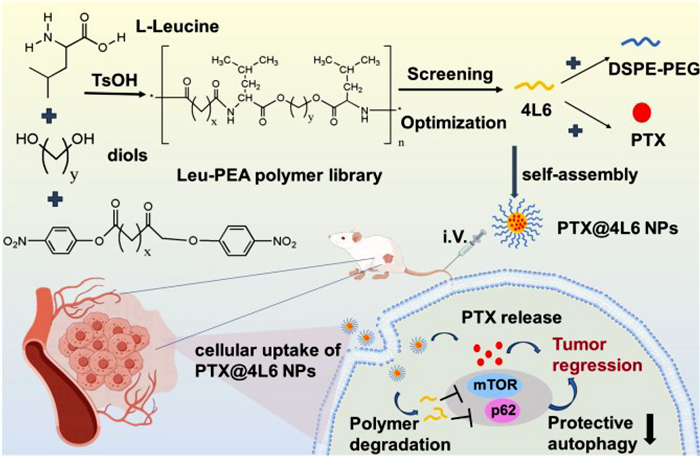
Scheme 1.
Schematic illustration of a PTX loaded nanoplatform based on Leu-PEA polymers with self-anticancer effect for CRC treatment.
Establishment of a leucine-based poly(ester amide)s library with self-anticancer effect as nano-drug carrier for colorectal cancer treatment
Tong Tong , Lezong Chen , Siying Wu , Zhong Cao , Yuanbin Song , Jun Wu

Nowadays, colorectal cancer (CRC) has become one of the most prevalent cancers worldwide and seriously threatens public health. Recent statistics indicate that CRC comprises approximately 7.84% of all new cancer cases in 2022 [1]. As the main traditional treatment method of CRC, chemotherapeutic drugs are limited by poor aqueous solubility and reduced tumor targeting, which lead to low drug loading efficiency and high toxicity to normal organs such as harm to digestive system, bone marrow, liver and kidney [2]. In recent years, in order to reduce these side effects, a variety of nanocarriers including liposomes, nano-capsules and microspheres have been developed [3–5]. However, current nanodrug systems still face challenges such as low drug efficiency, high drug toxicity and short circulation time, which greatly restricted the further application of nanocarriers [6,7].
Natural amino acids-based polymers have been widely used in drug delivery systems [8–12]. Some positive results have been achieved for example phenylalanine-based nanodrug delivery systems for cancer treatment [13–15] and other diseases like myeloid leukemia [16] and kidney injury [17], etc. Here, we imported natural leucine into poly(ester amide)s and established a polymer library. Leucine-based poly(ester amide)s (Leu-PEAs) are biodegradable and biocompatible owing to the unique "amide" and "ester" unit in their structures [18,19]. Moreover, Leu-PEAs are also structure-adjustable. Through changing the carbon chain length of diacids and diols, Leu-PEAs library containing 9 kinds of different polymers was established. Previous research has shown that carbon chain length can influence the physicochemical properties of the polymer, e.g., hydrophobic and hydrophilic performance thereby influence drug loading and surface properties of nanocarriers [9]. Leucine, a branched-chain amino acid, plays an important role in a variety of body metabolisms such as synthesis of muscle protein and insulin secretion [20–22]. Recent research suggests that leucine can activate the mammalian target of rapamycin (mTOR) and influence phosphorylation of its downstream proteins [23,24]. It is worth noting that traditional chemotherapeutic drugs usually bring many side effects, for example, paclitaxel (PTX), as a frequently-used and eutherapeutic anti-tumor drug in clinical, has been found to potentially induce protective autophagy in tumor cells, reducing drug sensitivity and thereby diminishing the apoptotic effects on tumor cells [25,26]. It was might owing to the induce of PTX to the dephosphorylation of mTOR and its downstream factors. Therefore, the addition of leucine might inhibit the protective autophagy and improve therapeutic effects through activating mTOR.
In this study, a library of leucine-based polymer was synthesized. The Leu-PEAs could self-assemble into nanoparticles and load PTX with the existence of stabilizer through nanoprecipitation method. Then through the characterization of Leu-PEAs and PTX-loaded nanoparticles, the relationship of polymer structure and drug loading could be investigated. Also, the optimal polymer and nanoparticle formula were picked out for the subsequent experiments. In vitro and in vivo experiments were conducted to evaluate the drug release profile, cellular uptake, drug distribution, anticancer efficacy, toxicity, and the potential antitumor mechanism of PTX-loaded nanoparticles (Scheme 1).
Leu-PEAs were synthesized through a well-established condensation polymerization method. The synthetic route was presented in Fig. S1 (Supporting information). Leu-PEA was obtained by the polymerization reaction between natural l-leucine, diacids and diols. In the structure of Leu-PEAs, x represents the carbon length of diacid while y represents the carbon length of diols. Through changing x and y, a series of Leu-PEAs could be obtained. In this study, the chain length of diacid (x) was set as 2, 4, or 8 carbon atoms, and for diols (y), it was set as 2, 4, or 6 carbon atoms. Therefore, the Leu-PEA library contained 9 kinds of Leu-PEA polymers. To characterize the Leu-PEA library, several material characterization tests were performed. The 1H NMR spectrum of a typical Leu-PEA polymer (4L6) was presented in Fig. S2 (Supporting information). The appearance of protons belonging to the CONH group at 8.16 ppm indicated the successful formation of amide group. Additionally, the FT-IR spectrum shown in Fig. 1A also confirmed the synthesis of the polymer. The stretching vibration peak of the N—H bond (3300 cm−1) implied the formation of -NH2 and -COOH through the amide group. In addition, no matter y was set 2, 4 or 6, when y increased, the peak intensity of CH2 (2930, 2856 cm−1) enhanced obviously, indicating that the longer carbon chain appeared accompanying with the higher x value. As for comparing the thermostability of Leu-PEAs, thermogravimetric analysis (TG) spectrum was shown in Fig. 1B. When y = 2 or 4, the thermostability increased with x growing. However, when y = 6, the polymer with a mid-range x value (4L6) exhibited the highest thermal stability. It started to lose weight at about 350 ℃, which is higher than 8L2 and 8L4 (300 ℃). That is to say, the polymer 4L6 possessed the most excellent stability against heating. In addition, the gel permeation chromatography (GPC) test results of Leu-PEAs were presented in Fig. 1C and Fig. S3 (Supporting information). Generally, the weight average molecular weight (Mw) ranged from 1674 to 24,913 with a polydispersity index (PDI) of 1.1 to 1.4, indicating relative narrow molecular weight distribution. When y = 6, the polymer (4L6, 8L6) presented intermediate Mw, which is suitable for drug loading and they could be more easily degraded in comparison to other polymers with different molecular weights.

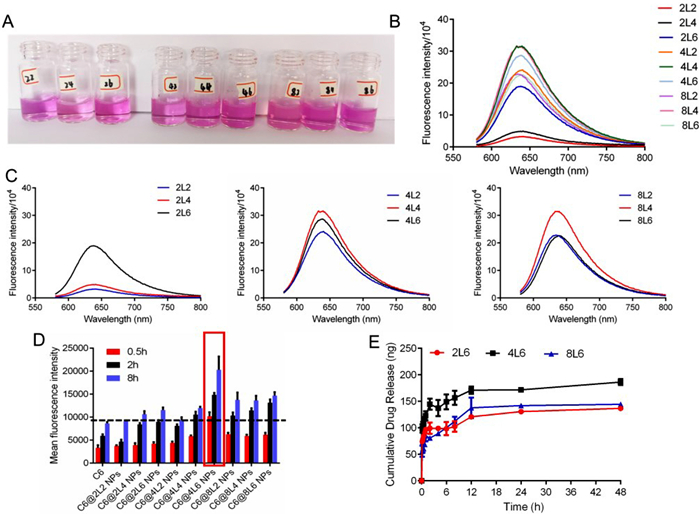
Following a series of characterization tests, including thermal and molecular weight analysis, screening of these Leu-PEAs is necessary in order to identify the optimal polymer for the subsequent drug loading. First and foremost, change of carbon chain length of polymers was probable to influence their physicochemical properties such as solubility, hydrophobicity, degradability and so on. Furthermore, when drugs were loaded with the polymers, the drug loading efficiency, surface properties of nano carriers and even the therapeutic effects both in vitro and in vivo could be affected. A lipophilic fluorescent dye, Nile red, was loaded into Leu-PEAs assembled nanocarriers and nine different types of nanoparticles were prepared corresponding to the various combinations of x and y values. Owing to the lipophilic characterization of Nile red, the loading capacity is positively related to the hydrophobicity of polymers. Fig. 2A presented the images of Nile red loaded Leu-PEAs nanoparticles. The shade of color indicated the dosage of loaded dye. To analyze the results more directly, the fluorescence spectra from the comparison of hydrophobic and hydrophilic performance tests for Leu-PEA polymers loaded with Nile red were showed in Figs. 2B and C. The results suggested that an increase in carbon chain length correlated with an increase in the hydrophobicity of the Leu-PEAs. Interestingly, for x values of 4 and 6, the polymers with the highest y value (4L6 and 8L6) exhibited intermediate hydrophobicity, which aligned with the GPC test results. Moreover, coumarin 6 (C6) loaded with Leu-PEAs was utilized for cellular uptake exploration. The results in Fig. 2D implied that the cellular uptake was positively related with the carbon chain length to a great degree. However, C6@4L6 NPs presented the highest cellular uptake whether at 0.5, 2 or 8 h. From the comparisons above, it is inferred that the polymer with mid-range x and y values (4L6) could potentially be the most effective nanocarrier for drug loading and therapeutic efficacy. When y was set as 6, Dil was loaded into 2L6, 4L6, and 8L6 to further investigate their drug release patterns. Then the cumulative Dil release in phosphate buffer saline (PBS) solution (pH 5.0) during 48 h was detected and compared. As anticipated in Fig. 2E, based on its optimal hydrophobicity and molecular weight characteristics, Dil@4L6 NPs exhibited the highest drug release. In conclusion, through the screening of Leu-PEAs, the polymer 4L6 presented the most promising drug loading potentials for the subsequent disease treatment.
Following the exploration results above, 4L6 was picked out for drug loading. Here, a typical chemotherapeutic drug, PTX, was selected to be loaded. Both blank 4L6 NPs and PTX@4L6 NPs were prepared through the nanoprecipitation method.
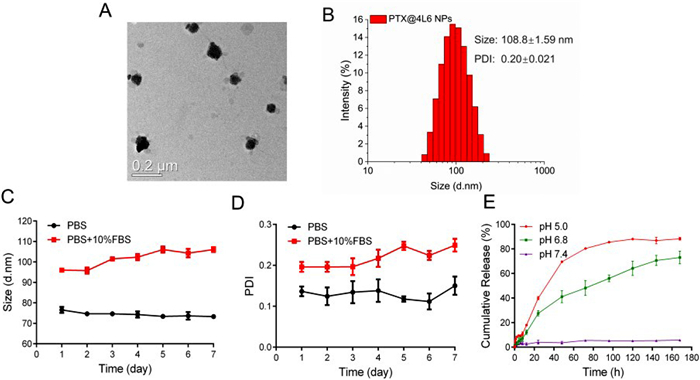
To verify the drug loading capacity, size distribution, PTX loading capacity (LC), and encapsulation efficiency (EE) tests were performed on the 9 kinds of Leu-PEAs, and the results are presented in Fig. S4 (Supporting information). From the results, PTX@4L6 NPs presented appropriate size and PDI. Moreover, the LC of PTX was 8.5% ± 0.01% and the EE was 85.5% ± 0.11%, both of which were significantly higher than the other synthesized polymers. The TEM images and size distribution of PTX@4L6 NPs in Figs. 3A and B confirmed its spherical structure and uniform size distribution at about 108 ± 1.59 nm. The TEM image and size distribution of 4L6 NPs in Fig. S5 (Supporting information) confirmed a smaller size of approximately 91.8 ± 0.73 nm. In addition, the zeta potential of PTX@4L6 NPs changed to −26.8 ± 1.65 mV while the zeta potential of blank 4L6 NPs was −36.2 ± 1.65 mV. The change of surface charge also confirmed the drug loading. The size and PDI variation of PTX@4L6 NPs in PBS and PBS supplemented with 10% fetal bovine serum (FBS, to simulate physiological conditions) over 7 days were shown in Figs. 3C and D, indicating good stability in a physiological-like environment. As for the condition of PTX release, three kinds of environment (pH 5.0, 6.8 and 7.4, PBS solution) were set and the cumulative release of PTX during 7 days was calculated. The results were shown in Fig. 3E. The results indicated that the release of PTX was less than 20% at pH 7.4 within 7 days, while it increased to more than 60% at pH 6.8 and over 80% at pH 5.0 during the same period. The reason was probably related to the structure of nanoparticle. In acidic environment, the acid amide bond of 4L6 was broken, resulting in the destruction of the nanoparticle structure and the release of the loaded drug. Therefore, compared with the neutral environment at pH 7.4, acid environment at pH 6.8 and pH 5.0 presented more destruction of nanoparticle structure and induced the constant release of PTX.
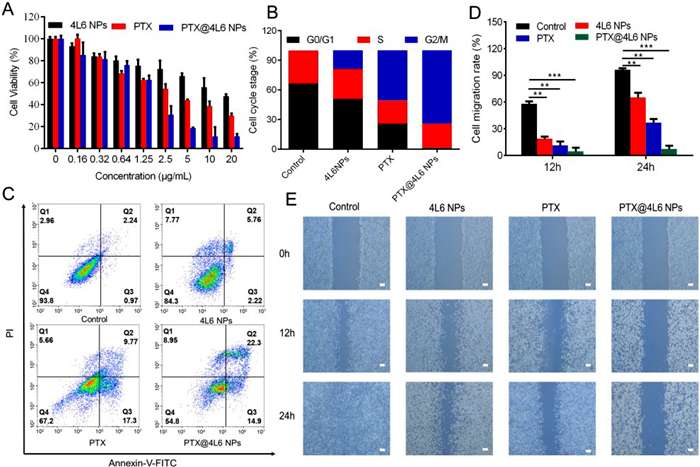
To investigate the antitumor activity of PTX, 4L6 NPs, and PTX@4L6 NPs on the viability of CT26 cells, an MTT assay was performed. Blank DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin was served as the control group. As presented in Fig. 4A, after incubated with PTX, 4L6 NPs or PTX@4L6 NPs at the equivalent PTX concentration gradient for 24 h, the cell viability decreased gradually with the increasing PTX concentration. From the calculation results, the half maximal inhibitory concentration (IC50) of free PTX was approximately 3.804 µg/mL while IC50 of PTX@4L6 NPs was roughly 1.533 µg/mL. It meant that PTX@4L6 NPs presented stronger tumor cell killing effects compared with PTX along. This enhanced efficacy might be attributed to the higher cellular uptake and targeted distribution of the drug within the tumor tissue by the nanoparticles. Interestingly, 4L6 NPs alone also demonstrated antitumor activity in vitro, with cell viability dropping to below 60% at a concentration equivalent to 20 µg/mL of PTX. Furthermore, as shown in Figs. S6 and S7 (Supporting information), 4L6 NPs did not exhibit significant cytotoxicity toward normal NIH 3T3 cells and showed minimal hemolysis, indicating the biocompatibility of the polymer. In addition, further exploration of cell cycle and cell apoptosis test results were presented in Figs. 4B and C. The proportion of apoptotic and necrotic cells in the PTX group increased from 6.2% to 32.8%, compared to the control, indicating the tumor cell killing effects of PTX. However, in the PTX@4L6 NPs group, this number increased to 45.5%, confirming the enhanced antitumor effects. Consistent with MTT assay results, 4L6 NPs groups also presented a higher proportion apoptotic and necrotic cells at about 15.7%. As for cell cycle investigation, about 50.69% CT26 cells with PTX treatment group stayed at the G2/M stage and the number grew to 74.08% in PTX@4L6 NPs group, which was significantly higher than that in control group. The results implied that PTX could inhibit the tumor cell proliferation through mitotic suppression. Moreover, the cell migration assay results shown in Figs. 4D and E demonstrated that PTX@4L6 NPs had superior cell migration inhibition compared to the PTX group, and 4L6 NPs also exerted an effect in this regard.
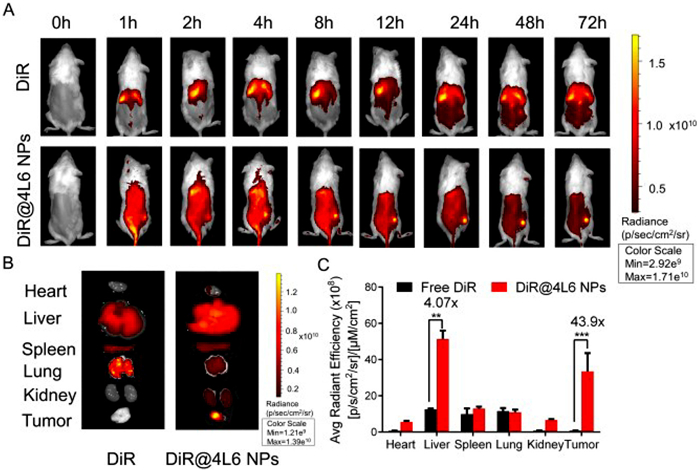
As demonstrated in previous sections, PTX@4L6 NPs appeared to show better antitumor effects than free PTX. This improved efficacy is likely due to the enhanced accumulation and targeting of PTX@4L6 NPs at tumor sites. To verify this hypothesis, an in vivo biodistribution study was conducted. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Sun Yat-sen University and approved by the Animal Ethics Committee of Sun Yat-sen University (approval No. SYSU-IACUC-2022-001158). The fluorescent dye, DiR, was used as a probe to trace the biodistribution of PTX. Male CT26 tumor-bearing BALB/c mice were injected intravenously with free DiR or DiR@4L6 NPs (at 1 equiv. DiR dosage of 0.5 mg/kg). The fluorescent distribution images of the mice at various time points are shown in Fig. 5A. For the DiR@4L6 NPs group, a gradual increase in DiR fluorescence accumulation at the tumor site could be observed while there was no obvious fluorescence gathering at tumor in free DiR group. To quantitatively evaluate the results, organs (heart, liver, spleen, lung and kidney) and tumors were harvested from mice in both the DiR and DiR@4L6 NPs groups and imaged for fluorescence at 72 h. Images and quantitative analysis were shown in Figs. 5B and C. The results showed that the fluorescence intensity of the DiR@4L6 NPs group was markedly higher at the tumor site, which was about 43.9 folds greater than that of the free DiR group, indicating the longer circulation time and tumor-targeting effects of DiR@4L6 NPs. In addition, DiR@4L6 NPs group also possessed much higher fluorescence signals in liver and kidney than free DiR group. This is likely because the liver and kidneys are the primary organs responsible for metabolism and excretion of nanoparticles in the body.
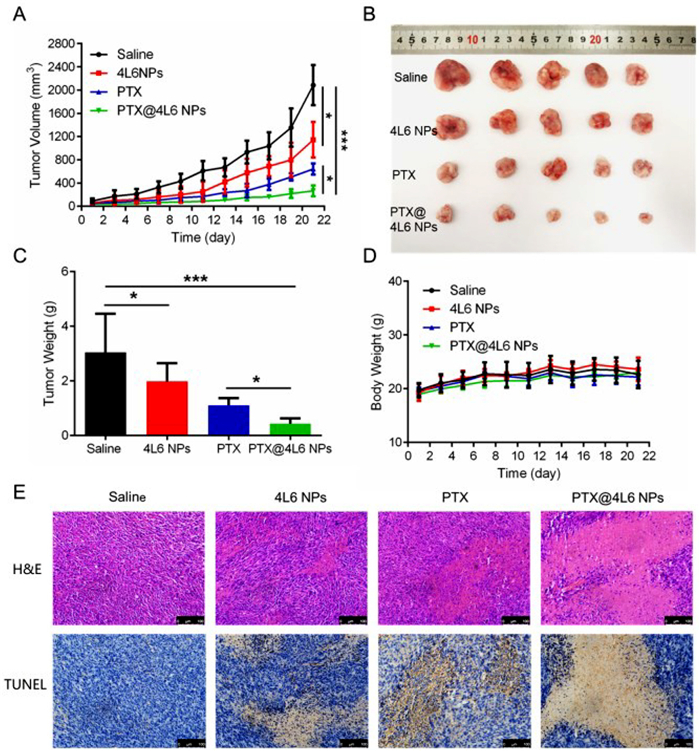
As presented above, PTX@4L6 NPs was shown to accumulate at the tumor site and induce tumor cells apoptosis. Furthermore, the in vivo antitumor effects of PTX@4L6 NPs were investigated. For this study, 20 male CT26 tumor-bearing BALB/c mice with a tumor volume of approximately 80–100 mm3 were randomly divided into four groups (n = 5). Mice were intravenously injected with saline, 4L6 NPs, PTX, and PTX@4L6 NPs, respectively, every other day at a PTX-equivalent dose of 5 mg/kg. From the results in Fig. 6A, tumors of saline group grew extremely rapidly within 21 days and tumor volume reached nearly 2000 mm3. The free PTX group showed moderate antitumor efficacy (~600 mm3) while PTX@4L6 NPs group presented superior tumor inhibition effects (~300 mm3) in comparison. Interestingly, 4L6 NPs also showed tumor inhibition effects (~1000 mm3), a finding that aligns with previous in vitro results. At the end of experiment, mice were sacrificed and the tumors were collected for imaging and weight measurement. The tumor weight and imaging results, shown in Figs. 6B and C, were consistent with the tumor volume measurements. The tumor weight of PTX@4L6 NPs was significantly lower than free PTX group (P < 0.05), indicating the superior antitumor efficacy of PTX@4L6 NPs. Body weight monitoring, as depicted in Fig. 6D, revealed no significant changes within 21 days, suggesting that the nanocarriers did not induce systemic toxicity. In addition, hematoxylin-eosin (H&E) and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay (TUNEL) staining of tumors were also investigated for antitumor effects assessment. As presented in Fig. 6E, compared with the neat cell arrangement and clear chromatin of saline group, tumor tissues of PTX and PTX@4L6 NPs groups exhibited cell necrosis. However, the PTX@4L6 NPs group exhibited more pronounced nuclear rupture and cytoplasmic damage. An increased positive brown area in TUNEL staining results for the PTX@4L6 NPs group, which indicated enhanced cell apoptosis, further confirmed the superior antitumor effects of PTX@4L6 NPs. In the further biosafety tests shown in Fig. S8A (Supporting information), no obvious tissue toxicity was observed in the H&E staining results of main organs (heart, liver, spleen, lung and kidney). Biochemical index test results, presented in Figs. S8B–F (Supporting information), showed no significant changes in the primary hepatic indicators, aspartate aminotransferase (ALT) and alkaline phosphatase (ALP), while alanine transaminase (AST) levels in the PTX group showed a slight increase, suggesting minimal liver inflammation. But AST of the PTX@4L6 NPs group maintained normal level as comparison, confirming the safety. The main renal function index, blood urea nitrogen (BUN) and creatinine (CREA) showed that PTX@4L6 NPs possessed no obvious toxicity to kidney. In conclusion, based on the evaluation of mouse body weight, H&E staining of major organs, and biochemical index tests of hepatic and renal indicators, the safety and biocompatibility of the established nano system (PTX@4L6 NPs) in vivo can be confirmed.
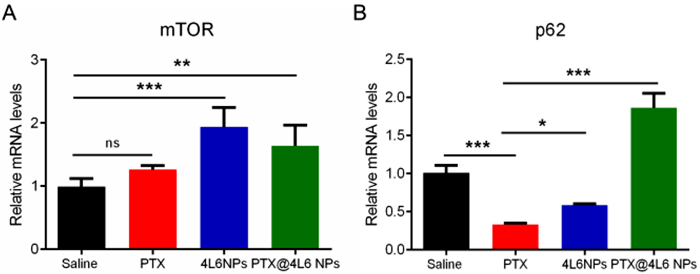
As previously discussed, PTX@4L6 NPs demonstrated significantly enhanced efficiency in inhibiting tumor growth compared to free PTX, both in vitro and in vivo. In addition to the prolonged accumulation and passive targeting of the nanocarrier to the tumor site, a possible reason might be related to the autophagy inhibition in CT26 cells. PTX, a traditional chemotherapeutic drug, has been found to induce protective autophagy through down-regulating the phosphorylation of mTOR in tumor cells, resulting in decreased apoptosis and reduced therapeutic efficacy against cancer. Interestingly, leucine has been shown to activate the mTORC1 complex, which inhibits autophagy. To verify this hypothesis, tumors from mice treated with saline, PTX, 4L6 NPs, and PTX@4L6 NPs were harvested, and RNA was extracted from these tissues for q-PCR analysis. The results, as presented in Fig. 7A, indicate that the leucine- containing groups (4L6 NPs and PTX@4L6 NPs) exhibited notably higher mTOR expression compared to the free PTX group. Regarding p62, a representative autophagy-related protein, its expression tends to increase when autophagy is inhibited. In Fig. 7B, the relative mRNA level of p62 was significantly reduced in the PTX group compared to the saline group, while it was greatly increased in the leucine-containing groups, suggesting that leucine-containing nanocarriers inhibit tumor cell autophagy.
In general, amino acid metabolism is now related with the treatment of various diseases. Recent studies have reported that the introduction of amino acids into nanodrug delivery system might enhance the therapeutic effect through regulating signal pathways relevant to cell metabolism [27]. In our study, noting the relationship between leucine and cell autophagy metabolism, we introduced natural leucine into biodegradable polymers and synthesized nanocarriers for drug delivery. The self-anticancer effects have been proved and the influence could be explored continuously to further validate its therapeutic potential.
In conclusion, a leucine-based poly(ester amide)s biomaterial library was successfully synthesized through the polycondensation of natural l-leucine, diacids and diols. The obtained Leu-PEAs were then characterized and compared. An optimal polymer 4L6 was picked out for the establishment of PTX loaded nano system. Characterization of PTX@4L6 NPs showed its uniform nanosize (~109 nm), spherical structure, good stability, continuous drug release in tumor microenvironment and no obvious cytotoxicity to normal cells. In vitro and in vivo antitumor tests demonstrated that PTX@4L6 NPs could accumulate at tumor site and presented the superior antitumor efficacy than any other groups while blank 4L6 NPs was also helpful for tumor therapy probably owing to its inhibition of tumor cell autophagy. Therefore, structure-adjustable Leu-PEAs were promising carriers for drug loading and could be applied in the treatment of various cancers.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This research was accomplished with the support of the National Natural Science Foundation of China (Nos. 52173150, 52073313), the Guangzhou Science and Technology Program City-University Joint Funding Project (No. 2023A03J0001), and the Shenzhen Science and Technology Program (No. KCXFZ 202002011010232).
Supplementary material associated with this article can be found, in the online version, at doi:
R.L. Siegel, K.D. Miller, H.E. Fuchs, A. Jemal, CA Cancer J. Clin. 71 (2021) 7–33. doi: 10.3322/caac.21654
D.S. Shewach, R.D. Kuchta, Chem. Rev. 109 (2009) 2859–2861. doi: 10.1021/cr900208x
H. Mao, Y. Xie, H. Ju, et al., ACS Appl. Mater. Interfaces 10 (2018) 33923–33935. doi: 10.1021/acsami.8b11159
R. Mahmoudi, S.A. Mirahmadi-Babaheidri, H. Delaviz, et al., J. Biomater. Appl. 35 (2020) 743–753.
X.R. You, Y. Kang, G. Hollett, et al., J. Mater. Chem. B 4 (2016) 7779–7792. doi: 10.1039/C6TB01925K
N. Bertrand, J. Wu, X.Y. Xu, N. Kamaly, O.C. Farokhzad, Adv. Drug Deliv. Rev. 66 (2014) 2–25. doi: 10.1016/j.addr.2013.11.009
F.M. Kievit, M.Q. Zhang, Adv. Mater. 23 (2011) 217–247.
J. Wu, L.L. Zhao, X.D. Xu, et al., Angew. Chem. Int. Ed. 54 (2015) 9218–9223. doi: 10.1002/anie.201503863
X.R. You, Z.P. Gu, J. Huang, et al., Acta Biomater. 74 (2018) 180–191. doi: 10.1016/j.actbio.2018.05.040
Q.J. Yuan, J. Huang, C.H. Xian, ACS Appl. Mater. Interfaces 13 (2021) 2165–2178. doi: 10.1021/acsami.0c15133
S.Y. Han, J. Wu, ACS Appl. Mater. Interfaces 14 (2022) 55944–55956. doi: 10.1021/acsami.2c16492
H.H. Hu, Z. Zhang, Y.F. Fang, L. Chen, J. Wu, Chin. Chem. Lett. 34 (2023) 107953. doi: 10.1016/j.cclet.2022.107953
X.R. You, L.Y. Wang, J.F. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107720. doi: 10.1016/j.cclet.2022.07.063
Z. Gao, J. Huang, Z.X. Xie et al., Biomater. Sci. 10 (2022) 5187–5196. doi: 10.1039/d2bm00697a
Y.B. Meng, J. Wu, ACS Appl. Mater. Interfaces 13 (2021) 49658–49670. doi: 10.1021/acsami.1c13013
Y.X. Zeng, X.Y. Zhang, D.J. Lin et al., J. Hematol. Oncol. 14 (2021) 189. doi: 10.1109/cloud53861.2021.00032
J.W. Zhang, J.H. Yan, Y.N. Wang et al., Chin. Chem. Lett. 35(2024) 108434. doi: 10.1016/j.cclet.2023.108434
M. Winnacker, B. Rieger, Polym. Chem. 7 (2016) 7039–7046. doi: 10.1039/C6PY01783E
H.L. Sun, R. Cheng, C. Deng, et al., Biomacromolecules 16 (2015) 597–605. doi: 10.1021/bm501652d
Q. Yin, J.M. Brameld, T. Parr, A.J. Murton, Amino Acids 52 (2020) 477–486. doi: 10.1007/s00726-020-02829-0
B.H. Boehmer, P.R. Baker, L.D. Brown, S.R. Wesolowski, P.J. Rozance, J. Endocrinol. 247 (2020) 115–126. doi: 10.1530/joe-20-0243
L.Y. Zhang, F.N. Li, Q.P. Guo, et al., Nutrients 12 (2020) 1299. doi: 10.3390/nu12051299
J. Chen, Y.H. Ou, R. Luo, et al., Nature 596 (2021) 281–284. doi: 10.1038/s41586-021-03768-w
X. Xu, J. Wang, T. Tong et al., Haematologica 107 (2022) 2344–2355. doi: 10.3324/haematol.2021.280290
G.M. Xi, X.Y. Hu, B.L. Wu, et al., Cancer Lett. 307 (2011) 141–148. doi: 10.1016/j.canlet.2011.03.026
Q. Zhang, S.H. Si, S. Schoen, et al., J. Exp. Clin. Cancer Res. 32 (2013) 99. doi: 10.1186/1756-9966-32-99
C.Z. Li, X.R. You, X. Xu et al., Adv. Sci. 9 (2022) 2104134. doi: 10.1002/advs.202104134

Scheme 1 Schematic illustration of a PTX loaded nanoplatform based on Leu-PEA polymers with self-anticancer effect for CRC treatment.

Figure 1 Characterization of Leu-PEA polymers. (A) FT-IR spectrum of Leu-PEAs. (B) Thermogravimetric analysis (TG) spectrum of Leu-PEAs. (C) Gel permeation chromatography (GPC) test results of Leu-PEAs.

Figure 2 Screening of Leu-PEAs. (A) Picture and (B) fluorescence spectra comparison results of hydrophobic and hydrophilic performance test of Leu-PEA polymers Nile red loading. (C) Fluorescence spectra of N2, N4 and N8 series. (D) Cellular uptake of C6 loaded Leu-PEA NPs for 0.5, 2 and 8 h. (E) Cumulative release of Dil from 2L6, 4L6 and 8L6 NPs in PBS solution (pH 5.0). The data are presented as means ± standard deviation (SD) (n = 3).

Figure 3 Characterization of PTX@4L6 NPs. (A) TEM picture and (B) size distribution of PTX@4L6 NPs. (C) Size and (D) PDI stability of PTX@4L6 NPs under PBS or PBS + FBS environment in 7 days. (E) The cumulative release of PTX from PTX@4L6 NPs. The data are presented as means ± SD (n = 3).

Figure 4 In vitro antitumor activity. (A) Cell viability test, (B) cell cycle test, (C) cell apoptosis test and (E) cell migration test of CT26 cells treated with control group, 4L6 NPs, PTX and PTX@4L6 NPs at different concentrations for 24 h. (D) Cell migration rate statistics. Scale bar: 100 µm. The data are presented as means ± SD (n = 3). **P < 0.01, ***P < 0.001. Data were all analyzed by ANOVA.

Figure 5 In vivo biodistribution. (A) Representative picture of the distribution of free DiR and DiR@4L6 NPs in mice during 72 h. (B) Representative picture and (C) quantitative analysis of DiR and DiR@4L6 NPs in heart, liver, spleen, lung, kidney and tumor at 72 h. The data are presented as means ± SD (n = 3). **P < 0.01, ***P < 0.001. Data were all analyzed by ANOVA.

Figure 6 In vivo antitumor effects. (A) Tumor volume, (B) tumor images, (C) tumor weight and (D) body weight of saline, 4L6 NPs, PTX and PTX@4L6 NPs treated BALB/c mice during 21 days. (E) H&E and TUNEL staining results of tumors from saline, 4L6 NPs, PTX and PTX@4L6 NPs treated BALB/c mice on the 21st day. The data are presented as means ± SD (n = 5). *P < 0.05, ***P < 0.001. Data were all analyzed by ANOVA. Scale bar: 100 µm.

Figure 7 The mechanism of the antitumor effect of leucine-based polymer. mRNA expression levels of (A) mTOR and (B) p62 (autophagy related protein). The data are presented as means ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001, ns represents no significant differences. Data were all analyzed by ANOVA.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: