The Fifth Affiliated Hospital, Guangdong Provincial Key Laboratory of Molecular Target & Clinical Pharmacology, the NMPA and State Key Laboratory of Respiratory Disease, the School of Pharmaceutical Sciences, Guangzhou Medical University, Guangzhou 511436, China
b.
Department of Pulmonary and Critical Care Medicine, Zhujiang Hospital, Southern Medical University, Guangzhou 510280, China
Received Date:
18 December 2023 Accepted Date:
23 February 2024 Revised Date:
20 February 2024 Available Online:
15 December 2024
Abstract:
The anti-oxidative characteristic and immunosuppressive microenvironment contribute to a high resistance of tumor to many treatments. In this work, a glutathione (GSH)-responsive metal-coordinated oxidative stress amplifier (designated as CuPA) is fabricated to suppress tumor growth through elevating the cellular level of reactive oxygen species (ROS) and eliminating M2 macrophages. Among which, cooper ion (Cu2+) is capable of coordinating with thioredoxin (Trx) inhibitor of PX-12 and signal transducer and activator of transcription 6 (STAT6) inhibitor of AS1517499 with the assistance of distearoyl phosphoethanolamine-PEG2000 (DSPE-PEG2000), which can extensively increase the stability to enhance drug delivery in vitro and in vivo. Furthermore, CuPA can upregulate intracellular ROS to cause tumor cell death through restraining Trx and degrading GSH. Also, CuPA-mediated STAT6 inhibition results in the elimination of M2 macrophage to reverse the immunosuppressive tumor microenvironment. Finally, the elevated oxidative stress and increased immune activation amplify the synergistic antitumor effect without causing obvious side effect. This work provides a new sight for synergistic tumor suppression through chemo-immunotherapy in consideration of the complex resistant tumor microenvironment.
Malignant proliferation of tumor cells generates an increased oxidative stress, but tumor cells can develop a unique anti-oxidative microenvironment to combat the oxidative damage and cause therapeutic resistance [1,2]. Based on the scenario, some antitumor drugs have been developed to break the redox homeostasis of tumor cells, making them sensitive to oxidative drugs through downregulation of the antioxidant capacity [3–9]. As one of the most important anti-oxidative enzymes, tumor overexpressed thioredoxin (Trx) system can catalytically eliminate the intracellular reactive oxygen species (ROS) [10,11]. Therefore, a series of Trx inhibitors have been developed to interrupt the anti-oxidative characteristic of tumor cells and restore the oxidative damage ability of reactive oxygen species (ROS) [12–14]. Among which, PX-12 (PX for short) is a Trx specific inhibitor in preclinical investigation, which can inhibit the activity of Trx to elevate intracellular oxidative stress for tumor chemotherapy [15,16]. Moreover, recent studies indicate that copper ions (Cu2+) could initiate a Fenton-like reaction and deplete intracellular glutathione (GSH) [17]. Briefly, Cu2+ could convert hydrogen peroxide into a more oxidative hydroxyl radical (•OH) to enable chemodynamic therapy [18,19]. However, PX is strongly hydrophobic with low bioavailability, and the synergistic effect between PX and Cu2+ has been rarely investigated.
Apart from the anti-oxidative characteristics, tumor cells also reshape the microenvironment into an immunosuppressive phenotype to avoid immune elimination [20–22]. Upon the microenvironment, tumor associated macrophages (TAMs) are the dominant immune suppressive cells to accelerate the tumor growth and suppress the therapeutic efficiency [23]. Especially, TAMs could be roughly divided into immunosuppressive M2 type and immunotherapeutic M1 type [24]. In the past decades, many efforts have been devoted to discovering the therapeutic target of M2 macrophage to restore the antitumor effect [25–27]. Some recent studies found that signal transducer and activator of transcription 6 (STAT6) is greatly activated in M2 macrophage, and STAT6 inhibition could reduce the proportion of M2 macrophage [28–30]. In detail, STAT6 inhibition could impede the activation of STAT6 in response to interleukin 4 (IL-4) and deplete M2 macrophages. The activation of STAT6 could promote the expression of pro-tumor genes of Arg1, Fizz1 and Ym1, contributing to the conversion of macrophages to the M2 phenotype. Moreover, the inhibition of STAT6 could reduce the polarization of TAMs, leading to an antitumor effect. Of special note, AS1517499 (AS for short) as a specific inhibitor of STAT6 could not only boost tumor cell apoptosis, but also activate immunotherapeutic T cells to amplify the tumor suppressive effect [31–35]. Consequently, the combination of STAT6 inhibition with chemotherapeutic elimination of tumor cells is waited to be discovered in view of the complex resistant tumor microenvironment.
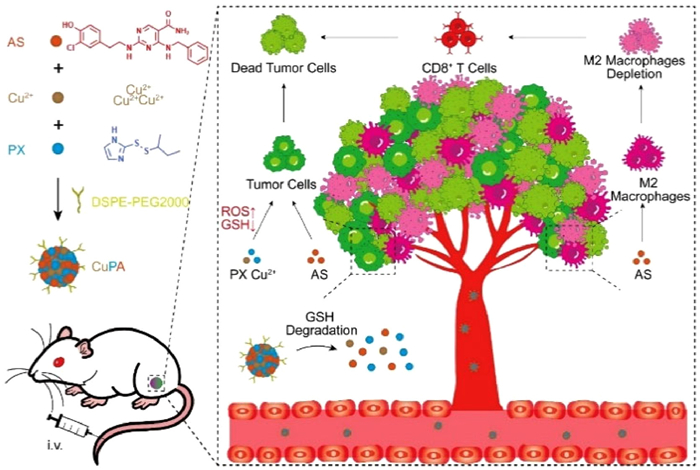
To further amplify the synergistic effect, therapeutic drug combination is preferred to concentrate in tumors to increase the treatment efficiency while reduce the drug exposure induced side effect. Instead of the complex drug carrier design, a simple but applicable nanomedicine is urgently needed in consideration of the drug structure and the synergistic mechanism [36–38]. As reported, the imidazole ring nitrogen atom could coordinate with metal ions to form coordination compounds [39]. Moreover, such metal-coordinated compound could be endowed with the ability of loading other drugs. Herein, a GSH-responsive metal-coordinated oxidative stress amplifier (designated as CuPA) was fabricated to suppress tumor growth by increasing intracellular ROS levels and eliminating M2 macrophages (Scheme 1). In detail, Cu2+, PX and AS could self-assemble into nanomedicine of CuPA in the presence of distearoyl phosphoethanolamine-PEG2000 (DSPE-PEG2000). The spherical CuPA exhibited an elevated stability and GSH responsiveness. Moreover, the improved cellular uptake and tumor targeting behaviors of CuPA enhanced the drug delivery efficiency. Meanwhile, CuPA could efficiently inhibit the activity of Trx to upregulate intracellular ROS, thereby enhancing the oxidative cell damage to decrease tumor proliferation. Importantly, CuPA was found to eliminate M2 macrophage to reverse the immunosuppressive microenvironment, contributing to the activation of cytotoxic T cells to synergistically suppress tumor growth. This work provided a new sight to develop metal-coordinate nanomedicine for enhancing the tumor suppression behavior of drugs regardless of the complex resistant tumor microenvironment.
Scheme 1
Scheme 1.
Preparation of metal-coordinated oxidative stress amplifier (CuPA) for tumor therapy based on the assembly of Cu2+, PX and AS in the presence of DSPE-PEG2000. The GSH responsiveness of CuPA facilitated the release of Cu2+ and PX to elevate the oxidative stress causing cell death. Moreover, AS could deplete M2-type macrophages to improve immunosuppressive microenvironment, promoting the activation of cytotoxic T cells for synergistic tumor treatment.
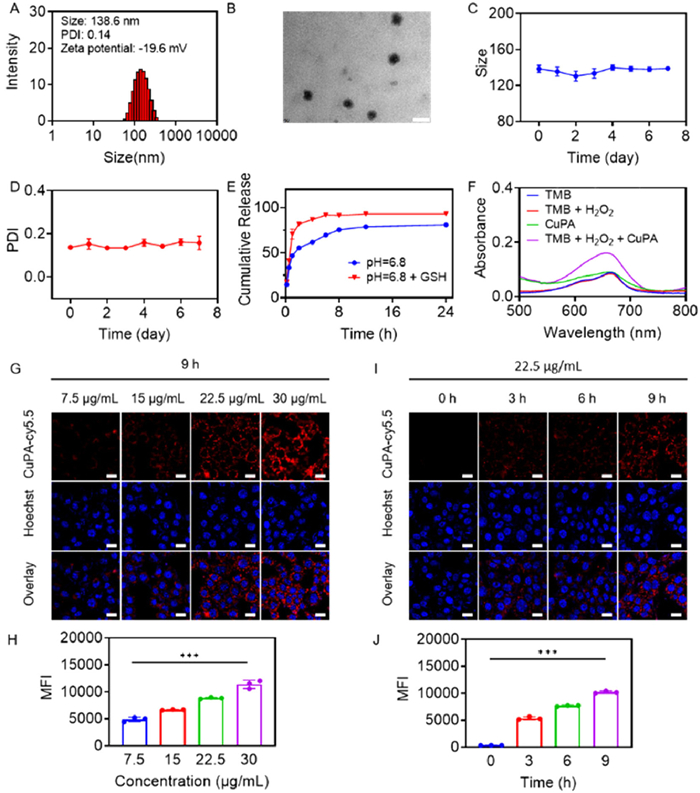
Nanomedicines, characterized by their nano size and unique drug properties, offer numerous advantages in the field of tumor therapy. Cu2+ had been found to form complexes with drugs that contained imidazole groups, thereby enabling the loading of a third drug. Herein, Cu2+ was expected to coordinate with PX and load AS to create an oxidative stress amplifier. In order to achieve a stable nanomedicine, CuPA was synthesized through the self-assembly of Cu2+, PX, and AS in the presence of DSPE-PEG2000 under alkaline condition. Excitingly, CuPA had favorable particle size of 138.6 nm and low polymer dispersity index (PDI) of 0.14 (Fig. 1A), which helped penetrate into target tissues and organs. The zeta potential of CuPA was measured to be −19.6 mV, which contributed to the reduction of protein adsorption and the prolongation of circulation time. In the course of conducting transmission electron microscopy (TEM) image analysis, it was determined that CuPA exhibited spherical shape and homogeneity (Fig. 1B). To further validate the properties of oxidative stress amplifier, the particle size and PDI of CuPA were analyzed by dynamic light scattering (DLS) for a duration of 7 days (Figs. 1C and D). Notably, the particle size and PDI of CuPA underwent a marginal change in 7 days. The particle size exhibited fluctuations ranging from 130 nm to 140 nm, while PDI remained consistently stable at approximately 0.14. CuPA demonstrated a good stability over a period of 7 days, which was beneficial for conducting further research on drug treatment. Subsequently, the concentrations of PX and AS were detected by high performance liquid chromatography (HPLC). Inductively coupled plasma-mass spectrometry (ICP-MS) was used to test the concentration of Cu2+. Finally, the drug loading rates of Cu2+, PX and AS were identified to be 5.9%, 42.2% and 29.9%. Moreover, due to the binding of Cu2+ with GSH, CuPA was able to release drugs in the presence of GSH owing to the competitive coordination effect. And the stable nanostructure of CuPA was also disrupted (Fig. S1 in Supporting information). In a simulated acidic tumor environment, the release of CuPA occurred rapidly in the presence of GSH. After 6 h, the CuPA release rate in the presence of GSH reached 91%, while 69% of the CuPA was released without GSH by that time (Fig. 1E). The release of CuPA facilitated rapid attainment of a high drug concentration to expedite treatment effectiveness. Some studies had found that copper mediated the catalytic synthesis of •OH for tumor treatment by the Fenton reaction. Tetramethylbenzidine (TMB) could undergo ultraviolet and visible spectrophotometry (UV–vis) absorption changes in the presence of •OH, which was commonly used as a Fenton reaction detection reagent. As demonstrated in Fig. 1F, CuPA was confirmed to catalyze the production of •OH from H2O2, leading to a pronounced enhancement in the absorption of TMB at 650 nm. No significant changes of the spectra were observed in the case of TMB under other conditions. Overall, CuPA, a stable metal-coordinated oxidative stress amplifier, had been successfully constructed with the ability of GSH response and generating Fenton reaction.
Figure 1
Figure 1.
Characterizations of CuPA. (A) The hydrodynamic size distribution and (B) TEM image of CuPA. Scale bar: 200 nm. (C) The particle size variations and (D) PDI changes of CuPA for 7 days (n = 3). (E) Drug release behaviors of CuPA in the absence or presence of GSH at pH 6.8 (n = 3). (F) UV–vis absorbance of TMB with or without H2O2 and CuPA. (G) CLSM images and (H) flow cytometry analysis of 4T1 cells after treatment with CuPA-cy5.5 at the concentration of 7.5, 15, 22.5 and 30 µg/mL (n = 3). (I) CLSM images and (J) flow cytometry analysis of 4T1 cells after treatment with CuPA-cy5.5 for 0, 3, 6 and 9 h (n = 3). Scale bar: 20 µm. ***P < 0.001 was tested via a Student's t-test.
To further evaluate the excellent performance of nanosized CuPA, confocal laser scanning microscope (CLSM) and flow cytometry were employed as detection tools to test the cellular uptake behavior. Making this approach possible, cy5.5 was added during the synthesis of CuPA, resulting in the formation of fluorescent CuPA named CuPA-cy5.5. As pictured in Fig. 1G, the incubation of 4T1 cells with CuPA-cy5.5 for 9 h led to an observable red fluorescence within the cells. The fluorescence elevation demonstrated a positive correlation with the concentration of CuPA-cy5.5, indicating a concentration-dependent uptake of CuPA by the cells. The same result was obtained using flow cytometry, showing a 2.32-fold increase in fluorescence intensity between the groups with the highest and lowest drug concentrations (Fig. 1H). The exceptional cellular uptake ability of CuPA could be attributed to its internalization via endocytosis, a cellular uptake mechanism commonly associated with nanomedicines. Definitely, 4T1 cells cultured with CuPA-cy5.5 also showed a rise in fluorescence over time (Figs. 1I and J), indicating an excellent cellular uptake property. It was supposed that CuPA could keep stable in aqueous phase but it would be decomposed to release drugs after internalized by tumor cells with overexpressed GSH.
To verify whether the excellent uptake capacity of CuPA had any effect on the generation of ROS in cells, 2′, 7′-dichlorodihydrofluorescein diacetate (DCFH-DA) and CellROX as the cell membrane permeable fluorescent probes were utilized as a detecting agent to measure the intracellular oxidant stress. In theory, the Fenton reaction could facilitate the generation of cytotoxic •OH by Cu2+, and PX, an inhibitor of thioredoxin-1, could inhibit the removal of ROS. However, CLSM was unable to detect the generation of ROS after treatment of Cu2+, PX, Cu2+ + PX or Cu2+ + PX + AS. In contrast, 4T1 cells treated by CuPA displayed bright DCF fluorescence owing to the generation of ROS (Fig. 2A). The drugs other than CuPA failed to generate ROS, possibly because of limitations in low-dose administration and the cellular uptake of small-molecule drugs. Besides, the cellular uptake of CuPA through endocytosis enabled it to function as an amplifier of oxidative stress, effectively regulating intracellular ROS levels. Furthermore, the mean fluorescence intensity (MFI) in Fig. 2B displayed that the group of CuPA was 3.55 times higher than that of Cu2+ + PX + AS. Moreover, CellROX was used as a probe to repeat the experiment in order to further confirm the excellent ROS generation of CuPA (Fig. 2C). As a result, 4T1 cells treated with Cu2+, PX, AS, Cu2+ + PX and Cu2+ + PX + AS displayed weak red fluorescence by CLSM. However, self-assembly amplifier of CuPA significantly contributed to the augmentation of intracellular oxidative stress, resulting in the highest fluorescence intensity. Besides, the MFI differed notably between blank control and CuPA group, and a 1.45-fold increase of MFI was observed following treatment with CuPA when compared to the three free drugs (Fig. 2D). These results further confirmed nanosized drug had better efficacy. Mitochondria served as the primary producer of ROS and played a vital role in the metabolic reprogramming of tumors, which were intricately linked to the tumor growth and progression. Elevated ROS levels triggered the activation of the mitochondrial membrane permeability transporter pore, ultimately resulting in cellular apoptosis. Following treatment, PX, as a Trx inhibitor, was found to elevate mitochondrial ROS levels, but no distinguishable difference was observed from the control group (Figs. 2E and F). Nevertheless, CuPA was confirmed to increase the content of ROS produced by mitochondria, which caused a 1.35-fold raise in MFI compared with the group of three free drugs (Fig. 2F). It was evident from the above findings that CuPA exhibited a proficient ability to generate cytotoxic ROS due to robust cellular uptake.
Figure 2
Figure 2.
Detection of ROS and GSH levels in 4T1 cells. (A) CLSM images and (B) MFI of 4T1 cells after treatment with Cu2+, PX, AS, Cu2+ + PX, Cu2+ + PX + AS or CuPA to evaluate ROS generation by DCFH-DA. (C) CLSM images and (D) MFI of 4T1 cells incubated with those drugs to test ROS generation by CellROS. (E) CLSM images and (F) MFI of 4T1 cells after those treatments to detect mitochondrial ROS using MitoROS 580. (G) CLSM images and (H) MFI of 4T1 cells after those treatments to detect GSH levels using CMFDA (n = 3). Scale bar: 20 µm. *P < 0.05, **P < 0.01 and ***P < 0.001 were tested via a Student's t-test. Error bar was computed using mean ± standard deviation (SD).
Besides, the GSH exhaustion by CuPA was also assessed by CLSM. 5-Chloromethylfluorescein diacetate (CMFDA) was used as a GSH indicator to measure intracellular redox homeostasis, which could emit a strong green fluorescence in the presence of GSH. As could be seen from Fig. 2G, 4T1 cells in blank group fluoresced brightly when incubated with CMFDA only, which illustrated an original high level of GSH in tumor cells. Superabundant GSH not only caused cellular resistance but also promoted tumor metastasis. However, 4T1 cells that dealt with Cu2+ and AS, had a negligible reduction in fluorescence of CMFDA (Fig. 2H). After treatment with PX, Cu2+ + PX or Cu2+ + PX + AS, the cellular fluorescence was slightly reduced. These findings implied that the GSH consumption ability of CuPA should be primarily endowed by PX. Better yet, a 0.53-fold reduction of the fluorescence intensity was detected in CuPA group compared with that of Cu2+ + PX + AS group, suggesting a significantly better effect of CuPA on GSH decline owing to the increasing drug uptake. Depletion of GSH was likely to induce oxidative stress in tumor, leading to increased drug sensitivity. In sum, CuPA could regulate intracellular redox state nicely than free medication by increasing ROS and exhausting GSH.
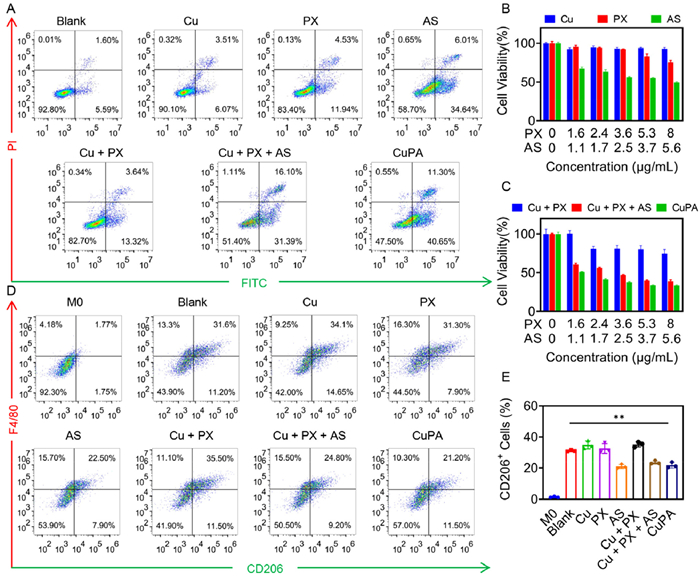
After confirming the excellent ability of CuPA to regulate oxidative stress, further investigation was conducted to examine its potential antitumor toxicity. The Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis assay was performed firstly. As shown in Fig. 3A, Cu2+ as a trace element did not cause a significant cytotoxicity when incubated at low levels. The Fenton effect produced in this case had minimal impact on the tumor cells. Similarly, the limited cellular penetration of PX hindered its ability of modulating oxidative levels for tumor inhibition. Despite an enhanced incidence of apoptosis compared to treatment alone, there was no discernible therapeutic benefit when Cu2+ and PX were administered together. The cytotoxicity induced by Cu2+ and PX was not satisfactory owing to the weak ROS levels. Interestingly, AS had some inhibitory effect on tumor proliferation. The apoptotic rate of 4T1 cells reached about 40.65% when AS was administered alone. The combination of three drugs was administered, and it was discovered that Cu2+ and PX induced oxidative stress improved the antitumor effectiveness of AS, resulting in a 47.49% of the cells to apoptosis. The findings of this study indicated that disrupting the oxidative balance enhanced the therapeutic efficacy of the medication. Noteworthy, more than half of the cells underwent apoptosis after CuPA treatment. It was inferred that the delivery characteristics of the pharmaceuticals had been changed by combining the three drugs as nanomedicine, which improved the tumor-inhibiting action even more.
Figure 3
Figure 3.
Antiproliferation and macrophage regulation effect of CuPA. (A) Flow cytometry apoptotic analyses of 4T1 cells after treatment with Cu2+, PX, AS, Cu2+ + PX, Cu2+ + PX + AS or CuPA. CCK8 analysis of 4T1 cell viabilities after treatment with gradient concentrations of (B) Cu2+, PX, AS, and (C) Cu2+ + PX, Cu2+ + PX + AS or CuPA. (D) Flow cytometry and (E) quantitative analysis of M2 polarization of RAW 264.7 cells after treatment with different drugs (n = 3). M2 macrophages were induced by IL-4 in advance. **P < 0.01 was tested via a Student's t-test. Error bar was computed using mean ± SD.
The cell counting kit 8 (CCK8) assay was then performed to further validate the therapeutic effect of CuPA. Cu2+ and PX were not observed to significantly limit cell growth at either low or high doses (Fig. 3B). The outcomes for the combined administration of Cu2+ and PX were similar (Fig. 3C). However, the cell survival was significantly decreased when Cu2+, PX and AS were added at the same time. It could be concluded that without the administration of drugs that reduced the integrity of tumors, the efficient functioning of oxidative stress became challenging. The cytostatic effects of CuPA were more effective at low doses, but there was little distinction between the three-part mixture and CuPA at high doses. This indicated that oxidative stress enhanced the cytotoxicity of AS at low concentrations, whereas at high concentrations, AS became the primary contributor to cytotoxicity. However, the viability of 3T3 cells was more than 80% after treatment with CuPA, which verified its low toxicity against normal cells and the biocompatibility in the short term (Fig. S2 in Supporting information).
Meanwhile, AS could remove tumor-promoting M2 macrophages to regulate the tumor microenvironment. As shown in Fig. 3D, RAW 264.7 cells were induced to differentiate into M2 macrophages by IL-4 in blank group, resulting in a population of 31.6% F4/80+CD206+ M2 macrophages. After treatment with Cu2+, PX or Cu2+ + PX, the number of M2 macrophages remained essentially constant. This might be due to these medications' insufficient capacity to produce ROS intracellularly, failing to depletion by M2 macrophages. As opposed to this, AS had an extremely powerful capacity to remove M2 macrophages whether it was provided solely in combination, or in a nanoscale form (Fig. 3E). Following the treatment of AS, Cu2+ + PX + AS or CuPA, the respective rates of M2 macrophages were 22.5%, 24.8% and 21.2%. Among which, the rate of M2 macrophages after treatment with CuPA had decreased to 0.7 times compared with that of without any processing. The pronounced reduction of M2 macrophages by AS effectively would alleviate the immunosuppressive characteristics of the tumor microenvironment. In conclusion, CuPA could both prevent the growth of tumor cells and eliminate M2 macrophages.
The antitumor effect of CuPA was further assessed through in vivo tests. All animal experiments were performed with the approval of Institutional Animal Care and Use Committee (IACUC) of the Animal Experimental Center of Guangzhou Medical University (approval No.: GY2023–106). To enhance in vivo antitumor studies, the metabolic properties of the drug were first investigated for biodistribution in mice. CuPA was labeled with cy5.5 for fluorescence imaging and an equal amount of cy5.5 was used as a control. As shown in Fig. 4A, CuPA-cy5.5 had a favorable cycling effect in the organism. The drug concentration in vivo was higher after 2 h and then underwent metabolism by the liver and kidney. The stronger tumor retention effect of CuPA-cy5.5 compared to free cy5.5 might be attributed to the enhanced permeability and retention (EPR) effect of the nanomedicine, which favored the effective delivery of CuPA. After 24 h of administration, the fluorescence of isolated tumor was significantly stronger in the CuPA-cy5.5 group than in the cy5.5 group, which strongly verified the preferable accumulation of nanomedicine at tumor site. The quantified fluorescence of the isolated tissues further showed that CuPA had a much better tumor retention ability than cy5.5 and it was mainly metabolized by liver and kidney (Fig. S3 in Supporting information).
Figure 4
Figure 4.
Tumor inhibitory effect of CuPA. (A) Real time fluorescence images of 4T1 tumor bearing mice after intravenous injection of CuPA-cy5.5 for 2, 4, 6, 8, 10, 12 and 24 h. 1–6 represented heart, liver, spleen, lung, kidney and tumor. (B) The body weight and (C) tumor volume changes of mice after treatment with PX, AS, Cu2+ + PX, Cu2+ + PX + AS or CuPA for 13 d. (D) The sacrificed tumor images at the end of treatment. (E) The tumor inhibitory rate and (F) the tumor weight of mice after treatment with different drugs. (G) H&E staining and immunofluorescence of (H) TUNEL, (I) CD3+CD8+ T cells of tumor tissues treated with PX, AS, Cu2+ + PX, Cu2+ + PX + AS or CuPA at the 13th day (n = 3). Scale bar: 100 µm. *P < 0.05 and **P < 0.01 were tested via a Student's t-test. Error bar was computed using mean ± SD.
Antitumor study was then carried out by tail vein administration. The mice were randomly divided into six groups and underwent four treatments. Mice weights and tumor volumes were recorded every two days until the mice were dissected on day 13th (Figs. 4B and C). Afterward, the tumor tissues were collected and imaged (Fig. 4D). PX, which regulated cellular oxidative stress, exerted a weak tumor suppressive effect. Despite of the good antiproliferative ability at the cellular level, AS did not exhibit an excellent therapeutic effect in vivo. Also, neither Cu2+ + PX attempted to enhance oxidative stress nor Cu2+ + PX + AS combination met the desired inhibitory expectations. Therapeutic outcomes had demonstrated that the limitation in delivering small molecule drugs hindered their therapeutic application. In contrast, the tumor inhibition rate of CuPA was calculated to be 56.4% (Fig. 4E). The utilization of nanosized CuPA enhanced the efficacy of the drug in vivo by improving the drug delivery and promoting drug aggregation at the tumor site, leading to a slowdown in tumor growth. Solid evidence was offered from the weight analysis of tumors (Fig. 4F). The CuPA treated tumors were the lightest and there was a 0.34-fold decline in tumor weight compared with blank control. Moreover, because of the excellent physicochemical properties, CuPA had significant superiority on tumor treatment over the group administered with a mixture of the three drugs. But it should be noted that CuPA could not completely remove the tumors. The main cause should be that CuPA suppressed tumor growth through the interruption of the intracellular redox balance and depletion of M2 macrophages to reverse the immunosuppressive microenvironment. However, tumor cells developed complex mechanisms to defend and repair ROS induced cellular damage, which also possessed a complex mechanism to suppress immune activation. Consequently, the tumor suppression ability of CuPA was moderate, but it could be improved by the combination use of drugs and multi-synergistic mechanisms.
To further analyze the cellular status in tumor tissues after drug treatment, the isolated tumors were assessed by hematoxylin and eosin (H&E) staining, terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL), and CD3CD8 staining. H&E staining revealed that the tumor cells exhibited cytoplasmic shrinkage and nuclear condensation following CuPA treatment (Fig. 4G). In addition, the degree of nuclear crumpling in the group of Cu2+ + PX + AS was inferior to that of CuPA group, whereas no significant changes were seen in the other treatment groups. Meanwhile, fluorescence of TUNEL showed that the CuPA group had a bright green fluorescence (Fig. 4H). The available evidence suggested that CuPA played a role in inducing significant apoptosis through disrupting redox equilibrium and causing toxicity associated with AS. Moreover, the immunofluorescence of CD3+CD8+ T cells proved activation of toxic T cells in the CuPA-treated group (Fig. 4I), which should be associated with the depletion of M2 macrophages mediated by AS. Taken together, the induction of oxidative stress and modulation of M2 macrophages were the key of CuPA with enhanced antitumor ability.
In the process of bringing drugs from the lab to the clinic, good biosafety was essential. Major organs were removed for H&E staining following the in vivo antitumor studies to evaluate the drug's biosafety (Fig. S4 in Supporting information). As results were analyzed, no abnormal cellular morphology was observed in these tissues. CuPA did not cause significant harm to other tissues or organs despite of its robust antitumor effect. Moreover, serum samples were collected for blood biochemical analysis at the conclusion of various drug treatments (Fig. S5 in Supporting information). The experimental findings of alanine transaminase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and uric acid (UA) demonstrated that following CuPA therapy, the biochemical indices of serum did not deviate from the reference range. These results indicated that CuPA had minimal negative impact on the liver and kidney. In addition, CuPA would not cause hemolysis over a wide dose range (Fig. S6 in Supporting information), indicating a good blood compatibility of CuPA.
In summary, we had designed a GSH-responsive metal-coordinated oxidative stress amplifier called CuPA to increase the intracellular ROS and eliminate M2 macrophages, amplifying the antitumor effect of chemotherapeutic agents. Based on the assembly of Cu2+, PX and AS in the presence of DSPE-PEG2000, the prepared CuPA was advantageous for its good stability and GSH responsiveness. By virtue of the improved delivery efficiency, CuPA was able to act as an oxidative stress amplifier to alter intracellular ROS and GSH levels, thereby increasing the oxidative damage for tumor suppression. Additionally, the remarkable in vivo antitumor activity of CuPA was deemed to be associated with the activation of cytotoxic T cells by scavenging M2 macrophages. Importantly, this exemplary metal-coordinated oxidative stress amplifier also exhibited a good biosafety profile. This study might advance the development of chemotherapeutic nanoplatforms for synergistic tumor suppression despite the complex resistant tumor microenvironment.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful for the financial support of National Key R&D Program of China (No. 2021YFD1800600), the National Natural Science Foundation of China (No. 32371394), the Guangdong Basic and Applied Basic Research Foundation (No. 2021B1515020043), and the Special Projects in Key Areas of Colleges and Universities in Guangdong Province (No. 2022ZDZX2046).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109696.
Scheme 1
Preparation of metal-coordinated oxidative stress amplifier (CuPA) for tumor therapy based on the assembly of Cu2+, PX and AS in the presence of DSPE-PEG2000. The GSH responsiveness of CuPA facilitated the release of Cu2+ and PX to elevate the oxidative stress causing cell death. Moreover, AS could deplete M2-type macrophages to improve immunosuppressive microenvironment, promoting the activation of cytotoxic T cells for synergistic tumor treatment.
Figure 1
Characterizations of CuPA. (A) The hydrodynamic size distribution and (B) TEM image of CuPA. Scale bar: 200 nm. (C) The particle size variations and (D) PDI changes of CuPA for 7 days (n = 3). (E) Drug release behaviors of CuPA in the absence or presence of GSH at pH 6.8 (n = 3). (F) UV–vis absorbance of TMB with or without H2O2 and CuPA. (G) CLSM images and (H) flow cytometry analysis of 4T1 cells after treatment with CuPA-cy5.5 at the concentration of 7.5, 15, 22.5 and 30 µg/mL (n = 3). (I) CLSM images and (J) flow cytometry analysis of 4T1 cells after treatment with CuPA-cy5.5 for 0, 3, 6 and 9 h (n = 3). Scale bar: 20 µm. ***P < 0.001 was tested via a Student's t-test.
Figure 2
Detection of ROS and GSH levels in 4T1 cells. (A) CLSM images and (B) MFI of 4T1 cells after treatment with Cu2+, PX, AS, Cu2+ + PX, Cu2+ + PX + AS or CuPA to evaluate ROS generation by DCFH-DA. (C) CLSM images and (D) MFI of 4T1 cells incubated with those drugs to test ROS generation by CellROS. (E) CLSM images and (F) MFI of 4T1 cells after those treatments to detect mitochondrial ROS using MitoROS 580. (G) CLSM images and (H) MFI of 4T1 cells after those treatments to detect GSH levels using CMFDA (n = 3). Scale bar: 20 µm. *P < 0.05, **P < 0.01 and ***P < 0.001 were tested via a Student's t-test. Error bar was computed using mean ± standard deviation (SD).
Figure 3
Antiproliferation and macrophage regulation effect of CuPA. (A) Flow cytometry apoptotic analyses of 4T1 cells after treatment with Cu2+, PX, AS, Cu2+ + PX, Cu2+ + PX + AS or CuPA. CCK8 analysis of 4T1 cell viabilities after treatment with gradient concentrations of (B) Cu2+, PX, AS, and (C) Cu2+ + PX, Cu2+ + PX + AS or CuPA. (D) Flow cytometry and (E) quantitative analysis of M2 polarization of RAW 264.7 cells after treatment with different drugs (n = 3). M2 macrophages were induced by IL-4 in advance. **P < 0.01 was tested via a Student's t-test. Error bar was computed using mean ± SD.
Figure 4
Tumor inhibitory effect of CuPA. (A) Real time fluorescence images of 4T1 tumor bearing mice after intravenous injection of CuPA-cy5.5 for 2, 4, 6, 8, 10, 12 and 24 h. 1–6 represented heart, liver, spleen, lung, kidney and tumor. (B) The body weight and (C) tumor volume changes of mice after treatment with PX, AS, Cu2+ + PX, Cu2+ + PX + AS or CuPA for 13 d. (D) The sacrificed tumor images at the end of treatment. (E) The tumor inhibitory rate and (F) the tumor weight of mice after treatment with different drugs. (G) H&E staining and immunofluorescence of (H) TUNEL, (I) CD3+CD8+ T cells of tumor tissues treated with PX, AS, Cu2+ + PX, Cu2+ + PX + AS or CuPA at the 13th day (n = 3). Scale bar: 100 µm. *P < 0.05 and **P < 0.01 were tested via a Student's t-test. Error bar was computed using mean ± SD.

 DownLoad:
DownLoad:



 下载:
下载:






 下载:
下载:
