
Scheme 1.
Palladium-catalyzed decarboxylation of vinyl cyclic carbamates for generating aza-π-allylpalladium 1,n-dipoles.
Palladium-catalyzed enantioselective decarboxylation of vinyl cyclic carbamates: Generation of amide-based aza-1,3-dipoles and application to asymmetric 1,3-dipolar cycloaddition
Xiaohui Fu , Yanping Zhang , Juan Liao , Zhen-Hua Wang , Yong You , Jian-Qiang Zhao , Mingqiang Zhou , Wei-Cheng Yuan

Catalytic asymmetric dipolar cycloaddition reaction is considered to be one of the most powerful and widely used strategies for the construction of chiral carbo- and heterocyclic compounds with elaborate ring frameworks [1–3]. In this research area, an important aspect is the design and exploration of effective dipole intermediates, which should have high reactivity and facilitate the control of reaction stereoselectivity in the asymmetric transformations. Actually, chiral transition-metal catalysts acting on reaction substrates to generate appropriate metal-containing zwitterionic dipoles for sequential asymmetric dipolar cycloaddition represents a remarkable advance in modern organic synthesis, and a number of elegant synthetic methods have been developed [4–8]. Despite this fact, the growing demand for structurally diverse three-dimensional cyclic scaffolds in the field of medicinal chemistry still inspires synthetic chemists to develop new and practical methods for complementing the existing approaches [9]. Therefore, the design and development of novel precursors to produce metal-mediated highly active dipole intermediates for new reaction discovery is still highly desirable.
Transition metal-catalyzed asymmetric decarboxylation of cyclic carbonates/carbamates, leading to the in situ formation of active metal-containing zwitterionic intermediates for the construction of chiral cyclic structures, has become a research hotspot in organic synthetic chemistry [10–18]. In particular, the palladium-mediated decarboxylation of vinyl-substituted cyclic carbamates, which gives rise to aza-π-allylpalladium 1,n-zwitterionic dipoles containing a nucleophilic nitrogen anion and a ligand ligated π-allylpalladium cation counterpart, has been widely applied in asymmetric dipolar cycloaddition to access nitrogen-containing heterocycles [19–23]. In fact, to date, only very few vinyl cyclic carbamates have been used as aza-π-allylpalladium 1,4- or 1,3-dipole precursors for asymmetric reactions via a decarboxylation process. For example, the vinyl benzoxazinanones V1 were first reported as aza-π-allylpalladium 1,4-dipole precursors for enantioselective decarboxylative [4 + 2] cycloaddition by Tunge (Scheme 1a) [24–30]. Another type of aza-π-allylpalladium 1,4-diploes generated from cyclic vinyl carbamates V2 for asymmetric transformations were reported by Harrity's and our group, respectively (Scheme 1b) [31–33]. In addition, Ooi and co-workers realized the first Pd-catalyzed asymmetric decarboxylative cycloaddition of vinyl oxazolidinones V3, which acted as highly reactive aza-π-allylpalladium 1,3-dipoles precursors (Scheme 1c) [34,35]. Moreover, Shi's and Deng's group independently discovered that the vinyl indoloxazolidones V4 could be used as indolyl-based Pd-allyl zwitterionic species precursors for asymmetric cycloaddition (Scheme 1d) [36,37].
Inspired by the precedents mentioned above, and as a continuation of our interest in catalytic asymmetric decarboxylative cycloaddition of cyclic carbonates/carbamates [33,38–44], we envisioned that introducing a vinyl motif at the α-position of amide compounds and then converting them into vinyl-substituted cyclic carbamates V5, which should undergo a decarboxylation process in the presence of a palladium catalyst thus leading to the formation of active amide-based aza-π-allylpalladium 1,3-dipoles V5-Int (Scheme 2). If successful, we speculated that the in situ generated amide-based aza-π-allylpalladium 1,3-dipoles would be applied to asymmetric 1,3-dipolar cycloadditions with various dipolarophiles including C=C, C=N, and C=O double bonds. Indeed, we have successfully synthesized a series of 5-vinyloxazolidine-2,4-diones V5 acting as reactive precursors of aza-π-allylpalladium 1,3-dipoles and further realized enantioselective cycloaddition reactions by a catalytic system consisting of a readily available phosphoramidite ligand and Pd(0) catalyst [45,46], affording highly functionalized optically active pyrrolidin-2-ones, imidazolidin-4-ones, and oxazolidin-4-ones (Scheme 2). Notably, due to the existence of the nucleophilic amide anion site in the in situ generated 1,3-dipole intermediates V5-Int, this would provide an efficient access to previously unknown structural opportunities, such as highly functionalized lactam-type heterocyclic skeletons, which are ubiquitous ring systems in many bioactive compounds and are a fundamental unit of important candidates in pharmaceutical research (Scheme 2, bottom) [47–50]. More importantly, this unique strategy not only is complementary to the well-established palladium-mediated dipolar cycloadditions but also significantly expands the reaction range of the amide-based aza-π-allylpalladium 1,3-dipoles compared to the precedents [45,46]. Herein, we hope to report the results from this study.

We started our study with the model reaction of 5-vinyloxazolidine-2,4–dione 1a and 3-cyanochromone 2a in CH2Cl2. As shown in Table 1, Pd2(dba)3‧CHCl3 was used as a precatalyst to investigate the chiral phosphoramidite ligands L1–4 (entries 1–4). It was found that the readily available (R)-Feringa ligand L2 gave relatively satisfactory results for the cycloadducts in 98% yield with 1.2:1 dr and 90% ee/75% ee for the two diastereoisomers 3a and 3a' (entry 2). Fortunately, the two diastereoisomers could be isolated by column chromatography. And then, changing the ratio of precatalyst to L2 showed a detrimental effect on the enantioselectivities (entries 5 and 6). Solvent screening revealed that ethyl acetate afforded the best ee values for the two diastereoisomers (entry 9). The addition of 3 Å molecular sieves slightly improved the ee values to 96% ee and 97% ee (entry 10). To our delight, the reaction also proceeded smoothly with a reduced catalyst loading, providing 94% yield with 1.2:1 dr and 95% ee and 98% ee (entry 11).
With the optimal reaction conditions in hand, a series of 5-vinyloxazolidine-2,4-diones and 3-cyanochromones were tested to evaluate the generality of the Pd-catalyzed asymmetric decarboxylative 1,3-dipolar cycloaddition. As shown in Scheme 3, the 5-vinyloxazolidine-2,4-diones bearing either electron-withdrawing or electron-donating substituent on the benzene ring, such as F-, Cl-, Br-, Me-, or MeO- group, reacted smoothly with 2a to generate the corresponding products 3b/b'−3f/f' in excellent yields with very high ee values. Likewise, the doubly substituted phenyl group is tolerated under the reaction conditions to give the products 3g and 3g′ in 98% yield with 2.6:1 dr and 93% ee/92% ee. In addition, the vinyl cyclic carbamates bearing naphthyl and thiophen moieties were also compatible well with the developed reaction conditions, thus giving the cycloadducts 3h/h′ and 3i/i′ with acceptable results. Moreover, it was found that the reactivity and stereoselectivity were hardly affected by the incorporation of different substituents on the nitrogen atom of the amide group, such as Me-, MeO-, EtO, Bn-, allyl-, and PMP-substituents, the desired products 3j/j'−3o/o' could be smoothly obtained in good to excellent yields with good diastereoselectivites and excellent enantioselectivities. Notably, non-terminal alkenyl-substituted cyclic carbamate could also be used for the cycloaddition reaction, affording 3p in 23% yield with 14:1 dr and 89% ee. Although the catalytic system is less effective for the reactivity of substrate containing a non-terminal double bond, the results further proved that the reaction undergoes the palladium-catalyzed asymmetric decarboxylation to form aza-π-allylpalladium dipole intermediates for the nucleophilic attack the unsaturated electrophiles and the subsequent intramolecular nucleophilic cyclization. On the other hand, 3-cyanochromones bearing different steric and electronic natures could react well with 1a to furnish the desired products 3q/q'−3y/y' in good to excellent yields with moderate diastereoselectivities and excellent enantioselectivities. In the case of 3-cyano-6,8-dimethyl chromone as a dipolarophile, the reaction with 1a proceeded smoothly under the catalytic system to furnish products 3z and 3z' in 97% yield with 1:1.2 dr and 92% ee/99% ee. The reaction of 1a with 3-cyanobenzochromone also performed well to generate corresponding cyclic products 3aa and 3aa' in high yield (94%) with high enantioselectivity (91% ee/93% ee) and 1:1 diastereoselectivity. Replacing the cyano group of the chromone with another electron-withdrawing group, such as ester, formyl, or carboxyl group, was also allowed for the occurrence of the 1,3-dipolar cycloaddition reaction, as shown by products 3ab/ab'−3ae/ae'. It should be pointed out that the formyl (CHO) group and the carboxyl (CO2H) group in products 3ad/ad' and 3ae/ae' were eliminated. Furthermore, the palladium-catalyzed asymmetric decarboxylative cycloaddition reaction of 1a with other unsaturated electrophiles was also examined. The reaction of 1a with isocyanate 4 proceeded smoothly and furnished chiral imidazolidine-2,4–dione 3af in 97% yield with 58% ee. Nevertheless, it was also found that some other acceptors bearing different electron-withdrawing groups, such as malononitrile (5), nitrile acetate (6), barbiturate (7), 1H-indene-1,3(2H)–dione (8), and oxindole (9), were also suitable in the developed catalytic system to give the desired cycloadducts with moderate to good results. In particular, the spirooxindole product 3ak could be obtained with >20:1 dr and 72% ee. Notably, the two diastereoisomers 3 and 3′ in Scheme 3 could be isolated by column chromatography. The absolute configurations of products (S,S,R)−3a and (S,R,S)−3a' were unambiguously confirmed by X-ray crystallography, and assigned to all of other products by analogy.
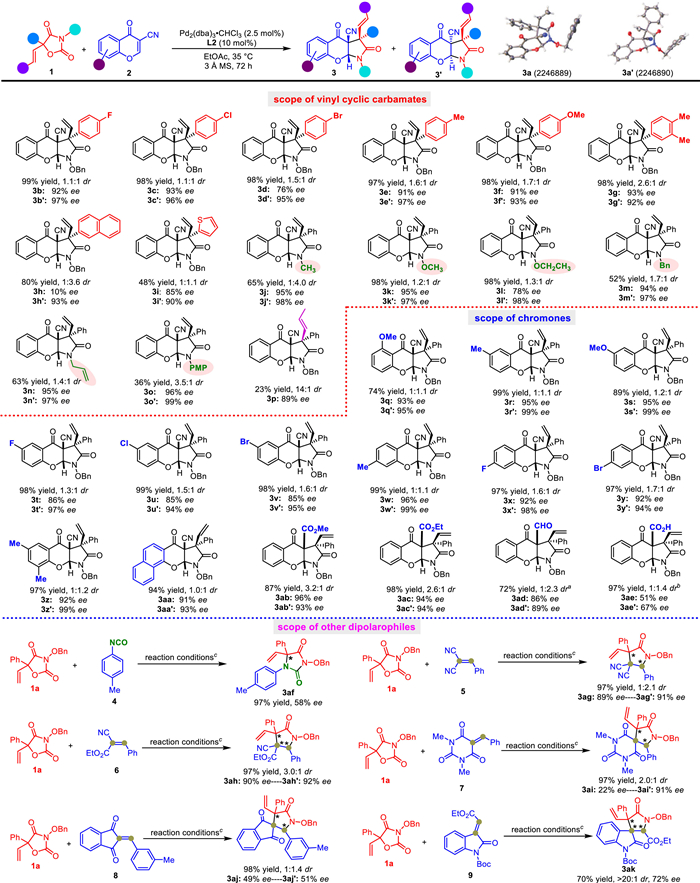
To our surprise, the catalytic system developed above could be successfully extended to imines as dipolarophiles. As shown in Scheme 4, at first, the reaction of 1a with (E)-N-benzylidene-4-methylbenzenesulfonamide (10a) in chlorobenzene as solution proceeded well, thereby delivering the cycloadduct 11a in 93% yield with >20:1 dr and 96% ee. Nevertheless, different vinyl cyclic carbamates 1 bearing a variation of substituents including electron-withdrawing as well as electron-donating functionalities on the aryl moiety could also undergo decarboxylation for 1,3-dipolar cycloaddition with 10a, providing the corresponding chiral imidazolidin-4-one products 11b-g in 23%−82% yield with 93%−99% ee. 5-Vinyloxazolidine-2,4-diones bearing different protecting groups (Me-, EtO-, allyl-, and PMP-) at the nitrogen atom were well tolerated in the reaction conditions to give the cycloadducts 11h-k in good yields (77%−98%) with excellent enantioselectivities (96%−99% ee). In addition, the non-terminal alkenyl-substituted cyclic carbamate could react with 10a to produce 11l in 98% yield with 7:1 dr and 98% ee. On the other hand, imines 10 derived from arylaldehydes bearing different substitution patterns and electronic properties could react well with 1a under the standard conditions, affording products 11m-p in moderate to good yields with high to excellent ee values. In the case of imine derived from 1-naphthaldehyde, the cycloaddition reaction gave product 11q in 48% yield with >20:1 dr and 98% ee, but the imine from 2-naphthaldehyde was able to produce the corresponding cycloadduct 11r in 82% yield with >20:1 dr and 98% ee. The results of this pair of examples indicate that the steric hindrance of the aldimines has an obvious influence on the reactivity of the cycloaddition. Moreover, the pyridin-2-ylsulfonyl group was also compatible with the catalytic system, as shown by the products 11s and 11t. The absolute configuration of (R,S)−11a was unambiguously confirmed by X-ray crystallography, and assuming a common reaction pathway, the configuration of the other cycloadducts 11 was assigned by analogy. Notably, all products except 11l could be obtained in >20:1 diastereoselectivity through the asymmetric decarboxylative 1,3-dipolar cycloaddition reaction.

More excitingly, we also found that the formaldehyde (37% aqueous solution) 12, an abundant feedstock and a reactive one carbon electrophile, could be used for the palladium-catalyzed decarboxylative 1,3-dipolar cycloaddition with vinyl cyclic carbamates 1 under the developed catalytic system. As shown in Scheme 5, a series of 5-vinyloxazolidine-2,4-diones with different substitution patterns and electronic properties performed well, affording the oxazolidin-4-one products 13a-i in 54%−90% yield with 72%−98% ee. In addition, the reaction conditions were also effective for the NOMe, NOEt, and N-Bn substituted 5-vinyloxazolidine-2,4-diones, thus delivering the corresponding products 13j-l in moderate to high yields (42%−99%) with 83%−92% ee. Furthermore, non-terminal alkenyl-substituted cyclic carbamate could also react with formaldehyde to afford product 13m in 44% yield with 95% ee under the standard reaction conditions.

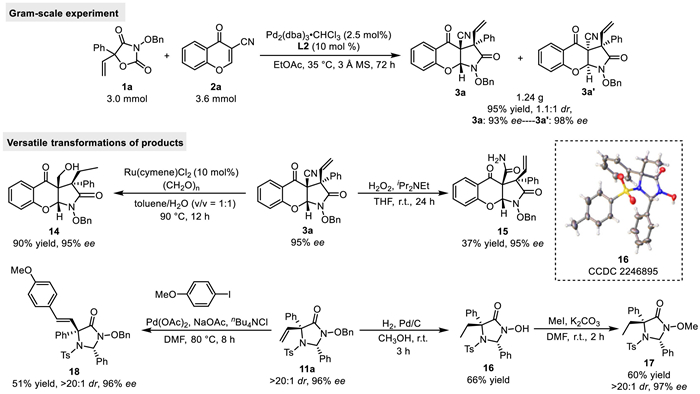
To investigate the synthetic applications of the Pd-catalyzed asymmetric decarboxylative 1,3-dipolar cycloaddition of 5-vinyloxazolidine-2,4-diones, we performed the gram-scale experiment and the versatile derivatizations of the products. As shown in Scheme 6, the reaction of 1a and 2a could be scaled up to 3.0 mmol for 1a and proceeded smoothly under the standard conditions, affording the products 3a and 3a' in 95% yield with 1.1:1 dr and 93% ee/98% ee (Scheme 6, top). Luckily, the diastereoisomers 3a and 3a' could be readily separated by flash column chromatography on silica gel. And then, a variety of synthetic transformations of the products were explored. At first, under Ru-catalyzed reductive conditions, 3a was readily converted to compound 14 in 90% yield with 95% ee in the presence of paraformaldehyde with a mixture of toluene and water as solvent. In addition, the nitrile group of 3a was hydrolyzed to the amide group by using a 10-fold molar excess of 30% aqueous H2O2 in THF at room temperature, resulting in the formation of 15 in 37% yield without deterioration of enantiomeric purity. On the other hand, we also found that product 11a could be sequentially transformed into compounds 16 and 17 in good yields without loss of the diastereo- and enantioselectivities. Moreover, the Pd-catalyzed Heck coupling reaction of 11a with 1-iodo-4-methoxybenzene occurred smoothly in DMF at 80 ℃, leading to the generation of 18 in 51% yield with >20:1 dr and 96% ee. The structure of 16 was unambiguously assigned by X-ray crystallography.
Taking into account the previous related reports [26,36,37,51–53] and reconciling them with the results of our study, a possible reaction mechanism was proposed and outlined in Scheme 7. The chiral PdL* complex from Pd2(dba)3⋅CHCl3 and ligand L2 activates vinyl cyclic carbamates 1 through an oxidative addition and decarboxylation process to release CO2 and generate a Pd-containing amide-based zwitterionic intermediate A, which could be detected with the help of ESI-HRMS analysis and characterized by peaks at m/z 1450.4208 as the [A + H]+ species and at m/z 1472.4036 as the [A+Na]+ species. This observation clearly demonstrates that not only the in situ formation of aza-π-allylpalladium 1,3-dipoles A in the catalytic system, but also the chiral palladium complex ligated 1,3-dipole intermediate A is critical for the asymmetric 1,3-dipolar cycloaddition. And then, the nucleophilic nitrogen anion site of intermediate A attacks the dipolarophiles 2 (C=C double bond), 10 (C=N double bond), and 12 (C=O double bond) via aza-Michael addition, Mannich-type addition, and aldol-type addition to form intermediates B1, B2, and B3, respectively. These intermediates then undergo independently intramolecular allylic alkylation (B1), intramolecular allylic amination (B2), and intramolecular allylic oxidation (B3) for the cyclization to generate their corresponding optically pure cycloadducts pyrrolidin-2-ones 3 + 3′, imidazolidin-4-ones 11, and oxazolidin-4-ones 13, along with releasing the palladium catalyst into the next catalytic cycle.

In conclusion, we have successfully developed a series of vinyl cyclic carbamates containing an oxazolidine-2,4–dione fragment, which could be used as reactive precursors for the in situ generation of amide-based aza-π-allylpalladium 1,3-dipoles via decarboxylation process with a palladium catalyst. This type of amide-based aza-π-allylpalladium 1,3-dipoles could be used for asymmetric decarboxylative 1,3-dipolar cycloaddition with different types of dipolarophiles including C=C, C=N, and C=O double bonds. A catalytic system consisting of a readily available chiral phosphoramidite ligand and a Pd(0) catalyst displays high efficiency and excellent stereocontrol in the cycloaddition reactions. This strategy provides an opportunity for the synthesis of previously unusual structures, such as highly functionalized optically pure pyrrolidin-2-ones, imidazolidin-4-ones, and oxazolidin-4-ones. This protocol also has significant features including wide substrate scope, mild reaction conditions, simple operation, and good to excellent results (70 examples, up to 99% yield, >20:1 dr and 99% ee). The synthetic utility of the developed asymmetric decarboxylative 1,3-dipolar cycloaddition was showcased by gram-scale experiment and versatile derivatizations of the products. This unique strategy significantly expands the reaction range of the amide-based aza-π-allylpalladium 1,3-dipoles compared to the precedents. Further applications of amide-based aza-π-allylpalladium 1,3-dipoles in diverse asymmetric cycloaddition reactions and in the synthesis of biologically active compounds are currently underway.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are grateful for the National Natural Science Foundation of China (Nos. 22271027, 22171029, 21901024, 21871252, 21801024, and 21801026), the Sichuan Science and Technology Program (No. 2021YFS0315), and the Talent Program of Chengdu University (Nos. 2081919035 and 2081921038).
Supplementary material associated with this article can be found, in the online version, at doi:
S. Kobayashi, K.A. Jorgensen, Cycloaddition Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2001.
L.M. Harwood, R.J. Vickers, A. Padwa, W.H. Pearson, Org. Proc. Res. Dev. 8 (2004) 293–300.
T. Hashimoto, K. Maruoka, Chem. Rev. 115 (2015) 5366–5412. doi: 10.1021/cr5007182
B.D.W. Allen, C.P. Lakeland, J.P.A. Harrity, Chem. Eur. J. 23 (2017) 13830–13857. doi: 10.1002/chem.201702486
N. De, E.J. Yoo, ACS Catal. 8 (2018) 48–58. doi: 10.1021/acscatal.7b03346
T.R. Li, Y.N. Wang, W.J. Xiao, L.Q. Lu, Tetrahedron Lett. 59 (2018) 1521–1530. doi: 10.1016/j.tetlet.2018.02.081
J. Wang, S.A. Blaszczyk, X. Li, W. Tang, Chem. Rev. 121 (2021) 110–139. doi: 10.1021/acs.chemrev.0c00160
M.M. Zhang, B.L. Qu, B. Shi, et al., Chem. Soc. Rev. 51 (2022) 4146–4174. doi: 10.1039/D1CS00897H
R.D. Taylor, M. MacCoss, A.D.G. Lawson, J. Med. Chem. 57 (2014) 5845–5859. doi: 10.1021/jm4017625
J.D. Weaver, A. Recio Ⅲ, A.J. Grenning, J.A. Tunge, Chem. Rev. 111 (2011) 1846–1913. doi: 10.1021/cr1002744
R. Shintani, Bull. Chem. Soc. Jpn. 85 (2012) 931–939. doi: 10.1246/bcsj.20120171
A. Khan, Y.J. Zhang, Synlett 26 (2015) 853–860. doi: 10.1055/s-0034-1380170
W. Guo, J.E. Gómeȥ, À. Cristòfol, et al., Angew. Chem. Int. Ed. 57 (2018) 13735–13747. doi: 10.1002/anie.201805009
J. Zhang, Y. Chen, Y. Liu, et al., Chin. J. Org. Chem. 42 (2022) 3051–3101. doi: 10.6023/cjoc202206028
Y. You, Y.P. Zhang, Z.H. Wang, et al., Chem. Commun. 59 (2023) 7483–7505. doi: 10.1039/D3CC01401K
Zuo.W.Guo L, Synlett 33 (2022) 903–906. doi: 10.1055/a-1741-8898
B. Yan, W. Guo, Synthesis 54 (2022) 1964–1976. doi: 10.1055/a-1715-7413
T. Liu, Y. Fang, L. Zuo, et al., Org. Chem. Front. 8 (2021) 1902–1909. doi: 10.1039/D1QO00070E
L. Zuo, T. Liu, X. Chang, W. Guo, Molecules 24 (2019) 3930–3945. doi: 10.3390/molecules24213930
Q.Z. Li, Y. Liu, T. Qi, et al., Org. Biomol. Chem. 18 (2020) 3638–3648. doi: 10.1039/D0OB00458H
B. Niu, Y. Wei, M. Shi, Org. Chem. Front. 8 (2021) 3475–3501. doi: 10.1039/D1QO00273B
Y. Tian, M. Duan, K. Dong, et al., Adv. Synth. Catal. 363 (2021) 4461–4474. doi: 10.1002/adsc.202100715
Y. You, Y.P. Zhang, Z.H. Wang, et al., ChemCatChem 14 (2022) 1–34.
C. Wang, J.A. Tunge, Org. Lett. 8 (2006) 3211–3214. doi: 10.1021/ol0610744
C. Wang, J.A. Tunge, J. Am. Chem. Soc. 130 (2008) 8118–8119. doi: 10.1021/ja801742h
T.R. Li, F. Tan, D.Q. Shi, et al., Nat. Commun. 5 (2014) 5500–5510. doi: 10.1038/ncomms6500
C. Guo, D. Janssen-Müller, M. Fleige, et al., J. Am. Chem. Soc. 139 (2017) 4443–4451. doi: 10.1021/jacs.7b00462
M.M. Li, Y. Wei, L.Q. Lu, et al., J. Am. Chem. Soc. 139 (2017) 14707–14713. doi: 10.1021/jacs.7b08310
C. Guo, M. Fleige, C.G. Daniliuc, et al., J. Am. Chem. Soc. 138 (2016) 7840–7843. doi: 10.1021/jacs.6b04364
L.A. Leth, F. Glaus, E.A. Bitsch, et al., Angew. Chem. Int. Ed. 55 (2016) 15272–15276. doi: 10.1002/anie.201607788
B.D.W. Allen, M.J. Connolly, J.P.A. Harrity, Chem. Eur. J. 22 (2016) 13000–13003. doi: 10.1002/chem.201602586
J. Han, L. Hoteite, J.P.A. Harrity, Chem. Eur. J. 28 (2022) 1–6.
S.P. Yuan, Y.P. Zhang, M.Q. Zhou, et al., Org. Lett. 24 (2022) 8348–8353. doi: 10.1021/acs.orglett.2c03383
K. Ohmatsu, N. Imagawa, T. Ooi, Nat. Chem. 6 (2014) 47–51. doi: 10.1038/nchem.1796
K. Ohmatsu, S. Kawai, N. Imagawa, T. Ooi, ACS Catal. 4 (2014) 4304–4306. doi: 10.1021/cs501369z
Q.Q. Hang, Y.C. Zhang, G.J. Me, et al., Chin. J. Chem. 38 (2020) 1612–1618. doi: 10.1002/cjoc.202000104
F. Tian, W.L. Yang, J.Z. Zhang, et al., Sci. China Chem. 64 (2021) 34–40. doi: 10.1007/s11426-020-9854-3
T.T. Li, Y. You, Z.H. Wang, et al., Org. Lett. 24 (2022) 5120–5125. doi: 10.1021/acs.orglett.2c01959
Y. You, Q. Li, Y.P. Zhang, et al., ChemCatChem 14 (2022) e202101887. doi: 10.1002/cctc.202101887
Y. You, T.T. Li, J.Q. Zhao, et al., Org. Lett. 24 (2022) 7671–7676. doi: 10.1021/acs.orglett.2c03244
J.Q. Zhao, H.W. Rao, Z.H. Wang, et al., Org. Chem. Front. 9 (2022) 6172–6178. doi: 10.1039/D2QO01297A
T. Wang, Y. You, B.D. Cui, et al., Org. Lett. 25 (2023) 1274–1279. doi: 10.1021/acs.orglett.3c00075
Y. You, G.Y. Gan, S.Y. Duan, et al., Org. Chem. Front. 10 (2023) 5421–5427. doi: 10.1039/D3QO00961K
Y.P. Zhang, Y. You, J.Q. Yin, et al., Eur. J. Org. Chem. (2023) e202300728.
K. Li, S. Zhen, Y. Wu, et al., Chem. Sci. 14 (2023) 3024–3029. doi: 10.1039/D3SC00112A
A. Scuiller, N. Casaretto, A. Archambeau, J. Org. Chem. 88 (2023) 9941–9945. doi: 10.1021/acs.joc.3c00707
S. Omura, T. Fujimoto, H. Tanaka, et al., Antibiot 44 (1991) 113–116. doi: 10.7164/antibiotics.44.113
S. Flohr, S. Stengelin, M. Gossel, T.D.E. Klabunde, Patent, WO2004072076 (A1), 2004.
M. Tadesse, M.B. Strom, K. Stensvag, et al., Org. Lett. 12 (2010) 4752–4755. doi: 10.1021/ol101707u
J. Caruano, G.G. Muccioli, R. Robiette, Org. Biomol. Chem. 14 (2016) 10134–10156. doi: 10.1039/C6OB01349J
A. Khan, R. Zheng, J. Xing, et al., Angew. Chem. Int. Ed. 53 (2014) 6439–6442. doi: 10.1002/anie.201403754
L. Yang, A. Khan, L.Y. Jin, et al., Org. Lett. 17 (2015) 6230–6233. doi: 10.1021/acs.orglett.5b03218
A. Khan, C. Zhao, Y.J. Zhang, Chem. Commun. 54 (2018) 4708–4711. doi: 10.1039/C8CC02456A

Scheme 1 Palladium-catalyzed decarboxylation of vinyl cyclic carbamates for generating aza-π-allylpalladium 1,n-dipoles.

Scheme 2 Design and exploration of amide-based aza-π-allylpalladium dipole precursors for asymmetric 1,3-dipolar cycloaddition, and the selected bioactive compounds containing lactam-type heterocyclic skeleton.

Scheme 3 Substrate scope for vinyl cyclic carbamates and diverse dipolarophiles containing C=C double bond. Reaction conditions: 1 (0.1 mmol), 2 (0.12 mmol), Pd2(dba)3⋅CHCl3 (2.5 mol%), L2 (10 mol%), and 3 Å MS (30 mg) in 1.0 mL EtOAc at 35 ℃ for 72 h under argon atmosphere. a The formyl (CHO) group was eliminated in the product. b The carboxyl (CO2H) group was eliminated in the product. c Reaction conditions: 1a (0.1 mmol), 4–9 (0.12 mmol), Pd2(dba)3⋅CHCl3 (5 mol%), L2 (20 mol%) in 1.0 mL CH2Cl2 at 35 ℃ for 168 h under argon atmosphere.

Scheme 4 Substrate scope for vinyl cyclic carbamates and imines. Reaction conditions: 1 (0.1 mmol), 10 (0.12 mmol), Pd2(dba)3⋅CHCl3 (5 mol%), L2 (20 mol%) in 1.0 mL PhCl at 35 ℃ for 168 h under argon atmosphere.

Scheme 5 Substrate scope for vinyl cyclic carbamates and formaldehyde. Reaction conditions: 1 (0.1 mmol), 12 (1.0 mmol, 37% aqueous solution), Pd2(dba)3⋅CHCl3 (5 mol%), L2 (20 mol%) in 1.0 mL PhCl at 35 ℃ for 168 h under argon atmosphere.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: