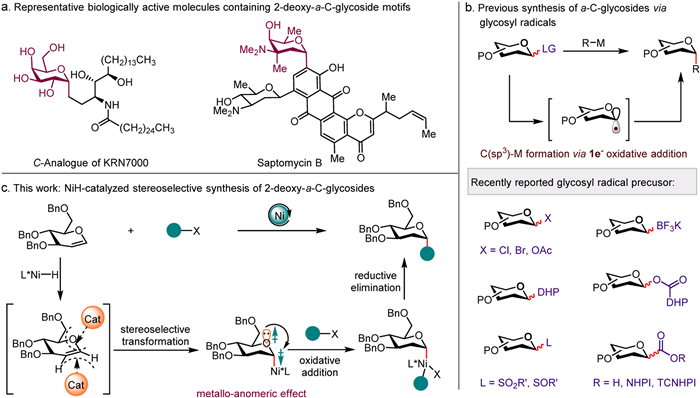
Figure 1.
Stereoselective synthesis of α-C-glycosides. (a) Representative structures of 2-deoxy-α-C-glycosides. (b) Metal-mediated synthese of α-C-glycosides via glycosyl radicals. (c) Reaction design (this work).

Glycosylation, a fundamental process in living organisms, entails the attachment of a sugar moiety to a target molecule. This pivotal reaction is facilitated by enzymes called glycosyltransferases, which play crucial roles in various biological processes, including cell signaling, protein folding, and immune response [1–6]. Among the different types of glycosylation reactions, carbon glycosylation, also known as carbon glycosidation or carbon glycosylation, has garnered significant attention in recent years [7–11]. Carbon glycosylation reactions have been extensively investigated due to their potential applications in the synthesis of natural products, pharmaceuticals, and bioactive compounds [12–14]. Especially for α-C-glycoside motifs are prominently prevalent in a wide range of bioactive molecules, exemplified by the C-Analogue of KRN7000 and saptomycin B (Fig. 1a) [15,16]. They have also displayed enhanced stability and pharmacokinetic properties in biological studies, making them valuable tools for investigating the biological functions of polysaccharides [17–20]. Therefore, the development of efficient and highly stereoselective carbosylation methods has garnered significant interest among synthetic chemists [21–26].
Within the realm of oligosaccharide synthesis, the precise control of stereochemistry during glycosidic bond formation presents a persistent challenge [27,28]. This crucial reaction in glycochemistry significantly constrains the advancement of glycochemical synthesis. In recent years, various strategies and catalysts have been explored to improve the efficiency and selectivity of carbon glycosylation reactions. These include the use of organocatalysts [29–35] and transition metal catalysts [14,36–43]. Among these strategies, transition-metal-catalyzed glycosylation reactions involving anomeric C(sp3)-metal bond formation via 1e− oxidative addition have emerged as a powerful tool for the synthesis of various glycosides and glycoconjugates due to their versatility, efficiency, and stereoselectivity with various glycosyl radical precursors (Fig. 1b). For instance, the Gong's group [44] and Koh's group [45,46] have successfully employed glycosyl chlorides as glycosyl radical precursors, enabling the synthesis of α-stereodefined C-glycosides under reductive conditions using nickel and iron catalysis, respectively. The emergence of metallaphotoredox catalysis has expanded the chemical space of carbon glycosylation [47–51]. Molander and co-workers introduced a novel class of glycosyl radical precursors, namely 4-pyranosyl/glycosyl DHPs, enabling the synthesis of non-classical arylated C-glycosides [52,53]. Glycosyl trifluoroborates have also been utilized as radical precursors in photoredox/nickel synergistic-catalyzed α-C-glycosylation reactions with high α-selectivity, as demonstrated by Walczak's group [54] and Hirai's group [55] respectively. Additionally, Niu's group recently succeeded in utilizing glycosyl sulfinates or glycosyl sulfoxides as radical precursors for the efficient and highly stereoselective synthesis of α-C-glycosides [56–58].
Despite the significant progress made in transition-metal-catalyzed cross-coupling reactions for the synthesis of α-C-glycosides, these strategies still face certain limitations. One such limitation is the need to prepare glycosyl radical precursors in advance, which restricts the range of coupling partners that can be used. Additionally, these reactions involve radical processes that occasionally suffer from issues related to stereoselectivity [59,60]. Therefore, there remain several challenges that need to be addressed. We envision whether we can enable precise control over the stereoselectivity of these reactions by discovering suitable catalysts and combining the steric effects of the ligand with the metallo-anomeric effect. In parallel with our work, Liu with cooperators Fu, Lu [61], Liu and Wang [62] reported the ligand-controlled highly stereoselective synthesis of β-C-glycosides via cobalt catalysis, respectively.
Herein, we developed an approach for the ligand-controlled nickel-catalyzed stereoselective synthesis of 2-deoxy-α-C-glycosides from glycals and alkyl halides. The introduction of the alkyl group enhances the structural diversity of carbohydrate compounds and offers greater possibilities for investigating the biological functions of polysaccharides [63,64]. This reaction exhibits a broad substrate scope and excellent tolerance towards various functional groups (Fig. 1c).
We initiated our study by selecting commercially available glycal 1a and 4-fluorobenzyl chloride 2d as model substrates, using KF as the base and (MeO)3SiH as the hydrogen source (Table 1). Various bisoxazoline ligands with different backbones were evaluated in the C-alkyl glycosylation reaction. Encouragingly, the rigid 2,2-bis(2-oxazoline) ligand L1 was identified as the optimal ligand, demonstrating good α-selectivity (α/β = 10:1) and a high yield of 68% (entry 1). However, upon increasing the steric hindrance of the ligands by modifying the substituent adjacent to the nitrogen group, a significant decrease in both stereoselectivity and yield was observed (L2-L4). This observation highlights the crucial role of steric effects in the efficiency and stereoselectivity of this reaction. Furthermore, when flexibly bisoxazoline ligands were employed (L5-L8), the stereochemistry of the products became uncontrollable. These results collectively suggest that the ligands play a crucial role in determining the α-selectivity of this reaction. Subsequently, various nickel sources were tested, and it was found that NiBr2·DME led to an increased yield (entry 9). Finally, the optimal outcome was achieved by replacing trimethoxysilane with poly(methylhydrosiloxane) (PMHS) with 86% yield and 12:1 α-selectivity (entry 13).
After identifying the optimal conditions, we proceeded to investigate the substrate scope of this reaction, and the summarized results can be found in Scheme 1. Initially, we explored the benzyl halide derivatives, and our observations revealed that benzyl halides substituted with halogen groups (F, Br, Cl) underwent the reaction smoothly, leading to the formation of the desired products (3b, 3d, 3e, and 3f) with good yields and high α-selectivity. These results open up possibilities for diverse downstream transformations. This reaction demonstrated tolerance towards both electron-rich and electron-deficient groups, such as methoxy (3k and 3l), cyano (3g), and ester (3p), yielding moderate to high yields. Notably, polar groups such as aldehyde, alcohol and amide groups on benzyl halides (3o, 3m and 3r) were found to be compatible with this transformation, resulting in moderate yields and excellent α-selectivity. Additionally, the absolute α-anomeric configuration of the products was confirmed through X-ray crystal structures of 3g and 3n. Furthermore, additional unactivated alkyl halide, namely 1–bromo-3-phenylpropane and 1-iodo-3-phenylpropane, were also subjected to investigation. These compounds were observed to yield the desired products (3s) with moderate efficiency, albeit exhibiting lower levels of diastereoselectivity.

Subsequently, a diverse range of protected glycals with different substituents were subjected to the reaction, as depicted in Scheme 1. Various protected groups including Me-, Bn-, Bz-, TBS- and TIPS- were effectively employed in the C-alkyl glycosylation reaction, yielding products with yields ranging from 40% to 86%. Notably, even the unprotected primary alcohol group on the glycols yielded target products with high yields and good stereoselectivity (3x). However, when the 4-position substituent group on the glycals was positioned in the axial direction, lower efficiency was observed, likely due to significant steric effects (3u). It is worth noting that the aldehyde-protected glycals substrate can also obtain 2-deoxy-α-C-glycosides products with excellent stereoselectivity, which is convenient for further conversion (3y and 3z). Moreover, the structure of product 3z, derived from 4,6-O-benzylidene-d-glycal, was confirmed by X-ray crystallography. Notably, 3,6-dihydroglycal was also compatible, yielding the target product (3za) with moderate yield and excellent diastereoselectivity in this catalytic system.
By highlighting the application of this reaction, a scale-up experiment was conducted. As shown in Scheme 2a, reducing the amount of catalyst and ligand, the targeted product 3e was isolated in moderate yield and excellent diastereoselectivity, demonstrating the practicality of this transformation. Subsequently, we successfully achieved divergent synthesis for the formation of mono-C-glycoside and twice-C-glycoside products (3zb and 3zc), respectively, under the same conditions. This was accomplished by simply switching the amounts of glycals and benzyl chlorides, while utilizing specific benzyl chlorides (Scheme 2b). This method provides an efficient and feasible strategy for the skeleton splicing of different carbohydrate compounds, so that polysaccharides of different glycosyl fragments can be synthesized, it has very important synthetic value. Furthermore, we compareing with the previous method of synthesis of benzyl-glycosides, previous synthesis strategies focused on the synthesis of a single β-benzyl-glycosides, such as by reacted glycosyl halides with benzylmagnesium chloride [65], or hydrogenation of exo-glycals [66]. However, we developed nickel catalysis cross-coupling synthesis main α-benzyl-glycosides configuration from glycals and benzyl chlorides (Scheme 2c). In comparison to previous methods that predominantly resulted in β-selectivity products, our protocol demonstrated the ability to assemble α-selectivity products with the opposite configuration, exhibiting good efficiency (3a and 3u). This protocol fills the vacancy in the field of benzyl glycosidation and provides a new choice for the synthesis of carbohydrate compounds. α-Benzyl-glycosides with rich functional groups can be synthesized effectively, which provides a new possibility for the enrichment and diversity of the molecular library of carbohydrate compounds.
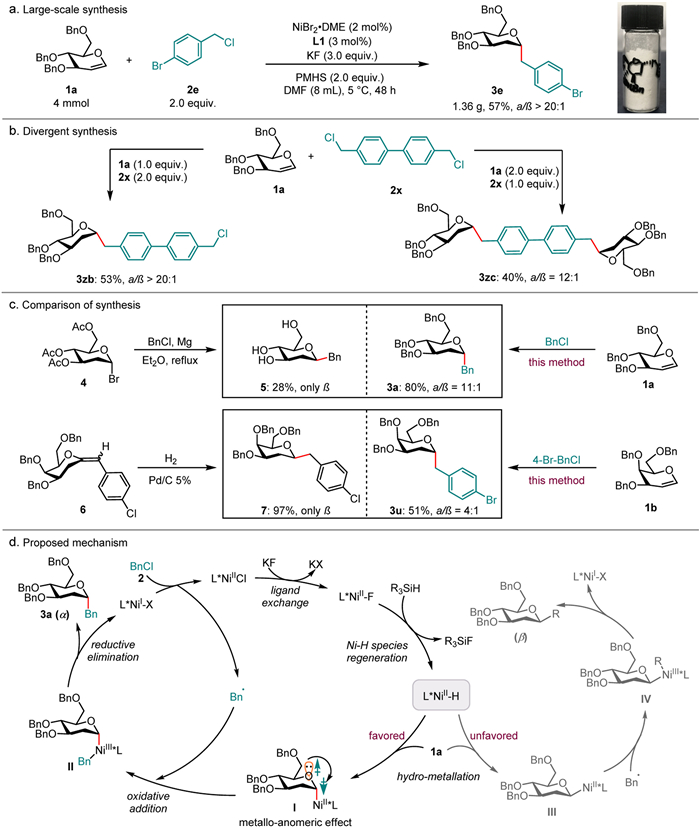
According to previous studies [65–77], a plausible mechanism was proposed, as demonstrated in Scheme 2d. Initially, the LNiII-H species was generated by reacting LNiII-F with silane, and then followed by glycals insertion, due to the steric effects of the ligand and the anomeric effect, which favored resulting in the formation of the syn-addition α-configured alkylnickel intermediate I, rather than the β-stereoselective alkylnickel intermediate III. Likely due to the LNiII complex, there is a strong thermodynamic propensity for the display of axial isomers. Furthermore, the lone pairs of the oxygen atom can effectively delocalize to the antibonding σ*(C−Ni) orbital, and the cancellation of the dipole-dipole interaction and the stabilization of the axial C—Ni bond. Then intermediate I was quickly trapped by a benzyl radical, leading to the formation of the highly active LNiIIIBn species II, and this high-valence nickel complex prefers the axial direction. Subsequently, reductive elimination occurred, producing the targeted products and the LNiI species. The LNiI species then reacted with benzyl chlorides, regenerating LNiII-Cl and a benzyl radical.
In summary, we have developed a Ni-catalyzed C(sp3)-C(sp3) coupling hydrobenzylation method for the efficient synthesis of α-configured C-glycosides. This transformation occurs under mild conditions and makes use of readily available glycals and commercially accessible benzyl chlorides. Notably, our methodology eliminates the requirement for a variety of glycosyl radical precursors. It demonstrates broad substrate scope, excellent α-stereoselectivity, and remarkable tolerance towards diverse functional groups. The success of the synthesis protocol has important scientific significance and application value, and is expected to provide new ideas and methods for the synthesis and research of glucosyl-containing drugs.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by grants from the Fundamental Research Funds for Central Universities (No. 2042021kf0190). We also thank the Core Facility of Wuhan University for providing the X-ray diffraction equipment.
V. Křen, J. Thiem, Chem. Soc. Rev. 26 (1997) 463–473. doi: 10.1039/CS9972600463
S. Hanashima, B. Castagner, D. Esposito, et al., Org. Lett. 9 (2007) 1777–1779. doi: 10.1021/ol0704946
S.A. Borisova, H.J. Kim, X. Pu, et al., ChemBioChem 9 (2008) 1554–1558. doi: 10.1002/cbic.200800155
Y. Fujimoto, T. Hattori, S. Uno, et al., Carbohydr. Res. 344 (2009) 972–978. doi: 10.1016/j.carres.2009.03.006
S. Muthana, H. Cao, X. Chen, Curr. Opinion Chem. Biol. 13 (2009) 573–581. doi: 10.1016/j.cbpa.2009.09.013
P. Nagorny, B. Fasching, X. Li, et al., J. Am. Chem. Soc. 131 (2009) 5792–5799. doi: 10.1021/ja809554x
M.H.D. Postema, Tetrahedron 48 (1992) 8545–8599. doi: 10.1016/S0040-4020(01)89435-7
D.Y.W. Lee, M. He, Curr. Topics Med. Chem. 5 (2005) 1333–1350. doi: 10.2174/156802605774643042
M. Ferrero, V. Gotor, Chem. Rev. 100 (2000) 4319–4348. doi: 10.1021/cr000446y
M. Adamo, R. Pergoli, Curr. Org. Chem. 12 (2008) 1544–1569. doi: 10.2174/138527208786786318
Y. Yang, B. Yu, Chem. Rev. 117 (2017) 12281–12356. doi: 10.1021/acs.chemrev.7b00234
P. Hultin, Curr. Top. Med. Chem. 5 (2005) 1299–1331. doi: 10.2174/156802605774643015
K. Kitamura, Y. Ando, T. Matsumoto, et al., Chem. Rev. 118 (2018) 1495–1598. doi: 10.1021/acs.chemrev.7b00380
W. Shang, R. Shi, D. Niu, Chin. J. Chem. 41 (2023) 2217–2236. doi: 10.1002/cjoc.202300153
E. De Clercq, J. Med. Chem. 59 (2016) 2301–2311. doi: 10.1021/acs.jmedchem.5b01157
H. Liao, J. Ma, H. Yao, et al., Org. Biomol. Chem. 16 (2018) 1791–1806. doi: 10.1039/C8OB00032H
P. Compain, O.R. Martin, Bioorg. Med. Chem. 9 (2001) 3077–3092. doi: 10.1016/S0968-0896(01)00176-6
W. Zou, Curr. Top. Med. Chem. 5 (2005) 1363–1391. doi: 10.2174/156802605774642999
M. Hocek, Chem. Rev. 109 (2009) 6729–6764. doi: 10.1021/cr9002165
E. Leclerc, X. Pannecoucke, M. Ethève-Quelquejeu, et al., Chem. Soc. Rev. 42 (2013) 4270–4283. doi: 10.1039/C2CS35403A
F. Zhu, M.J. Rourke, T. Yang, et al., J. Am. Chem. Soc. 138 (2016) 12049–12052. doi: 10.1021/jacs.6b07891
C.C.J. Loh, Nat. Rev. Chem. 5 (2021) 792–815. doi: 10.1038/s41570-021-00324-y
K.M. Hoang, N.R. Lees, S.B. Herzon, J. Am. Chem. Soc. 143 (2021) 2777–2783. doi: 10.1021/jacs.0c11262
V.U.B. Rao, C. Wang, D.P. Demarque, et al., Nat. Chem. 15 (2022) 424–435.
Q. Li, S.M. Levi, C.C. Wagen, et al., Nature 608 (2022) 74–79. doi: 10.1038/s41586-022-04958-w
C. Zhang, S.Y. Xu, H. Zuo, et al., Nat. Synth. 2 (2023) 251–260. doi: 10.1038/s44160-022-00214-1
L. Krasnova, C.H. Wong, J. Am. Chem. Soc. 141 (2019) 3735–3754. doi: 10.1021/jacs.8b11005
K. Kanagaraj, J. Rebek, Y. Yu, Green Synth. Catal. 4 (2023) 7–9. doi: 10.1016/j.gresc.2022.10.005
P.R. Schreiner, Chem. Soc. Rev. 32 (2003) 289–296. doi: 10.1039/b107298f
S.J. Connon, Chem. Eur. J. 12 (2006) 5418–5427. doi: 10.1002/chem.200501076
A.G. Doyle, E.N. Jacobsen, Chem. Rev. 107 (2007) 5713–5743. doi: 10.1021/cr068373r
Y. Takemoto, Chem. Pharm. Bull. 58 (2010) 593–601. doi: 10.1248/cpb.58.593
D. Larsen, L.M. Langhorn, O.M. Akselsen, et al., Chem. Sci. 8 (2017) 7978–7982. doi: 10.1039/C7SC03366D
Y. Park, K.C. Harper, N. Kuhl, et al., Science 355 (2017) 162–166. doi: 10.1126/science.aal1875
Q. Li, S.M. Levi, E.N. Jacobsen, J. Am. Chem. Soc. 142 (2020) 11865–11872. doi: 10.1021/jacs.0c04255
A. Chen, B. Yang, Z. Zhou, et al., Chem. Catal. 2 (2022) 3430–3470. doi: 10.1016/j.checat.2022.10.019
H. Gong, R. Sinisi, M.R. Gagné, J. Am. Chem. Soc. 129 (2007) 1908–1909. doi: 10.1021/ja068950t
H. Gong, M.R. Gagné, J. Am. Chem. Soc. 130 (2008) 12177–12183. doi: 10.1021/ja8041564
X. Li, J. Zhu, Eur. J. Org. Chem. 2016 (2016) 4724–4767. doi: 10.1002/ejoc.201600484
J. Ghouilem, M.D. Robichon, F.L. Bideau, et al., Chem. Eur. J. 27 (2021) 491–511. doi: 10.1002/chem.202003267
Q. Sun, H. Zhang, Q. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 19620–19625. doi: 10.1002/anie.202104430
X. Gou, Y. Li, W. Shi, et al., Angew. Chem. Int. Ed. 61 (2022) e202205656. doi: 10.1002/anie.202205656
N. Hussain, A. Hussain, RSC Adv. 11 (2021) 34369–34391. doi: 10.1039/D1RA06351K
J. Liu, C. Lei, H. Gong, Sci. China Chem. 62 (2019) 1492–1496. doi: 10.1007/s11426-019-9501-4
Y. Jiang, Q. Wang, X. Zhang, et al., Chem 7 (2021) 3377–3392. doi: 10.1016/j.chempr.2021.09.008
Q. Wang, Q. Sun, Y. Jiang, et al., Nat. Synth. 1 (2022) 235–244. doi: 10.1038/s44160-022-00024-5
Y. Ma, S. Liu, Y. Xi, et al., Chem. Commun. 55 (2019) 14657–14660. doi: 10.1039/C9CC07184A
M. Li, Y.F. Qiu, C.T. Wang, et al., Org. Lett. 22 (2020) 6288–6293. doi: 10.1021/acs.orglett.0c02053
W.Z. Shi, H. Li, G.C. Mu, et al., Org. Lett. 23 (2021) 2659–2663. doi: 10.1021/acs.orglett.1c00551
M. Zhu, S. Messaoudi, ACS. Catal. 11 (2021) 6334–6342. doi: 10.1021/acscatal.1c01600
W. Yao, G. Zhao, Y. Wu, et al., J. Am. Chem. Soc. 144 (2022) 3353–3359. doi: 10.1021/jacs.1c13299
S.O. Badir, A. Dumoulin, J.K. Matsui, et al., Angew. Chem. Int. Ed. 57 (2018) 6610–6613. doi: 10.1002/anie.201800701
A. Dumoulin, J.K. Matsui, Á. Gutiérrez-Bonet, et al., Angew. Chem. Int. Ed. 57 (2018) 6614–6618. doi: 10.1002/anie.201802282
E.M. Miller, M.A. Walczak, Org. Lett. 23 (2021) 4289–4293. doi: 10.1021/acs.orglett.1c01035
D. Takeda, M. Yoritate, H. Yasutomi, et al., Org. Lett. 23 (2021) 1940–1944. doi: 10.1021/acs.orglett.1c00402
W. Shang, S.N. Su, R. Shi, et al., Angew. Chem. Int. Ed. 60 (2021) 385–390. doi: 10.1002/anie.202009828
S. Xu, W. Zhang, C. Li, et al., Angew. Chem. Int. Ed. 62 (2023) e202218303. doi: 10.1002/anie.202218303
H. Zeng, Y. Li, R. Wu, et al., Org. Lett. (2023), doi: 10.1021/acs.orglett.3c00833.
Y. Jiang, Y. Zhang, B.C. Lee, et al., Angew. Chem. Int. Ed. 62 (2023) e202305138. doi: 10.1002/anie.202305138
L. Chen, Z. Zhao, S. Yu, Chin. Chem. Lett. 35 (2024) 109128. doi: 10.1016/j.cclet.2023.109128
B. Liu, D. Liu, X. Rong, et al., Angew. Chem. Int. Ed. 62 (2023) e202218544. doi: 10.1002/anie.202218544
M. Zeng, C. Yu, Y. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202300424. doi: 10.1002/anie.202300424
J.R. Clark, K. Feng, A. Sookezian, et al., Nat. Chem. 10 (2018) 583–591. doi: 10.1038/s41557-018-0020-0
P. Bhutani, G. Joshi, N. Raja, et al., J. Med. Chem. 64 (2021) 2339–2381. doi: 10.1021/acs.jmedchem.0c01786
J. Panigot, W. Curley, J. Carb. Chem. 13 (1994) 293–302. doi: 10.1080/07328309408009194
G. Díaz, A. Ponzinibbio, R.D. Bravo, Carb. Res. 393 (2014) 23–25. doi: 10.1016/j.carres.2014.04.009
Z. Wang, H. Yin, G.C. Fu, Nature 563 (2018) 379–383. doi: 10.1038/s41586-018-0669-y
D. Qian, S. Bera, X. Hu, J. Am. Chem. Soc. 143 (2021) 1959–1967. doi: 10.1021/jacs.0c11630
F. Zhu, M.A. Walczak, J. Am. Chem. Soc. 142 (2020) 15127–15136. doi: 10.1021/jacs.0c06882
Y. Wang, Y. He, S. Zhu, Acc. Chem. Res. 55 (2022) 3519–3536. doi: 10.1021/acs.accounts.2c00628
S.J. He, J.W. Wang, Y. Li, et al., J. Am. Chem. Soc. 142 (2020) 214–221. doi: 10.1021/jacs.9b09415
X.X. Wang, X. Lu, Y. Li, et al., Sci. China Chem. 63 (2020) 1586–1600. doi: 10.1007/s11426-020-9838-x
Y. Li, W. Nie, Z. Chang, et al., Nat. Catal. 4 (2021) 901–911. doi: 10.1038/s41929-021-00688-w
Y. Li, D. Liu, L. Wan, et al., J. Am. Chem. Soc. 144 (2022) 13961–13972. doi: 10.1021/jacs.2c06279
S.Z. Sun, Y.M. Cai, D.L. Zhang, et al., J. Am. Chem. Soc. 144 (2022) 1130–1137. doi: 10.1021/jacs.1c12350
X.X. Wang, Y.T. Xu, Z.L. Zhang, et al., Nat. Commun. 13 (2022) 1890. doi: 10.1038/s41467-022-29554-4
J.W. Wang, Z. Li, D. Liu, et al., J. Am. Chem. Soc. 145 (2023) 10411–10421. doi: 10.1021/jacs.3c02950

Figure 1 Stereoselective synthesis of α-C-glycosides. (a) Representative structures of 2-deoxy-α-C-glycosides. (b) Metal-mediated synthese of α-C-glycosides via glycosyl radicals. (c) Reaction design (this work).

Scheme 1 Substrate scope of alkyl halides and glycals. General reaction conditions: NiBr2·DME (5 mol%), L1 (6 mol%), KF (3.0 equiv.), PMHS (2.0 equiv.), 1 (0.2 mmol, 1.0 equiv.), 2 (2.0 equiv.), in DMF (0.4 mL) at 5 ℃ for 48 h. Yields are for the isolated product, α/β ratios were determined by 1H NMR analysis.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: