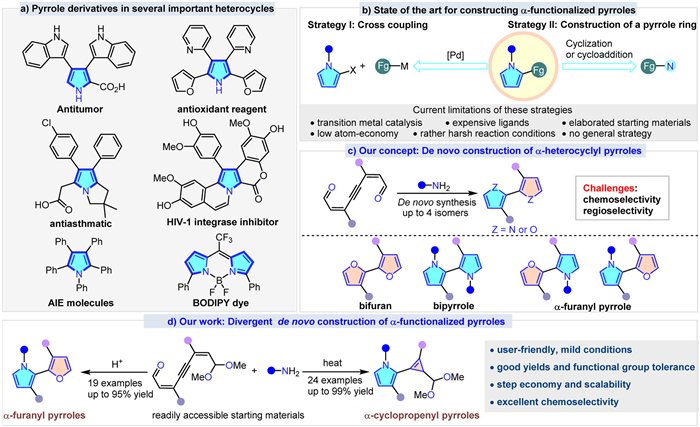
Scheme 1.
Pyrrole molecules and strategies for the synthesis of α-functionalized pyrrole derivatives.
Divergent de novo construction of α-functionalized pyrrole derivatives via coarctate reaction
Zhuwen Wei , Jiayan Chen , Congzhen Xie , Yang Chen , Shifa Zhu

The development of efficient and atom economical processes for the preparation of valuable heterocycles remains an important goal in synthetic organic chemistry. Arguably, pyrroles are one of the most prevalent five-membered heterocycles and are present in a large number of natural products [1–6], pharmaceuticals [7,8], functional materials [9–15], and ligands [16,17] (Scheme 1a). Even though there are many well-developed methods for the synthesis of pyrroles [18,5,19], the efficient synthesis of α-functionalized pyrroles is still challenging. Due to its electron-rich nature, the poor air stability of pyrroles towards atmospheric oxygen often makes it a great challenge to synthesize and further study the structure and properties for α-functionalized pyrrole derivatives, especially for these bearing electron-donating substituents [20]. Traditionally, transition-metal-catalyzed cross-coupling reaction between two prefunctionalized substrates is a common method for the synthesis of α-functionalized pyrroles (Scheme 1b, left), but it usually suffers from some drawbacks, such as expensive transition-metal catalysts and ligands, low atom-economy, low stability of some heteroaryl halides and poor tolerance of functional groups under acidic conditions [21–28]. Alternatively, regioselective functionalization of the preformed pyrrole rings offers the synthesis of multi-substituted α-functionalized pyrrole derivatives [29,30]. Meanwhile, to avoid the negative effects caused by the active pyrroles, the de novo construction of pyrrole rings through isomerization or cycloaddition represents another highly desirable and intriguing strategy for synthesizing α-functionalized pyrrole derivatives (Scheme 1b, right) [31–40]. However, the requirement of elaborated starting materials narrowed down the practical applicability of this method. Hence, due to its ubiquitous nature, the search for more efficient and sustainable procedures for the preparation of α-functionalized pyrrole derivatives and build up new α-functionalized pyrrole skeletons using readily available starting materials under mild reaction conditions would be highly desirable, especially if such methods rely upon state-of-art green credentials, which would meet the strong synthetic demand for green chemistry and sustainable development.
On the other hand, transition-metal-catalyzed cyclization reactions of conjugated enynals or enynones through a 5-exo-dig nucleophilic attack have become an efficient way to generate the α-furanyl carbene intermediates [41–54]. Despite several achievements in the synthesis of α-functionalized furan derivatives with structural diversity and atom economy by this way, reports on α-functionalized pyrrole derivatives are rare, which remain elusive and challenging [55,56].
Based on our previous work, in which enynals could efficiently give bifurans through the coarctate reactions [57,58] under the catalysis of acids [59], and as part of our ongoing interest in developing novel heterocyclic molecule frameworks [60–63], we envisioned that the simple and commercially inexpensive starting material enynals might undergo a tandem double cycloisomerization under the addition of amines to achieve the de novo synthesis of the desired α-functionalized pyrrole derivatives (Scheme 1c). It should be noted that our hypothesis faces some inherent challenges in terms of both chemo-and regioselectivity (up to 4 isomers): (1) Avoiding the direct competitive cyclization reaction before the condensation between amine and the formyl group to form imine, which may cause the reaction to produce the undesired bifuran byproducts; (2) As for the asymmetric enynals substrates, the reaction may result in the formation of two α-furanyl pyrroles isomers. If successful, this method would provide a general and modular platform for the de novo synthesis of α-functionalized pyrrole derivatives, which are otherwise challenging to access. In response to this assumption, we disclose a highly atom-economical, cost-effective, scalable, and regioselective coarctate reaction of enynals for the synthesis of α-furanyl and α-cyclopropenyl pyrrole derivatives in a programmed and diversity-oriented format under mild conditions (Scheme 1d).
To validate the feasibility of this hypothesis, we initiated our studies by using enynals 1a and tBuNH2 as the cyclization partner for reaction condition screening. As shown in Table 1, various Brønsted acid catalysts were tested for this reaction (entries 1–6). At the outset, when 0.1 equiv. pyridine p-toluenesulfonate (PPTS), which is a widely used and mild acidic catalytic reagent, was applied as the catalyst, the reaction virtually did not take place (entry 1). With the employment of 6.0 equiv. acetic acid (AcOH), we also have failed to detect the formation of target products at room temperature for 24 h (entry 2). We were delighted to discover that the desired α-furanyl pyrrole 2a could be successfully produced in 68% yield with the dropwise addition of 6.0 equiv. tetrafluoroboric acid (HBF4) within a time process of 10 min (entry 3). To our delight, trifluoromethanesulfonic acid (TfOH) could catalyze this reaction to furnish the desired product in high yield (entry 4, 72%). The yield of 2a could be significantly improved when 6.0 equiv. methanesulfonic acid (MsOH) was applied (entry 5, 92%). Increase in the equivalence of tBuNH2 failed to improve the product yield (entry 7). The further screening of the loading of tBuNH2 demonstrated that decreasing the loading of tBuNH2 had negative influence on the reaction efficiency (entries 8 and 9). Meanwhile, the reaction occurred most optimally with 5 equiv. of amines and a reduced number of amines would lead to the generation of bifuran byproducts. Besides, the examination of the solvent implied that THF was the most suitable one for the desired transformation (see Supporting information for details). It is worth mentioning that α-cyclopropenyl pyrrole 3b was surprisingly obtained in 97% yield without the addition of MsOH upon increasing the reaction temperature to 60 ℃ (entry 10). The unexpected formation of 3b was very exciting because cyclopropenes are a fascinating class of highly strained small molecules with applications in the fields of drug discovery, catalysis, and materials chemistry [64–66]. This interesting observation as well procedure to enable efficient access of α-cyclopropenyl pyrrole products from readily available enynals and amines. Our continuing reaction condition optimization revealed that the addition of 1.2 equiv. tBuNH2 is enough to led the desired product formation in 96% yield (entry 11). Moreover, the employment of ethyl acetate (EA) could also result in a good yield (90%, entry 12). A quick solvent screening revealed that THF was the solvent of choice (see Supporting information for details).
Having established an effective means of synthesizing α-functionalized pyrroles from hemi-protected enynals, we then examined the scope of substrates of this transformation for synthesizing α-furanyl pyrroles under the optimized conditions. The results indicated that this reaction system facilitated smoothly with a wide range of hemi-protected enynals 1 bearing different aryl groups, and afforded α-furanyl pyrroles with high chemo- and regioselectivity in moderate to good yields in very short reaction time (Scheme 2). For instance, when the two aryl substituents were the same, the target products 2a-2c could be obtained with good yields. In addition, the substrates substituted by two different aromatic groups are also compatible with these conditions, affording the corresponding products (2d-2m) efficiently despite various functionality. Both electron-withdrawing (2d-2i) and electron-donating groups (2j-2l) on enynals all reacted well to give the desired products in 37%–95% yields. Among them, when a nitro group was introduced to the substrate, this transformation could proceed smoothly to yield the desired product 2h in 66% yield. Moreover, α-furanyl pyrroles 2 m that was substituted by more sterically demanding naphthalene could also be obtained in a yield of 79%.
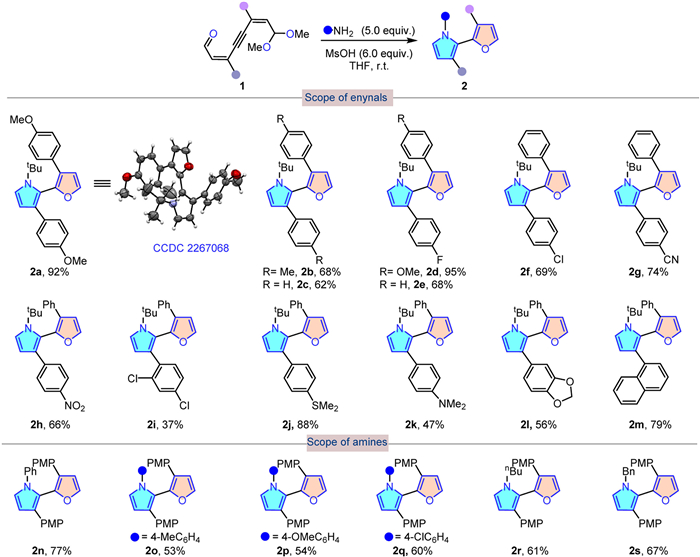
Next, the generality of the process was investigated with respect to amines. As summarized in Scheme 2, arylamines, regardless of the electronic properties, were compatible with the current reaction system to afford the expected products 2n-2q in moderate yields. Additionally, butylamine and benzylamine readily participated in this reaction, giving rise to 2r and 2s in 61% and 67% yield, respectively. In addition, the α-furanyl pyrrole structure was unambiguously confirmed by X-ray crystallographic analysis of 2a.
With the success of the coarctate reaction to give α-furanyl pyrroles under the catalysis of acids, we wondered whether enynals and amines could be used as readily available starting materials to obtain bipyrrole derivatives. It should be noted that bipyrroles represent the core structures of several marketed drug molecules [67–70]. However, the development of more practical and efficient procedures for synthesizing bipyrroles is still a challenging task. To explore the feasibility of coarctate reaction for giving the expected bipyrroles, we selected enynals 1a and aniline as benchmark substrates to optimize the reaction conditions (see Supporting information for details). After brief optimization studies, we were delighted to find that the symmetric bipyrrole product 4a could be obtained in 48% yield when 10 mol% of Bi(NO3)3·5H2O was used as Lewis acid catalyst in 1,4-dioxane at room temperature for 24 h (Scheme 3). Similarly, the electron-withdrawing hemi-protected enynals 1n were also amenable to the reaction, giving the asymmetric bipyrrole product 4n in 49% yield.

The substrate generality of the reaction to obtain α-cyclopropenyl pyrroles was then evaluated. As shown in Scheme 4, this reaction could be successfully applied to a variety of enynals 1 with different combinations of aromatic groups. Gratifyingly, either electron-rich or -poor substituents on the aryl ring were well tolerated (3a-3q). Subsequently, in order to further investigate the scope applicability of this green reaction, we surveyed readily available amine building blocks. And it is found that alkyl amine, benzylamine, aniline, ammonia, and even hydrazine were all well tolerated in this reaction, producing α-cyclopropenyl pyrrole products 3r-3x in 22%–82% yields. It is obvious that the electronic properties of anilines had no significant influence on the reaction, and the yields of the target product ranges from 64% to 82% (3r-3u). However, a considerable drop in yield was noticed when the disubstituted 2,6-dimethylaniline was applied, clearly suggesting that aniline with high steric resistance hampers the reaction outcome (3t, 40%). Furthermore, it is worth noting that the reactions functioned well when hydrazine was used as amine donor and give product 3w in 50% yield. Importantly, when ammonia was used as amine donor, and the desired N-unprotected pyrrole product 3x could also be furnished, albeit the yield was somewhat low. The α-cyclopropenyl pyrrole structure was unambiguously confirmed by X-ray crystallographic analysis of 3c.

Based on our previous reports [59], a plausible reaction mechanism was then proposed in Scheme 5. Initially, the condensation of enynal 1 with amine to form imine A. Under the condition of heating which was the absence of acid, the intermediate A may undergoes cycloisomerization directly to form α-cyclopropenyl pyrroles 3, and it is worth noting that a α-pyrroleyl free carbene intermediate B may be involved in this process. On the other hand, the intermediate A is deprotected by hydrolysis in the presence of acid to provide enynal-imine intermediate C. Then double 5-exo-dig cyclization of C will take place under the catalysis of MsOH to produce α-furanyl pyrroles 2. In this process, an excess of amines was required to completely inhibit the formation of bifuran byproduct, and 6 equiv. acids were consumed, some of which may react with the amine to form ammonium salt and others may be used to hydrolyze acetal-protected imine A to generate enynal-imine intermediate C.

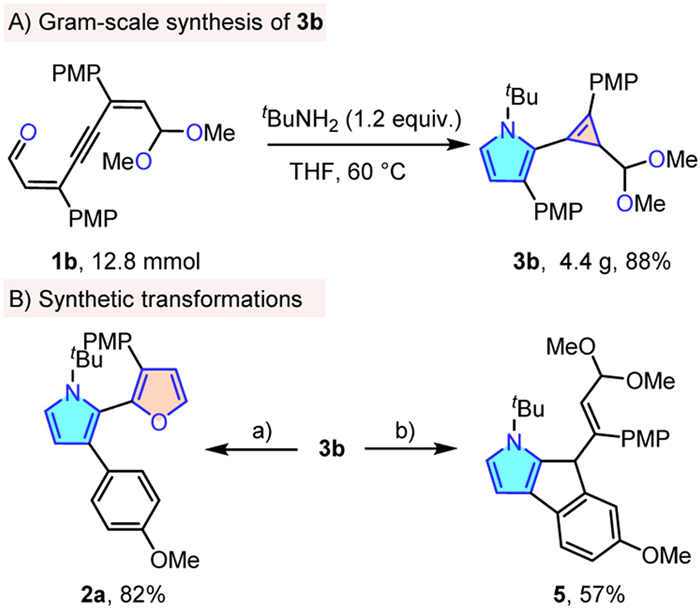
To further verify the potential application of this protocol, a gram-scale reaction of 1b was carried out. Pleasingly, the reaction could proceed smoothly as well, 4.4 g of 3b was isolated (88% yield). It is noteworthy that the α-cyclopropenyl pyrrole 3b could easily afford α-furanyl pyrrole product 2a in 85% yield with treatment of 2 equiv. HCl (1 mol/L) (Scheme 6). Furthermore, the cyclopropene moiety having abundant chemical transformations in the product also provided a convenient handle for further elaboration [71], as evidenced by the facile conversion of the product 3b into multi-fused pyrrole ring 5 in 57% yield through Au(I)-catalyzed C—H insertion reaction.
In summary, we have developed a method for efficiently and greenly synthesizing α-functionalized pyrrole derivatives from enynals and amines under mild conditions. This method provides an efficient way to construct α-cyclopropenyl pyrroles for the first time. In addition, under acidic conditions, enynals and amines could quickly deliver α-furanyl pyrroles in 10 min at room temperature. This approach features readily accessible starting materials, high functional group compatibility, step economy and scalability, which would complement previous methods and support expansion of the toolbox for the synthesis of valuable, but previously inaccessible, highly substituted and electron-rich α-functionalized pyrroles. The present method expands the application range of enynal molecules from the construction of α-functionalized furan derivatives to α-functionalized pyrrole derivatives, and providing a new type of pyrrole frameworks. We also hope this finding would build up a new platform for the development of green methods for the de novo construction of α-functionalized pyrrole derivatives via easy and available raw materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are grateful to the National Natural Science Foundation of China (Nos. 22071062 and 22271096) and the Fundamental Research Funds for the Central Universities, SCUT (No. 2022ZYGXZR107). This research achievement is funded by the National Postdoctoral Research Program (No. GZC20230839). We also thank Yanmei Chen for participating in the screening of reaction conditions of α-furanyl pyrroles.
Supplementary material associated with this article can be found, in the online version, at doi:
A. Fürstner, Angew. Chem. Int. Ed. 42 (2003) 3582–3603. doi: 10.1002/anie.200300582
H. Fan, J. Peng, M.T. Hamann, J. Hu, Chem. Rev. 108 (2008) 264–287. doi: 10.1021/cr078199m
A.D. Yamaguchi, K.M. Chepiga, H.M.L. Davies, et al., J. Am. Chem. Soc. 137 (2015) 644–647. doi: 10.1021/ja512059d
Q. Li, J. Jiang, Y. Jia, et al., Org. Lett. 13 (2011) 312–315. doi: 10.1021/ol1027877
C.T. Walsh, S. Garneau-Tsodikova, A.R. Howard-Jones, Nat. Prod. Rep. 23 (2006) 517–531. doi: 10.1039/b605245m
I.S. Young, P.D. Thornton, A. Thompson, Nat. Prod. Rep. 27 (2010) 1801–1839. doi: 10.1039/c0np00014k
M. Biava, G.C. Porretta, M. Anzini, et al., J. Med. Chem. 50 (2007) 5403–5411. doi: 10.1021/jm0707525
S. Ahmad, O. Alam, M. Iqbal, et al., Eur. J. Med. Chem. 157 (2018) 527–561. doi: 10.1016/j.ejmech.2018.08.002
W. Li, D. Chen, Y. Dong, et al., ACS Appl. Mater. Interfaces 7 (2015) 26094–26100. doi: 10.1021/acsami.5b07422
Y. Lei, Q. Liu, Y. Dong, et al., Chem. Eur. J. 24 (2018) 14269–14274. doi: 10.1002/chem.201803080
L. Dong, G. Shang, Y. Dong, et al., J. Phys. Chem. C 121 (2017) 11658–11664. doi: 10.1021/acs.jpcc.7b02125
I.S. Park, S.Y. Lee, C. Adachi, T. Yasuda, Adv. Funct. Mater. 26 (2016) 1813–1821. doi: 10.1002/adfm.201505106
Y. Chen, D.X. Zeng, N. Xie, Y.Z. Dang, J. Org. Chem. 70 (2005) 5001–5005. doi: 10.1021/jo050236r
S. Mabrouk, M. Zhang, S. Yang, et al., J. Mater. Chem. A 6 (2018) 7950–7958. doi: 10.1039/C8TA01773E
X. Jiang, S. Yue, K. Chen, et al., Chin. Chem. Lett. 30 (2019) 2271–2273. doi: 10.1016/j.cclet.2019.07.027
E.C. Hansen, D.J. Pedro, D.J. Weix, et al., Nat. Chem. 8 (2016) 1126–1130. doi: 10.1038/nchem.2587
K. Záruba, V. Setnička, V. Král, et al., Collect. Czech. Chem. Commun. 66 (2001) 693–769. doi: 10.1135/cccc20010693
S. Sadki, P. Schottland, N. Brodie, G. Sabouraud, Chem. Soc. Rev. 29 (2000) 283–293. doi: 10.1039/a807124a
B.R. Lee, A.C. Willis, M.G. Banwell, et al., Chem. Asian J. 15 (2020) 3059–3081. doi: 10.1002/asia.202000740
J.I. Setsune, Chem. Rev. 117 (2017) 3044–3101. doi: 10.1021/acs.chemrev.6b00430
D. Zhao, J. You, C. Hu, Chem. Eur. J. 17 (2011) 5466–5492. doi: 10.1002/chem.201003039
V.F. Slagt, A.H.M.D. Vries, J.G.D. Vries, R.M. Kellogg, Org. Process Res. Dev. 14 (2010) 30–47. doi: 10.1021/op900221v
A.E. Rubtsov, A.V. Malkov, Synthesis 53 (2021) 2559–2569. doi: 10.1055/s-0040-1706030
F. Bellina, Synthesis 53 (2021) 2517–2544. doi: 10.1055/a-1437-9761
Y. Yang, J. Lan, J. You, Chem. Rev. 117 (2017) 8787–8863. doi: 10.1021/acs.chemrev.6b00567
A. Cervantes-Reyes, A.C. Smith, M. Szostak, et al., Org. Lett. 24 (2022) 1678–1683. doi: 10.1021/acs.orglett.2c00267
F.S. Han, Chem. Soc. Rev. 42 (2013) 5270–5298. doi: 10.1039/c3cs35521g
Y.F. Zhang, Z.J. Shi, Acc. Chem. Res. 52 (2019) 161–169. doi: 10.1021/acs.accounts.8b00408
G. Sun, S. Ren, Y. Wan, et al., Org. Lett. 18 (2016) 544–547. doi: 10.1021/acs.orglett.5b03581
D. Sanchez-García, J.I. Borrell, S. Nonell, Org. Lett. 11 (2009) 77–79. doi: 10.1021/ol802380g
X. Gao, P. Wang, A. Lei, et al., Green Chem. 21 (2019) 4941–4945. doi: 10.1039/C9GC02118C
Y. Jiang, C.M. Park, Chem. Sci. 5 (2014) 2347–2351. doi: 10.1039/C4SC00125G
P.E. Simm, P. Sekar, J. Richardson, P.W. Davies, ACS Catal. 11 (2021) 6357–6362. doi: 10.1021/acscatal.1c01457
K. Dong, H. Zheng, M.P. Doyle, et al., ACS Catal. 11 (2021) 4712–4721. doi: 10.1021/acscatal.1c01051
J. Xuan, X.D. Xia, W.-J. Xiao, et al., Angew. Chem. Int. Ed. 53 (2014) 5653–5656. doi: 10.1002/anie.201400602
X. Wu, K. Li, A. Lei, et al., Org. Lett. 18 (2016) 56–59. doi: 10.1021/acs.orglett.5b03240
A. Douchez, A. Geranurimi, W.D. Lubell, Acc. Chem. Res. 51 (2018) 2574–2588. doi: 10.1021/acs.accounts.8b00388
Z. Shi, W. Mao, Z. Xu, et al., Chin. Chem. Lett. 34 (2023) 107488. doi: 10.1016/j.cclet.2022.05.002
S. Ren, K. Huang, G. Qiu, et al., Chin. Chem. Lett. 33 (2022) 4870–4873. doi: 10.1016/j.cclet.2022.02.028
L. Li, Q. Chen, M. Zhang, et al., Chin. Chem. Lett. 29 (2018) 1893–1896. doi: 10.1016/j.cclet.2018.09.004
D. Zhu, T. Cao, K. Chen, S. Zhu, Chem. Sci. 13 (2022) 1992–2000. doi: 10.1039/D1SC05374D
Q. Shi, Y. Chen, T. Cao, S. Zhu, Org. Lett. 24 (2022) 8142–8146. doi: 10.1021/acs.orglett.2c03185
Y. Chen, S. Zhu, Org. Chem. Front. 9 (2022) 6194–6199. doi: 10.1039/D2QO01324J
R. Wu, J. Lu, S. Zhu, et al., J. Am. Chem. Soc. 143 (2021) 14916–14925. doi: 10.1021/jacs.1c07556
H. Zhang, T. Cao, S. Zhu, et al., Org. Chem. Front. 6 (2019) 1118–1122. doi: 10.1039/C9QO00045C
L. Chen, Z. Liu, S. Zhu, Org. Biomol. Chem. 16 (2018) 8884–8898. doi: 10.1039/C8OB02348D
D. Zhu, L. Chen, S. Zhu, et al., Angew. Chem. Int. Ed. 57 (2018) 12405–12409. doi: 10.1002/anie.201805676
L. Chen, K. Chen, S. Zhu, Chem 4 (2018) 1208–1262. doi: 10.1016/j.chempr.2018.02.001
S. Mata, L.A. López, R. Vicente, Chem. Eur. J. 21 (2015) 8998–9002. doi: 10.1002/chem.201501155
Q. Wang, M. Rudolph, F. Rominger, A.S.K. Hashmi, Adv. Synth. Catal. 362 (2020) 755–759. doi: 10.1002/adsc.201901318
J. Ma, L. Zhang, S. Zhu, Curr. Org. Chem. 20 (2016) 102–118.
D. Zhu, L. Chen, S. Zhu, et al., Chem. Soc. Rev. 49 (2020) 908–950. doi: 10.1039/C9CS00542K
T. Murata, M. Murai, K. Ohe, et al., Org. Lett. 14 (2012) 2296–2299. doi: 10.1021/ol300718x
J.M. Yang, Z.Q. Li, M.L. Li, et al., J. Am. Chem. Soc. 139 (2017) 3784–3789. doi: 10.1021/jacs.6b13168
C.H. Oh, W. Park, M. Kim, Synlett 9 (2007) 1411–1415.
S. Li, G. Zeng, R. He, et al., New J. Chem. 45 (2021) 1834–1837. doi: 10.1039/D0NJ05253A
R. Herges, J. Org. Chem. 80 (2015) 11869–11876. doi: 10.1021/acs.joc.5b01959
B.S. Young, R. Herges, M.M. Haley, Chem. Commun. 48 (2012) 9441–9455. doi: 10.1039/c2cc34026g
Z. Wei, Y. Chen, S. Zhu, et al., Chem. Eur. J. 29 (2023) e202203444. doi: 10.1002/chem.202203444
Y. Chen, P. Shen, S. Zhu, et al., Nat. Commun. 12 (2021) 51 6165.
Y. Chen, Z. Liao, T. Cao, S. Zhu, Green Synth. Catal. 3 (2022) 89–94. doi: 10.1016/j.gresc.2021.11.001
S. Qiu, X. Gao, S. Zhu, Chem. Sci. 12 (2021) 13730–13736. doi: 10.1039/D1SC04961E
R. Wu, J. Lu, S. Zhu, et al., J. Am. Chem. Soc. 143 (2021) 14916–14925. doi: 10.1021/jacs.1c07556
M. Rubin, M. Rubina, V. Gevorgyan, Chem. Rev. 107 (2007) 3117–3179. doi: 10.1021/cr050988l
Z.B. Zhu, Y. Wei, M. Shi, Chem. Soc. Rev. 40 (2011) 5534–5563. doi: 10.1039/c1cs15074j
R. Vicente, Chem. Rev. 121 (2021) 162–226. doi: 10.1021/acs.chemrev.0c00151
V.J. Bauer, D.L.J. Clive, R.B. Woodward, et al., J. Am. Chem. Soc. 105 (1983) 6429–6436. doi: 10.1021/ja00359a012
J.L. Sessler, M.J. Cyr, J.A. Ibers, et al., J. Am. Chem. Soc. 112 (1990) 2810–2813. doi: 10.1021/ja00163a059
J.L. Sessler, J.M. Davis, Acc. Chem. Res. 34 (2001) 989–997. doi: 10.1021/ar980117g
E. Vogel, M. Kocher, H. Schmickler, J. Lex, Angew. Chem. Int. Ed. 25 (1986) 257–259. doi: 10.1002/anie.198602571
P. Li, X. Zhang, M. Shi, Chem. Commun. 56 (2020) 5457–5471. doi: 10.1039/D0CC01612H

Scheme 1 Pyrrole molecules and strategies for the synthesis of α-functionalized pyrrole derivatives.

Scheme 2 Substrate scope for the synthesis of α-furanyl pyrroles. Reaction conditions: 1 = 0.2 mmol, THF (1.0 mL), r.t., 10 min, the addition of acid was added dropwise over 10 min, isolated yield.

Scheme 4 Substrate scope for the synthesis of α-cyclopropenyl pyrroles. Reaction conditions: 1 (0.2 mmol), THF (2.0 mL), 60 ℃, 48 h, isolated yield.

Scheme 5 Plausible mechanism for the construction of α-functionalized pyrrole derivatives from enynals and amines.

Scheme 6 Gram-scale synthesis and transformations of α-cyclopropenyl pyrrole. (a) HCl (aq.) (2.0 equiv.), THF, r.t., 10 min; (b) Au(PPh3)Cl (5 mol%), NaBArF (5 mol%), DCM, r.t., 48 h.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: