects of chemotherapeutic drugs by influencing both extrinsic and intrinsic resistance pathways [17-20]. These resistance mechanisms contribute to chemotherapy failure, making MDR a formidable challenge for cancer therapy.
Peptide therapy has emerged as a new and promising modality for treating cancers. In contrast to conventional chemotherapeutics, which primarily affect tumor cell proliferation kinetics, peptides exhibit unique superiority through multiple mechanisms, irrespective of the genetic and epigenetic features of tumors. These mechanisms are believed to effectively overcome MDR in tumors [21]. Moreover, due to their structural diversity, facile synthesis, and natural functional properties as protein ligands, peptides can be easily designed, engineered, and utilized for precise targeting of MDR-associated proteins or signaling pathways without causing off-target toxicity or side effects on normal tissues. Concurrently, increasing preclinical evidence has demonstrated that peptides can enhance the sensitivity of tumor cells to existing anticancer drugs [22]. Significantly, there has been a persistent influx of peptide-based therapeutics entering clinical trials with promising outcomes. However, these peptides still encounter challenges regarding their inherent conformational instability, proteolytic susceptibility, and suboptimal therapeutic efficacy. This review comprehensively presents peptide-based strategies and their underlying mechanisms for treating MDR tumors. Additionally, the current clinical applications and potential development of these peptides in combination with existing anticancer drugs are also discussed in detail.
2.
Pharmacological mechanism of peptide-based strategies for overcoming MDR in cancer
The emergence of MDR stands as a prominent factor contributing to the failure of chemotherapy in tumor treatment and poses challenges to therapeutic efficacy [23]. Peptides have been used extensively in biomedical applications, including cancer/chronic disease therapy and drug/gene delivery, due to their excellent biocompatibility and biodegradability [24-26]. Peptide-based strategies in the fight against cancer and precision-targeted drug delivery systems enable the precise elimination of cancer cells while minimizing harm to normal cells [27-29]. Notably, compared to traditional chemotherapeutics, peptides exhibit lower susceptibility to developing drug resistance. Currently, the pharmacological mechanisms of peptides for treating MDR cancers can be summarized as inducing cancer cell death and mediating sensitization to chemotherapy/radiotherapy.
2.1
Peptides induce MDR cancer cell death
2.1.1
Cell membrane lysis
Natural antimicrobial peptides (AMPs) represent a versatile class of peptides with broad-spectrum antibacterial activity [30]. The net cationic charges and hydrophobic residues on AMPs enable them to selectively interact with negatively charged bacterial membranes through electrostatic binding. Due to the same anionic phospholipids, such as phosphatidylserine, on the surface of cancer cell membranes, AMPs can also preferentially bind to cancer cells through electrostatic and hydrophobic interactions, ultimately leading to cancer cell death [31]. In contrast, mammalian cell membranes predominantly consist of neutral phospholipids. As a result, AMPs rich in basic amino acids exhibit preferential targeting toward tumor cells rather than toward normal cells [21]. Furthermore, mammalian cell membranes contain abundant cholesterol, which is located primarily in the inner leaflet of the membrane. Cholesterol reduces membrane fluidity and increases membrane rigidity, thereby enhancing resistance against disruption by AMPs [21]. Therefore, membrane-lytic peptides derived from AMPs have attracted increased amounts of attention for treating MDR cancers due to their generalized mechanism of action, rapid killing effect, and low tendency to induce drug resistance.
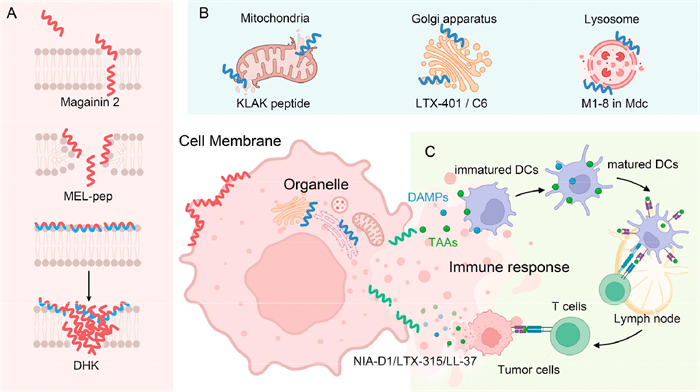
Magainin 2 (Fig. 1A, Table 1), an α-helical lytic peptide derived from the African clawed frog Xenopus laevis, was found to have the same antiproliferative effects on both chemonaive tumor cells and cell lines (human melanoma and small-cell lung carcinoma) with an MDR phenotype [32]. In contrast, normal fibroblasts did not show cell lysis in response to magainin 2 [33]. Melittin, the primary pharmacologically bioactive constituent of honeybee venom, is a well-studied antitumor peptide with membrane-lytic properties [34]. To enhance its antitumor effect, a derivative called MEL-pep was designed, synthesized, and evaluated in human 5-fluorouracil (5-FU)-resistant liver cancer cells (BEL-7402/5-FU) (Fig. 1A, Table 1) [35]. Remarkably, MEL-pep exhibited approximately 2.5-fold improved antiproliferative activity compared to melittin, and the cytotoxicity of MEL-pep toward BEL-7402/5-FU cells was more potent than that toward BEL-7402 cells [35]. Of note, MEL-pep further reversed the 5-FU resistance of BEL-7402/5-FU cells by downregulating P-gp expression, indicating that MEL-pep may be a promising candidate for the treatment of chemotherapeutic drug-resistant hepatocellular carcinoma (HCC) [35]. The acquisition of resistance by cancer stem cells and their role in tumor recurrence during treatment pose a formidable challenge to chemotherapy [36]. To evaluate the resistance formation of cancer stem cells to membrane-lytic peptides, Chen et al. [37] designed a peptide library consisting of 36 members and selected one specific peptide, called DHK (Fig. 1A, Table 1), to challenge the breast cancer stem cell line HMLER-shEcad at a half maximal inhibitory concentration (IC50) of either doxorubicin (DOX) or DHK. As expected, the DOX group exhibited an ∼7.8-fold increased IC50 value after 10 days. In contrast, treatment of HMLER-shEcad cells with DHK or its d-enantiomeric analog did not result in noticeable changes in morphology and IC50 values, indicating that membrane-lytic peptides may be less susceptible to cancer resistance than chemotherapeutics [37].
Figure 1
Figure 1.
Schematic representation of peptide-based therapeutic mechanisms inducing MDR-related cancer cell death. (A) Peptides lead to tumor cell membrane lysis. (B) Peptides selectively target various organelles, including mitochondria, the Golgi apparatus, and lysosomes. (C) Peptides induce anticancer immune responses.
In addition to membrane-disrupting effects, certain peptides employ an alternative mechanism by penetrating cancer cells without causing membrane disruption. These peptides can promote apoptosis/autophagy through targeted interactions with diverse organelles in cancer cells [38].
Mitochondria play pivotal roles in tumor development and progression, and mitochondrion-targeted therapy represents a promising strategy for overcoming MDR. Specifically, the pro-apoptotic peptide KLAK (KLAKLAK)2 has been shown to effectively target tumor cell mitochondria and disrupt their membrane integrity [39]. Bahmani et al. [40] investigated the synergistic effects of γ-irradiation and KLAK on monocytic leukemia (THP-1) radioresistant cells in vitro. The MTT assay results indicated that γ-irradiation alone exhibited slight cytotoxicity, while KLAK independently reduced THP-1 cell viability. Notably, the combination of KLAK and ionizing radiation induced mitochondrial damage and significantly decreased tumor cell viability, thereby promoting rapid apoptosis (Fig. 1B, Table 1). With a KLAK-based liposome (DKD-Lip) loaded with paclitaxel (PTX) designed for mitochondria-oriented delivery of chemotherapeutics to overcome MDR, this mitochondrion-targeted nanoparticles shown great potential as cancer-targeted therapies [41]. Due to the enhanced induction of apoptosis and accumulation of PTX facilitated by KLAK in human drug-resistant lung cancer A549/Taxol cells, DKD-Lip demonstrated superior efficacy for the treatment of A549/Taxol cells both in vitro and in vivo compared to free PTX and traditional liposome-loaded PTX [41]. Furthermore, a combinatory therapeutic system encapsulating the cytotoxic drug SN38 and KLAK also exhibited remarkable antitumor efficacy against DOX-resistant breast cancer cells (MCF-7/ADR) [42].
The Golgi is not only associated with the growth and metastasis of several cancers through the secretion of proteins, but also plays a vital role in the development of acquired drug resistance [43,44]. LTX-401 (Fig. 1B, Table 1), an amphipathic β (2,2)-amino acid, exhibited antitumor activity by specifically targeting the Golgi apparatus and disrupting its membrane structure [45]. Moreover, LTX-401-induced cell death in a rat model of JM1 HCC effectively suppressed tumor growth and elicited a durable protective immune response even after complete regression of the tumor, thereby mitigating tumor drug resistance [46]. In addition, the cysteine-rich targeting peptide C6 was identified to specifically target the Golgi apparatus in cancer cells. The C6 was combined with Flynn’s enzyme furan cleavage substrate and a short peptide helical fiber unit to form a nontoxic prodrug nanoparticle (Fig. 1B, Table 1). Upon reaching the Golgi apparatus through targeted transport, these nanoparticles undergone cleavage and deformation, transforming into helical nanofiber structures [43]. The resulting transformable helical self-assembly effectively disrupted the membrane of the cancer cell Golgi apparatus, leading to the inhibition of cytokine secretion and subsequent death of MCF-7 cells. Importantly, repeated stimulation of the tumor did not result in acquired drug resistance [43]. Therefore, physical disruption of the cancer cell Golgi apparatus may be an effective way to prevent drug resistance in cancer cells.
In cancer cells, the manipulation of lysosomal number, composition, and activity allows for the upregulation of metabolic pathways to support rapid growth and proliferation [47,48]. Consequently, therapeutic interventions targeting lysosomes hold significant potential in modulating cancer cell behavior such as proliferation rates and resistance to radiotherapy or chemotherapy [49]. Musca domestica cecropin (Mdc) is a linear peptide comprising 40 amino acids, featuring three α-helical structures at specific residue sites [50]. M1-8, the N-terminal 1-8 amino acids of Musca domestica cecropin, exhibited direct targeting ability to lysosomes in human HCC HepG2 cells. This led to disruption of lysosomal integrity and subsequent release of lysosomal tissue protease D (m-CTSD) into the cytoplasm [51]. Additionally, M1-8 triggered caspase activation, altered the mitochondrial membrane potential, induced apoptosis in cancer cells, and enhanced the expression of autophagy-related proteins such as microtubule-associated protein 1 light chain 3 Ⅱ (LC3 Ⅱ) in HCC cells by inhibiting autophagosome-lysosome fusion. (Fig. 1B, Table 1) [51].
2.1.3
Anticancer immune response
Inspired by the fact that the immune system fails to detect most cancers, immunotherapy has gained significant attention in recent years as an innovative approach to cancer treatment. This strategy aims to inhibit and eradicate tumor cells by reactivating and sustaining the cancer-immunity cycle (CIC) through eliciting specific immune responses against tumors [52]. The primary cascade of events in CIC involves the release of tumor antigens, tumor antigen presentation, and the initiation and activation of T cells, followed by their migration into the TME to kill tumor cells (Fig. 1C). Modulating each pivotal step within this intricate immune cycle holds immense promise for advancing cancer immunotherapy [53]. Immunotherapy represents a departure from the challenges associated with MDR in cancer treatment, as it stimulates the patient’s innate or adaptive immune system to combat cancer. Representative cancer immunotherapies include cancer vaccines, immune checkpoint inhibitors, and chimeric antigen receptor T-cell immunotherapy (CAR-T) [54-56].
Peptides also have the potential to stimulate the immune response by targeting CIC to effectively eliminate malignant cells. To overcome hypoxic tumor cell resistance to radiation, Zhu et al. [57] synthesized an amphiphilic peptide named NIA-D1 by coupling the hydrophobic radiotherapy sensitizer 2-(2-nitroimidazol-1-yl) acetic acid (NIA) with the hydrophilic PD-L1 antagonist D1 peptide, utilizing the hydrophobic matrix metalloproteinase-2 peptide substrate (PLGLAG) as a linker (Fig. 1C, Table 1). Subsequently, NIA-D1 was co-assembled with the hydrophobic Toll-like receptor (TLR)7/8 agonist R848 to form nanoparticles named NIA-D1@R848. In this context, NIA facilitated tumor cell apoptosis and antigen release, while R848 promoted dendritic cell (DC) maturation. Additionally, the D1 peptide blocked the programmed cell death protein-1/ligand 1 (PD-1/PD-L1) pathway to restore T-cell function, thereby initiating complete CIC [57]. This peptide-based approach concurrently targets three pivotal barriers encountered in CIC: insufficient antigen release within the TME, impaired antigen presentation by dendritic cells, and suppressed T-cell activation due to high PD-L1 expression levels on tumor cells.
The human AMP LL-37 (Fig. 1C, Table 1) not only possesses direct membrane lysis properties but also has a diverse range of immunomodulatory functions [58]. LL-37 can augment the effectiveness of chemotherapeutic drugs by facilitating their uptake in tumor cells and promoting apoptosis. Furthermore, LL-37 plays a pivotal role in enhancing tumor recognition and attack by regulating immune cell activity, thereby combating resistance to cancer drugs. Due to the high autophagy levels observed in pancreatic cancer, a majority of patients developed acquired resistance to gemcitabine within weeks of initial treatment. However, LL-37 inhibited pancreatic cancer autophagy and remodeled the tumor immune microenvironment by increasing the numbers of effector CD8+ and CD4+ T cells in the TME [59].
Thus, peptides targeting different stages of CIC offer the prospect for low toxicity, high specificity, sustained potency, and long-term survival in systemic tumor therapy.
2.2
Peptides mediate the chemosensitization of MDR cancers
Peptide-mediated sensitization to cancer chemotherapy is a strategy for increasing the accumulation of chemotherapeutic agents in cancer cells, thereby augmenting therapeutic efficacy through specific binding to receptors or molecules expressed on cancer cells [60-62]. Peptides can also target ABC transporter proteins, modulating their sensitivity to anticancer drugs and mitigating the MDR effect of cancers [63,64].
2.2.1
Peptides enhance the uptake of chemotherapeutics
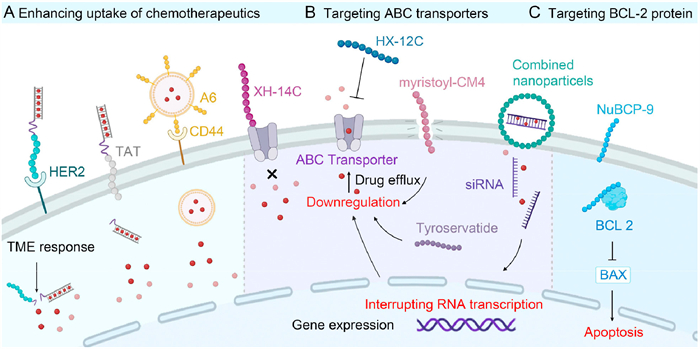
We have witnessed the remarkable success of antibody-drug conjugates (ADCs), which are combinations of antibodies and cytotoxic drugs that have been widely used to overcome MDR [65]. However, the complex structure of ADCs leads to high production costs, while their high molecular weight restricts effective penetration into solid tumors [66]. Owing to their enhanced tumor penetration, reduced immunogenicity, and lower production costs, peptide-drug conjugates (PDCs) have emerged as a novel class of drugs [67]. The PDC comprises a targeting peptide, linker, and cytotoxic payload. The peptide is designed to specifically target and deliver drugs to tumor cells, while the payload facilitates anticancer biological effects [68]. The peptides employed in PDCs can be categorized into two distinct groups: cell-targeting peptides (CTPs) and cell-penetrating peptides (CPPs). CTPs can selectively bind receptors or molecules on the surface of tumor cells. For instance, breast cancer cells often exhibit overexpression of the HER2 receptor. By combining HER2-targeting peptides with DOX, it is possible to significantly enhance DOX accumulation in HER2-positive breast cancer cells [69], thereby reducing drug resistance and toxicity to normal cells (Fig. 2A, Table 1). CPPs, characterized by their abundant cationic charges, exhibit a remarkable advantage in penetrating the cell membrane. This penetrability facilitates chemotherapeutic drugs in overcoming the barrier of the cell membrane [70].
Figure 2
Figure 2.
Schematic representation of the peptide-based therapeutic mechanisms mediating the chemosensitization of MDR cancers. (A) Peptides enhance the cellular uptake of chemotherapeutics. (B) Peptides targeting ABC transporters or downregulating their expression. (C) Peptides targeting BCL-2 family proteins.
The targeting property of CPPs facilitates the precise localization of chemotherapeutic drugs to cancer cells through endocytosis or other energy-dependent cellular processes [71]. A widely used CPP is the TAT peptide, which is derived from reverse transcription activators of human immunodeficiency virus type Ⅰ (HIV-1) (Fig. 2A, Table 1) [72]. In one study, a targeted nanoplatform based on TAT was engineered to achieve controlled release of DOX upon exposure to near-infrared light. The co-introduction of the targeting ligands cRGD and the CPP TAT significantly increased the intracellular accumulation of DOX [73]. In conclusion, the use of CPPs allowed for an increase in the local concentration of chemotherapeutic agents while reducing dosage requirements and toxic side effects. Therefore, CPPs hold significant therapeutic potential in drug delivery applications [74].
CTPs, ranging of 3–14 amino acids in length, are more favorable drug carrier molecules than CPPs. The latter face limitations in clinical development due to their lack of tissue targeting. In contrast, CTPs exhibit a remarkable affinity for receptors that are overexpressed on the surface of cancer cells, displaying characteristics similar to those of monoclonal antibodies [75]. Like CPPs, CTPs have also been extensively used to interact with biological membranes and facilitate the entry of chemotherapeutic agents into cancer cells, presenting promising therapeutic strategies for augmenting chemosensitivity. Multiple myeloma (MM) is a prevalent hematological malignancy characterized by the overexpression of the adhesion molecule CD44, and A6 (KPSSPPEE) is widely recognized as a CTP targeting CD44 (Fig. 2A, Table 1) [76]. Previous studies have demonstrated that the A6-functionalized polymersomal epirubicin exhibited enhanced targeting capabilities and antitumor effects in mice bearing MM tumors, resulting in a significant reduction in bone damage and improved survival rates compared to those of non-targeted nanoparticles and free epirubicin [77]. Furthermore, the safety of the A6 peptide has been demonstrated in phase Ⅰ and Ⅱ clinical trials, in which subcutaneous administration of this peptide at high doses (150–300 mg/kg) twice daily exhibited exceptional tolerability [78]. By utilizing these peptides, more precise drug delivery and enhanced tumor therapy can be achieved, ultimately offering patients more effective therapeutic options [68].
2.2.2
Targeting ABC transporters or downregulating their expression
The overexpression of ABC transporter proteins, which actively efflux various drugs from cells, is widely recognized as a major contributor to MDR in cancers [79]. MDR in cancer cells involves three key members of the ABC family: ABCB1/P-gp, ABCG2/BCRP, and ABCC1/MRP1. These members consist of a transmembrane structural domain and a cytoplasmic nucleotide-binding structural domain [80]. Their specific structure enables them to exhibit robust and sustained efflux of antitumor agents [81]. Therefore, the inhibition or reversal of ABC-mediated drug efflux pumps represents a pivotal strategy for combating MDR in cancer cells.
Several compounds have been identified as inhibitors of ABC transporter proteins, effectively blocking their activity. For instance, vatalanib, a multitargeted tyrosine kinase inhibitor, demonstrated anti-drug-resistant effects in colon cancer cells under hypoxic conditions by inhibiting both ABCG2 and ABCB1 efflux [82]. Competitive inhibitors such as lazertinib also interfered with the ATPase activity of the ABC family to reverse MDR mediated by either ABCB1 or ABCG2 [83,84]. However, ABC transporter protein inhibitors may interact with other synergistic chemotherapeutic agents, thereby impacting their metabolism and leading to severe side effects [85-87].
Peptides are a promising therapeutic strategy for targeting ABC transporter proteins due to their high selectivity, low toxicity, and good biocompatibility [22]. The peptide XH-14C has been shown to enhance the intracellular accumulation of PTX by obstructing the function of ABC transporter proteins [88]. Molecular dynamics simulation studies further demonstrated that the XH-14C-ABCB1 complex exhibited a stable binding conformation, indicating its potential for reversing ABC transporter-mediated MDR (Fig. 2B, Table 1) [88]. The peptide HX-12C (Fig. 2B, Table 1), an AMP derived from temporin-Pta, was also discovered to exhibit a remarkable inhibitory effect on ABCB1-mediated drug efflux, increasing the intracellular concentration and cytotoxicity of PTX against MDR cancer cells [89]. In addition to interacting with ABC transporter proteins to inhibit their function, some peptides can enhance drug efficacy by downregulating their expression. Previous studies have successfully modified the AMP CM4 (Table 1) by coupling a myristoyl group to its N-terminus, resulting in the development of myristoyl-CM4. The myristoylated CM4 demonstrated enhanced cytotoxicity against human chronic myeloid leukemia K562/MDR cells (Fig. 2B). Flow cytometry analysis further revealed that myristoylated CM4 could induce direct disruption of the cell membrane and significantly reduce the expression level of P-gp [90]. Tyroservatide (YSV) is a tripeptide compound with antitumor activity on HCC (Fig. 2B, Table 1), and it can reverse MDR in human HCC BEL-7402/5-FU cells to DOX and 5-FU by downregulating the expression levels of MDR1, MRP1, and lung resistance proteins while inhibiting their P-gp functions [91]. These findings implied the potential of peptide for overcome MDR, leading to improved outcomes during chemotherapy.
The expression of MDR-associated proteins can also be downregulated using RNA interference (RNAi) technology [92,93]. This technology uses small molecule RNAs (siRNAs or shRNAs) to interfere with the transcription and translation of target genes, thereby reducing the expression level of ABC transporter proteins (Fig. 2B). Given that PKM2 is frequently overexpressed in tumor cells and serves as an energy supplier for ABC transporters, a guanidine-rich spherical helical peptide was developed to load DOX and form nanocomplexes with PKM2 siRNA. This innovative approach not only hampered glycolytic metabolism but also reduced the energy supply for ABC transporter proteins, consequently impeding drug efflux associated with reverse drug resistance [94]. Thus, peptide-modified nanoplatforms can effectively co-deliver chemotherapeutic drugs and siRNAs into cells to enhance tumor therapeutic efficacy while simultaneously improving chemotherapeutic drug sensitivity and promoting synergistic application of chemotherapy and gene therapy.
2.2.3
Targeting BCL-2 family proteins
BCL-2, an oncogene that promotes cancer cell proliferation and inhibits apoptosis, is frequently overexpressed in various cancers. This uncontrolled overexpression has been associated with drug resistance to Taxol (cremophor-based formulation) and Nab-paclitaxel (Abraxane) [95]. To overcome this resistance mechanism, several peptides have been developed to target BCL-2 family members and modulate their activity and protein levels (e.g., TAT-IDPs, BIRD-2, and NuBCP-9). Among them, the peptide NuBCP-9 could bind to BCL-2 and alter its conformation to inhibit BAX. The nanoparticles loaded with PTX and NuBCP-9 exhibited approximately 64-fold improved efficacy compared to PTX alone on PTX-resistant MCF-7 cells, supporting the vital contribution of NuBCP-9 to the combination (Fig. 2C, Table 1) [95].
3.
Current clinical applications of peptides for overcoming MDR cancers
3.1
Oncolytic immunotherapy
High degrees of cancer heterogeneity are associated with genetic, epigenetic, transcriptional, and metabolic resistance to treatment and poor outcomes. In this context, oncolytic immunotherapies are an emerging class of therapeutic modalities for targeting cancer heterogeneity and overcoming MDR [96]. Notably, four oncolytic viruses have been approved for cancer treatment globally, with some promising success [97]. Nevertheless, the application of oncolytic viruses is still limited by the efficacy of monotherapy, undetermined biological safety profile, high cost, storage requirements, and administration challenges [98]. As one of the new alternative approaches, oncolytic peptides not only elicit robust anticancer immune responses against cancers through multiple mechanisms, but also offer several potential benefits over oncolytic viruses in terms of relative safety, cost-effective manufacturing, convenient handling, and moderate regulation [99]. In recent years, four oncolytic peptides have entered clinical trials, and alongside numerous peptides currently undergoing preclinical studies [96]. Clinical evidence has demonstrated that oncolytic peptides are valuable tools for inducing cancer immunotherapies [96]. Typically, the monotherapy of oncolytic peptide LTX-315 can effectively increase intralesional CD8+ T cells in 12 of 14 (86%) cancer patients and reduce tumor volume (≥30%) in 29% of patients, with an acceptable safety profile [100]. When combined with adoptive T-cell therapy, the best overall clinical response observed was stable disease for a duration of 208 days in a patient with metastatic soft tissue sarcoma [101].
In addition to peptide-mediated oncolysis and immunogenic cell death (ICD), oncolytic peptides exhibit multifunctionality in harnessing the TME. Indeed, although rapid membrane disruption and ICD stimulation are typical characteristics of oncolytic peptides, these processes represent only initial events in immunological pathways. New mechanisms underlying the ability of oncolytic peptides to overcome MDR are constantly being clarified, particularly those that are independent of cytolysis and accidental cell death. Cytotoxic T-lymphocyte antigen-4 (CTLA-4) and PD-1/L1 inhibitors have revolutionized cancer treatments with remarkable effects. However, these inhibitors still suffer from primary or acquired resistance, including a lack of tumor immunogenicity, abnormal expression of immune checkpoints, and an immunosuppressive TME [4,102]. Clinical studies have revealed that the majority of patients exhibiting microsatellite stability and proficient mismatch repair fail to elicit a response to ICB due to the inadequate presence of highly immunogenic tumor antigens. Oncolytic peptide-driven ICD provides abundant antigenic material to recruit APCs to the TME, thereby initiating CIC. Of note, unlike other ICD inducers, LTX-315-mediated protective antitumor effects did not rely on the type 1 interferon 1α/β receptor/TLR3 and high mobility group protein 1/TLR4 transduction pathways in vivo (Fig. 1C, Table 1) [103]. Thus, oncolytic peptides have been used to sensitize nonimmunogenic tumors (such as colorectal cancer) to PD-1/PD-L1 blockade treatment [104]. Moreover, alterations of immune checkpoints might also contribute to immune resistance. LTX-315 could increase CTLA-4 membrane levels in intratumoral CD8+ T cells in MCA205 sarcomas, thereby overcoming tumor resistance to CTLA-4 blockade [103], and downregulate PD-L1 expression in pancreatic tumor cells via ATP11B, which may reduce adaptive resistance to PD-L1 antibodies [105]. Additionally, the immunosuppressive TME is recognized as a primary source of resistance to ICB, and LTX-315 exhibited remarkable efficacy in reducing intratumoral regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs), both of which possess potent immunosuppressive properties. This reduction in Treg cells and MDSCs facilitates the infiltration of cytotoxic T lymphocytes into the TME and augments the response to PD-1 blockade in sarcoma models [103].
The aforementioned observations exemplify the multiple abilities of oncolytic peptides to overcome cancer resistance by oncolysis, inducing ICD, and reprogramming the tumor immune microenvironment via direct or indirect effects on tumor and immune cells.
3.2
Peptide-drug conjugates
Currently, there are approximately 100 ongoing clinical trials and 2 U.S. Food and Drug Administration (FDA)-approved PDC drugs, namely 177Lu-dotatate (lutathera) and Pepaxto (melphalan flufenamide/melflufen), all of which were designed to target overexpressed receptors on tumor cells [61,68]. As a first-in-class PDC drug, 177Lu-dotatate is a radiolabeled octreotide administered intravenously for treating gastrointestinal, pancreatic, and neuroendocrine tumors [106]. Octreotide, a peptide analog of somatostatin, has a high affinity for tumor cells overexpressing somatostatin receptor type 2. 177Lu-dotatate enters and accumulates within tumor cells via somatostatin receptor-mediated endocytosis, causing cytotoxic radiation to kill the cells [106]. Prostate cancer with neuroendocrine differentiation is associated with treatment resistance and poor prognosis, but an excellent response to 177Lu-dotatate in patients with metastatic castration-resistant prostate cancer with neuroendocrine differentiation was still observed after 177Lu-dotatate therapy [107,108]. Pepaxto is a dipeptide with the alkylating agent melphalan and 4-fluoro-l-phenylalanine targeting aminopeptidases, which are overexpressed in aggressive malignancies [109]. Pepaxto demonstrated significant antitumor activity against myeloma cells resistant to bortezomib, dexamethasone, and melphalan. In combination with dexamethasone, it received approval in 2021 for the treatment of relapsed or refractory MM [110]. Unfortunately, it was regrettably withdrawn from the U.S. market after 8 months because of the failure of the phase Ⅲ randomized controlled trial [111]. Nevertheless, the advancement of PDCs remains a vibrant and highly active area of research and development in the quest to combat MDR cancers.
4.
New peptide-based approaches and medicinal chemistry strategies for the treatment of MDR cancers
Two intrinsic pharmacological hurdles often restrict the clinical application of therapeutic peptides: proteolytic susceptibility and poor membrane permeability. Importantly, these drawbacks could lead to suboptimal therapeutic efficacy. To enhance the drug-like properties of peptides, diverse medicinal chemistry approaches have been developed to improve their metabolic stability, cellular membrane penetration, and bioactivity.
4.1
Cyclization
The past two decades have witnessed the approval of more than 60 peptide drugs by both the FDA and the European Medicines Agency. Approximately two-thirds of these peptides exist in a cyclic form [112]. Cyclic peptides with a preformed, constrained conformation exhibit improved proteolytic stability, target protein binding affinity, and cell membrane permeability when compared to linear peptides. Leveraging advancements in organic synthetic methods, the development of increasingly diverse cyclic peptides holds immense potential for overcoming challenges in drug discovery and development. In addition to head-to-tail cyclic peptides, stapled helical peptides have recently garnered significant attention and support due to their chemical diversity and improved drug-like properties [113]. In this approach, therapeutic peptides are stabilized by cross-linking either the side chain to the terminus or the side chain to another side chain in rational positioning, thereby forming pre-organized stable α-helical conformations with a lower entropic penalty for adapting to the cell environment and binding to targets [114].
To increase the structural stability and efficacy of oncolytic peptides, we developed a series of stapled anoplin peptides (StAnos) derived from an α-helical wasp venom peptide using a ring-closing metathesis-mediated all-hydrocarbon stapling strategy [52]. As expected, all the StAnos exhibited greater helicity (3.2%–40.0%) than did the linear anoplin (2.54%). Due to the stable α-helix topology and extended hydrophobic network on the nonpolar face of anoplin, StAnos also exhibited improved antiproliferative activity, ranging from 5.8- to 47.8-fold on mouse triple-negative breast cancer 4T1 and mouse melanoma B16F10 cells compared with anoplin. Of note, the optimal StAno variant, Ano-3, displayed superior oncolytic efficacy in both concentration- and time-dependent manners relative to that of LTX-315 [52]. To further improve the solubility and selectivity of Ano-3, we applied the side-chain-retention stapling strategy [115] to generate Ano-3s as a lead compound. The Ano-3s provided a wider tumor-specificity window than did Ano-3 without compromised oncolytic potency. Moreover, both Ano-3 and Ano-3s displayed more than 16-fold enhanced protease stability compared to anoplin (with a half-time of 12 min), whereas the half-life of LTX-315 was only 34 min. In vivo studies showed that Ano-3s could induce B16F10 tumor ablation in mice and achieve a 50% long-term survival rate through its oncolytic effect and stimulation of ICD [52]. Additionally, we found that stapling chemistry could serve as a potential strategy to expedite the development of oncolytic peptides based on other AMPs [104,116].
4.2
PEGylation
Polyethylene glycol (PEG)-related modifications have been widely applied to proteins or peptides (such as pegcetacoplan and PEGylated exenatide) in the past two decades after the FDA clinically approved the first PEGylated protein [117]. The PEG corona can sterically shield peptides from metabolic enzymes in the bloodstream and kidney clearance, thereby increasing bioavailability and decreasing immunogenicity/toxic side effects [118,119]. Previously, we successfully enhanced the drug-like properties of oncolytic peptides using cyclic methodology. However, undesired systemic toxicity profiles still limit the administration of these peptides via the intratumoral route, which poses an enormous challenge for inaccessible or metastatic tumors. Thus, we chose 4-arm PEG as a systemic formulation to synthesize a PEG-oncolytic peptide conjugate with a matrix metalloproteinase-2 cleavable spacer [104]. PEGylation drastically decreased the hemolytic activity of the stapled oncolytic peptide MP9. To achieve the targeted therapeutic effect of MP9 and ICB for colorectal cancer, our group further incorporated an anti-PD-L1 peptide with PEGylated MP9, forming a novel peptide-polymer conjugate named PEG-MP9-aPDL1 that specifically targets overexpressed PD-L1 on the surface of colorectal cancer cells. PEG-MP9-aPDL1 displayed a favorable pharmacokinetic profile with prolonged circulation, tumor targeting, and no obvious toxicity [104]. Significantly, this innovative application of a PEGylated strategy effectively addressed the insensitivity of colorectal cancer to anti-PD-L1 therapy caused by neoantigen deficiency [4,120]. This multifunctional polymer chimera offers a promising paradigm for enhanced immunotherapy.
4.3
Self-assembly
Owing to the same backbone structure and intermolecular noncovalent interactions, such as hydrogen bonds, hydrophobic, ionic, and π-π interactions, peptides can self-assemble into different units with stable structures and dynamic functions [121]. In comparison to monomeric peptides, Self-assembled peptides generally exhibit better resistance to proteases, flexible responsiveness, and biological function [122]. Moreover, peptide-based biomaterials formed through self-assembly also present distinct advantages in terms of biocompatibility, biodegradability, and safety when compared with other drug delivery systems, including PEG, liposomes, and poly(lactic-co-glycolic) acid [123].
KLVFFAE, a peptide derived from Alzheimer’s Aβ16-22 [124] that can self-assemble into typical β-sheet structures, has been widely utilized to efficiently trigger self-assembly processes in regions of interest, such as biological membranes and tumor-associated proteins [125]. To take advantage of self-assembled peptides, the Wang group [126-128] developed a series of in situ transformable self-assembled peptide strategies for overcoming renal cell carcinoma (RCC) resistance. RCC is one of the most aggressive types of kidney cancer, characterized by an inherent and acquired MDR or a low therapeutic response. Among these contributing factors, the extracellular matrix (ECM) [129] and P-gp [130] are closely associated MDR determinants that underlie the insensitivity of RCC to chemotherapeutics. Fibronectin, a vital ECM component, directly contributes to ECM-mediated anoikis resistance and cell adhesion-mediated drug resistance (CAM-DR) [131]. Therefore, Wang et al. [127] constructed a transformable ECM deprivation system based on fibronectin-targeting peptide and the KLVFF sequence. This system can target overexpressed fibronectin in RCC and transform into nanofibers to achieve prolonged ECM deprivation, which contributes to reversing anoikis resistance by blocking fibronectin-mediated signaling pathways and sensitizing RCC cells to chemotherapy by reducing CAM-DR. In addition, agents that change the efflux associated with P-gp did not significantly improve chemotherapeutic efficacy in RCC patients [132]; therefore, an alternative approach to enhancing the chemosensitivity of RCC involves focusing on increasing influx. Wang et al. [126] developed a recognition-reaction-aggregation cascade strategy involving three sequential steps: (1) a targeting peptide (P1) modified with dibenzocyclooctyne that specifically recognized overexpressed carbonic anhydrase Ⅸ on the RCC cell membrane; (2) another peptide (P2) containing KLVFFAE and azide reacted with P1 by reagent-free click chemistry to form the aggregable peptide P3; and (3) self-assembled P3 was stably retained on the cell membrane and perturbed its permeability, allowing more DOX to be passively taken up by renal RCC cells with a 3.5-fold reduced IC50 value compared to that of free DOX. This strategy also exhibited a 3.2-fold greater inhibition rate in SK-RC-52 tumor-bearing mice than in those treated with free DOX [126]. This self-assembly design provides not only a new strategy for developing peptides, but also an efficient avenue for sensitizing MDR cancers to chemotherapy.
5.
Conclusion
In conclusion, the dynamic landscape of peptide-based strategies represents a promising horizon for addressing multidrug-resistant cancers. The diverse pharmacological mechanisms by which these peptides act, ranging from direct cell membrane lysis and organelle targeting to eliciting an anticancer immune response, underscore their potential to overcome the barriers posed by MDR. Notably, their capacity to enhance the uptake of chemotherapeutics, target ABC transporters, and modulate BCL-2 family proteins provides a multifaceted approach to combat cancer resistance. These advances in clinical application, particularly in oncolytic immunotherapy and PDCs, further reinforce the viability of these peptides in clinical settings. These advancements are bolstered by innovative techniques such as cyclization, PEGylation, and self-assembly, which address vital therapeutic drug treatment challenges, including stability, solubility, and bioavailability.
Despite these promising developments, the journey toward fully harnessing the potential of peptides in MDR cancer treatment remains ongoing. Continued interdisciplinary collaboration, leveraging insights from medicinal chemistry and materials science, is crucial for advancing this field. This collaborative approach has been exemplified by the progress made in oral peptide drugs for managing type 2 diabetes and is pivotal for achieving translational success with peptide-based therapeutics in MDR cancers. These efforts portend a future where the versatility and efficacy of peptides can be maximized, thereby offering new avenues for cancer therapy in an era increasingly challenged by drug resistance.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the Science and Technology Innovation Program of Hunan Province (No. 2022RC1168), National Natural Science Foundation of China (Nos. 82322073, 82173846, 82304533), CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2023-I2M-3-009), Key project at central government level: The ability establishment of sustainable use for valuable Chinese medicine resources (No. 2060302), China Postdoctoral Science Foundation (No. 2021M702215), Oriental Scholars of Shanghai Universities (No. TP2022081), Jiangxi Province Thousand Talents Program (No. jxsq2023102168), Young Talent Lifting Project of China Association of Chinese Medicine (No. CACM-(2021-QNRC2-A08)), Shanghai Rising-Star Program (No. 22QA1409100), Shanghai Sailing Program (No. 22YF1445000), 2021 Shanghai Science and Technology Innovation Action Plan (No. 21S11902800), Three-year Action Plan for Shanghai TCM Development and Inheritance Program (Nos. ZY(2021-2023)-0208, ZY(2021-2023)-0401), High level Key Discipline of National Administration of Traditional Chinese Medicine (No. 71), Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (No. ZYYCXTD-D-202004), and Innovation team of high-level local universities in Shanghai: Strategic Innovation Team of TCM Chemical Biology. All figures were created with BioRender.com.
Figure 1
Schematic representation of peptide-based therapeutic mechanisms inducing MDR-related cancer cell death. (A) Peptides lead to tumor cell membrane lysis. (B) Peptides selectively target various organelles, including mitochondria, the Golgi apparatus, and lysosomes. (C) Peptides induce anticancer immune responses.
Figure 2
Schematic representation of the peptide-based therapeutic mechanisms mediating the chemosensitization of MDR cancers. (A) Peptides enhance the cellular uptake of chemotherapeutics. (B) Peptides targeting ABC transporters or downregulating their expression. (C) Peptides targeting BCL-2 family proteins.

 DownLoad:
DownLoad:


 下载:
下载:



 下载:
下载:
