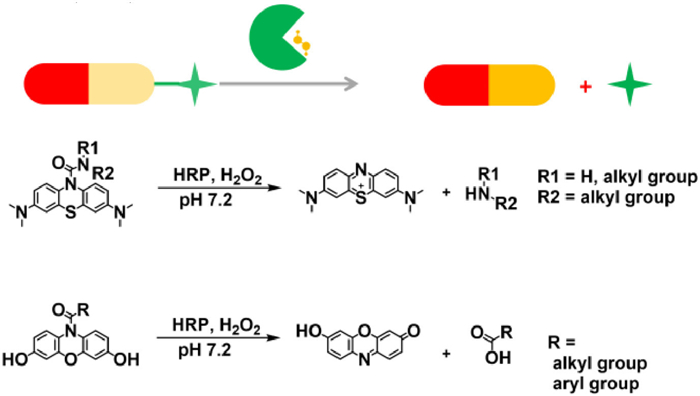
Scheme 1.
Cooperative activation of prodrug by exogenous enzyme (HRP) and endogenous biomarker (H2O2) based on bi-substrate (prodrug and H2O2) enzyme reaction.
Cooperative bond scission by HRP/H2O2 for targeted prodrug activation
Yunkang Tong , Haiqiao Huang , Haolan Li , Mingle Li , Wen Sun , Jianjun Du , Jiangli Fan , Lei Wang , Bin Liu , Xiaoqiang Chen , Xiaojun Peng

Systemic administration is a powerful approach in the armoury against cancer, but it is fraught with problems due to its global systemic toxicity. Prodrug strategies have been developed to address these problems [1,2]. The inactivated prodrugs could be obtained by modification of the active functional group of parent drugs, thus avoiding its toxicity and bioactivity [3]. When the protection group was cleaved by a specific stimulus at the tumor, parent drugs were released at the tumor site specifically, achieving targeted killing of tumor cells without systemic toxicity. To achieve an efficient and targeted cure effect, spatiotemporally controlled and fast activation of prodrugs is the key factor [4]. This means that spatiotemporally controlled cleavage chemistry with fast reaction dynamics is urged for prodrug strategy.
Specific small molecules, such as H2O2 [5,6], glutathione (GSH) [7-11], H+ [12], have a much higher abundance in tumors compared with normal tissues and organs. As a stimulus, their cleavage reaction with prodrug provides a method of drug activation. Romidepsin (Istodax), which contains a GSH-cleavable disulfide bond, has been approved by the Food and Drug Administration (FDA) for cutaneous T-cell lymphoma in 2009 [13]. However, the reaction rate between small molecules may be too slow to achieve an efficient maximum dose of the parent drug, which would attenuate the therapeutic effect.
The exogenous enzyme has been widely used for prodrug activation for its fast cleavage reaction rates and catalytic nature. The therapy with exogenous enzyme-activated prodrug was termed enzyme prodrug therapy (EPT) [14,15]. The vital aspect of this therapy is the pre-targeting of enzymes at the tumor site for selectively activating the prodrug. Antibody-directed enzyme prodrug therapy (ADEPT) was developed for this purpose [16,17]. The exogenous enzyme was modified by a specific antibody with high affinity with antigen overexpressed at tumor cells. After being administrated in the blood, enzymes accumulated at the tumor site. To avoid its unspecific activation, the prodrug must be administrated after the enzyme was cleaned in the blood. However, expensive antibody modification and complex two-step administration may hinder its clinical use.
Therefore, precise prodrug activation in a tumor-selective manner with a fast reaction rate is still a pressing need and full of challenges, prompting us to look for more suitable chemical tools. Toward this problem, we envision a synergistic approach to conduct targeted and efficient cleavage chemistry and prodrug activation via the merger of non-targeted exogenous enzyme and small molecule stimulus in the tumor microenvironment.
If successful, the developed methodology would be highly valuable, because it avoids not only the usage of expensive targeted enzymes but also the cumbersome procedures. The main challenge in this scenario lies in the synergistic work of enzyme and small molecule stimulus, namely, the development of enzyme-catalyzed bi-substrate reactions with prodrug and biomarker as substrate. Herein, we demonstrate horseradish peroxidase (HRP)/hydrogen peroxide (H2O2) pair cooperatively cleaves urea bonds in methylene blue (MB) derivatives and amide bonds in resorufin (RSF) derivatives for targeted therapy (Schemes 1 and 2).
HRP, a plant enzyme originating from horseradish, catalyzing nontoxic indole-3-acetic acid (IAA) to form cytotoxic radicals, has been widely explored for EPT [18,19]. Importantly, it could oxidize small organic molecules such as aniline [20], phenol [21], and phenothiazine [22,23] with H2O2 as a cosubstrate. In our previous work, we designed a prodrug platform for MB and drugs containing primary amines based on urea bonds [24]. Hydroxyl radicals generated by ultrasound could oxidize the urea bond in MB urea derivatives to release MB and primary amine-containing molecules simultaneously. We wonder whether HRP/H2O2 could oxidize urea-based MB due to its structural similarity toward phenothiazine.
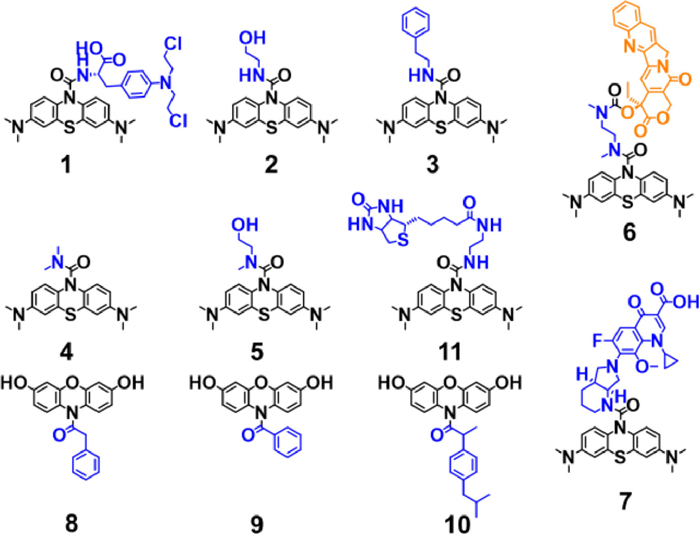
Hence, the feasibility of urea bond scission by HRP and H2O2 cooperatively was evaluated with compound 1 as a model molecule (Fig. 1A). A solution of 20 µmol/L compound 1 was coincubated with HRP (5 nmol/L) and H2O2 (50 µmol/L) in phosphate buffer saline (PBS) (pH 7.2) at 37 ℃ for 10 min before being analyzed by high-performance liquid chromatography-ultraviolet (HPLC-UV) (Fig. 1B), which revealed that the transformation from compound 1 to MB and melphalan (Mel) was quickly and cleanly with almost 100% conversion rate, demonstrating the feasibility of cooperative cleavage reaction for prodrug activation. In contrast, compound 1 treated with HRP or H2O2 solely keep its integrity even for 3 h, further confirming the cooperative nature of this bond scission. Liquid chromatography-high resolution mass spectrometry (LC—HRMS) was applied to identify the products (Fig. S19 in Supporting information). The corresponding peaks were highly identical to the predicted values of MB and Mel. Specifically, the cleavage rates strongly depended on the doses of H2O2 (Fig. 1C). Subsequently, the enzyme kinetics for the oxidation reaction was determined. The characteristic absorbance of MB at 664 nm enabled us to monitor the reaction by absorbance spectroscopy easily. The reaction follows an exponential reaction kinetics model (Fig. 1D), with the t1/2 as low as 2 min, which is substantially much faster than other reported H2O2-responsive prodrugs [5,25-27]. To acquire dynamic parameters, reaction rates at different substrate concentrations were obtained. Michaelis-Menten constants (Km) were calculated by plotting reaction rates against substrate concentrations (Figs. S20 and S21 in Supporting information) [28]. As an important characteristic physical constant of the enzyme, Km reflects the affinity of the catalyst to a substrate.
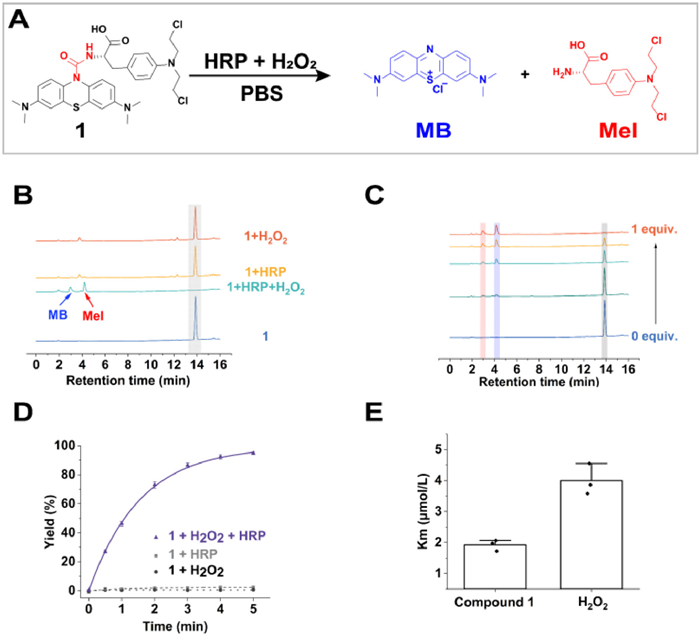
The lower the Km, the higher the affinity between the enzyme and the substrate. The Km for compound 1 and H2O2 was 1.92 ± 0.18 µmol/L and 4.00 ± 0.50 µmol/L, separately (Fig. 1E). The high concentration of H2O2 in the tumor (~50 µmol/L) is > 10 fold of Km for H2O2, enabling the reaction to proceed at a saturated concentration of H2O2. In addition, such a low Km for compound 1 implies that it is an excellent prodrug with a high affinity to HRP [29].
To gain more insight into this unique cooperative cleavage reaction, mechanistic studies were conducted. There are only two types of mechanism namely ordered and ping-pang mechanisms for a bi-substrate enzyme-catalyzed reaction according to whether a triad was formed or not during the reaction [30]. To elucidate this, we plotted [1/V] against [1/S] to acquire double reciprocal plots, the golden rule to determine the mechanism of this type of reaction [31,32]. Parallel curves suggest that this reaction followed a Ping-Pang route, similar to other substrates of HRP, for example 3,3,5,5-tetramethylbenzidine (TMB) (Figs. 2A and B).

Detailed studies indicated that the conversion was initiated by HRP compound I generated in situ from HRP and H2O2. After adding 1 equiv. of H2O2 into the solution of HRP, the characteristic absorption spectra of HRP compound I was observed immediately, which has two oxidizing equivalents (Fig. 2C, black line) [33]. Upon further addition of 1 equiv. of compound 1, HRP compound I was immediately recovered to the resting state of HRP, accompanied by the generation of MB with a conversion rate of 100% (Fig. 2C, blue line), revealing that the transformation is virtually initiated by HRP compound I. In contrast, adding compound 1 to the resting HRP solution change neither the absorbance of HRP nor of compound 1, indicating no direct interaction between the resting state of HRP and compound 1 (Fig. 2D).
Since HRP compound I carry out two-electron oxidations, whether the transformation involved a direct two-electron transfer process or two consecutive one-electron transfer steps with the formation of HRP compound II as an intermediate was further studied. The generated HRP compound I (5 µmol/L HRP + 5 µmol/L H2O2) in-situ decomposed into HRP compound II at 37.5 ℃ spontaneously after 30 min, as evidenced by the spectral change in soret regions (Fig. 2E) [34,35]. After further addition of 1 equiv. of compound 1, it was soon transformed to the resting state of HRP, accompanied by the generation of MB with a conversion rate of about 50%, indicating that single electron extraction is enough for the cleavage of the urea bond. Similar to amide bond cleavage for amplex red, the breakdown of the urea bond may be led by a non-enzyme process between free radicals [36]. As expected, vitamin C, a radical scavenger [37], inhibited this cleavage reaction in a dose-dependent manner, further revealing that the reaction proceeds via the free radical mechanism (Fig. 2F). After the possible reaction mechanism was proposed, the substrate scope for the cooperative cleavage strategy was explored. Consist to the above radical mechanism, MB precursors derived from primary and secondary amines were both responsive to HRP/H2O2 system were responsive toward HRP/H2O2 system, showing the scalability of this kind of cleavage reaction (Figs. S22–S25 in Supporting information). In theory, any drugs containing primary amines or secondary amines as active groups can be developed as prodrugs by forming this kind of MB urea derivatives. However, enzyme-catalyzed reaction was affected by a lot of factors, for example, molecular volume [38]. When substrate was too bulky, it may lose its responsivity toward the enzyme due to steric hindrance between the substrate and catalytic center.
To investigate the feasibility of bulky molecules as prodrugs, we designed and synthesized compound 6. Camptothecin (CPT), a topoisomerase I (TOP I) inhibitor with its hydroxyl as the bioactive group was selected as starting material [39,40]. It was linked with MB by N, N'-dimethyl-1,2-ethanediamine, a self-immolative linker, to form a bulky prodrug (compound 6) [41]. As expected, such a modification blocked the bioactivity of CPT with half-maximal inhibitory concentration (IC50) of > 50 µmol/L, as shown in Fig. S26 (Supporting information). However, CPT showed high toxicity toward HepG2 cells with IC50 of 143.6 nmol/L. To evaluate responsivity of blocked 6 toward HRP/H2O2, a solution of 20 µmol/L compound 6 was co-incubated with HRP (20 nmol/L) and H2O2 (50 µmol/L) in PBS at 37 ℃ for 10 min before being analyzed by HPLC, which revealed fortunately that the transformation from compound 6 to MB and amine-derivative CPT (Fig. S27 in Supporting information). Subsequently, the self-immolative linker leaves to give free CPT with its bioactive hydroxyl group as evidenced by LC-MS (Fig. S28 in Supporting information).
Interestingly, such a structure quenched the fluorescence of CPT. Due to the electron-rich nature of phenothiazines, this phenomenon can be attributed to intramolecular photo-induced electron transfer. However, upon the cleavage of the urea bond, CPT's fluorescence is restored, providing a visual means to track its release process and intracellular distribution (Fig. S29 in Supporting information). Moreover, the antibiotic moxifloxacin was conjugated with MB directly to form bulky prodrug 7 [42]. Similar reactive results were obtained (Figs. S30 and S31 in Supporting information). Collectively, these indicated that this cooperative cleavage reaction shows excellent tolerance toward bulky substrates, and the urea bond is an effective functional group for quenching the fluorescence of parent drug.
Efforts for screening possible substrates of HRP/H2O2 pair also has been made based on another profluorescent structure. Two analogs of amplex red were designed and synthesized where the phenyl acetyl (compound 8) or benzoyl (compound 9) group was used to cage the highly fluorescent resorufin (RSF, ΦF = 0.754) by an amide bond [36]. For the precursors of RSF, the characteristic red color of resorufin disappeared, forming colorless solutions (Figs. S32A and B in Supporting information). However, after being treated by HRP/H2O2, the characteristic fluorescence of RSF recovered from these two prechromophores with an obvious color change from colorless to pink, indicating the release of free RSF (Figs. S32C and D in Supporting information) [43]. Moreover, this indicated that such a lock-and-unlock strategy is both applicable to aromatic and alkyl carboxylic acids.
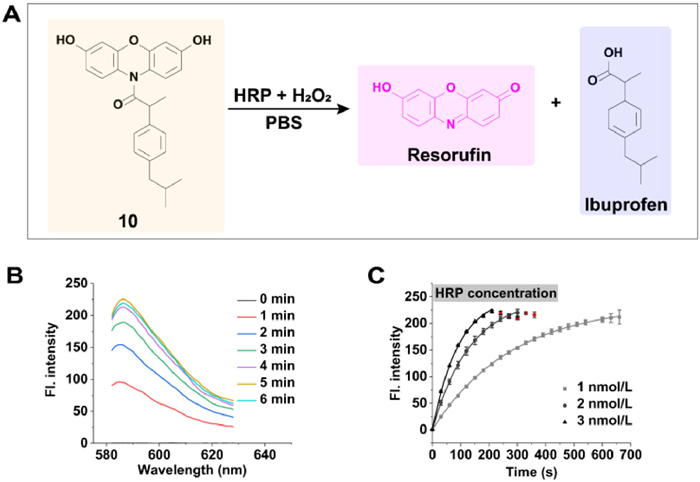
The high fluorescence of RSF and its modifiability by carboxylic acid will make it useful in theragnostic prodrug design since many drugs for ROS-relevant diseases such as non-steroidal anti-inflammatory drugs (NSAID), contains bioactive carboxyl functional group [44]. Subsequently, a theragnostic prodrug (compound 10) for ibuprofen was designed and synthesized [45]. When the amide bond was broken, the released RSF will report the release process of ibuprofen simultaneously by its strong fluorescence (Fig. 3A). As expected, free ibuprofen was released from compound 10 after being treated by HRP/H2O2, as determined by MS (Fig. S33 in Supporting information). The fluorescence of RSF initially rises rapidly and then gradually decreases, with the rate depending on the concentration of HRP (Figs. 3B and C). The decrease in fluorescence occurred because the released RSF continues to be a substrate for HRP, gradually oxidizing to become non-fluorescent resazurin in the presence of excess H2O2 [46]. However, the signal-noise ratio was extremely high with a value of 960 (from 0.23 to 222.38, HRP = 3 nmol/L), benefiting from the low background signal and high fluorescence quantum efficiency of RSF [47]. Since many drugs contain a bioactive carboxyl functional group, the kind of RSF derivatives could be an expandable prodrug platform, such as quinolone antibiotics.
Having established the chemical basis of the cooperative cleavage strategy, we next explored specific applications. Despite the virtue of efficiency and non-invasive feature, photodynamic therapy (PDT) is largely hampered by low selectivity between tumor and normal tissue which cause significant side-effects such as dermatitis [48]. To solve this problem, developing pre-photosensitizers selectively activated in tumors is a promising strategy [49-51]. Hence, compound 11 was designed and synthesized for biological application by attaching hydrophilic biotin to leuco-MB.
Not surprisingly, photosensitizer MB was released from compound 11 after being treated by HRP/H2O2 (Fig. S34 in Supporting information). In contrast, merely HRP or H2O2 led to no release of MB even after 3 h of treatment. Importantly, the yields of MB were linear related to the H2O2 level, and higher H2O2 concentration not only accelerate the release rate, but also the amount of MB, enabling targeted cell killing (Fig. 4A). Moreover, this cleavage reaction proceeded efficiently in the presence of different amounts of fetal bovine serum (FBS), showing the robustness toward other biomolecules (Fig. S35 in Supporting information).
Subsequently, cooperative cleavage strategy-induced singlet oxygen (1O2) sensitization recovery was evaluated by singlet oxygen sensor green (SOSG) in PBS [52,53]. SOSG is a specifically fluorescent 1O2 probe, which emits strong fluorescence at 525 nm after reacting with 1O2. As expected, the released free MB sensitized the generation of 1O2 effectively upon light irradiation (Figs. S36 and S37 in Supporting information). In contrast, nearly no generation of 1O2 was detected in HRP or H2O2 solely treated group.
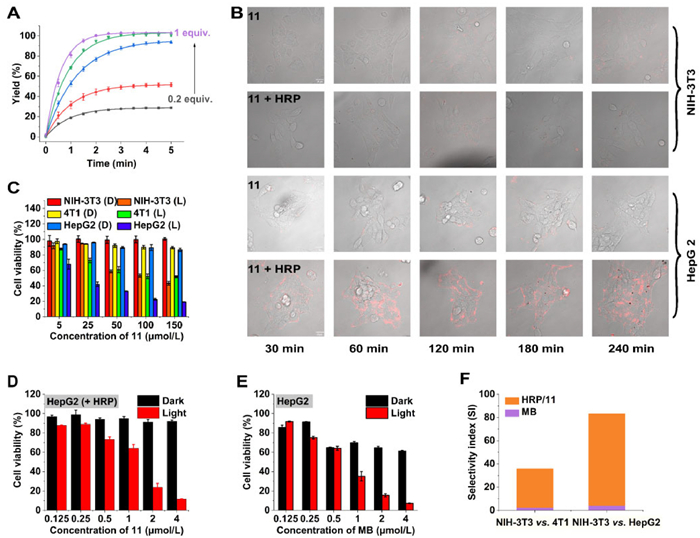
Encouraged by these results, the controllable cleavage chemistry was evaluated in living mammalian cells by fluorescent confocal imaging. HRP was incorporated in liposome by the reverse-phase evaporation vesicle method to improve its stability and bioavailability. The concentration was determined as 5 µmol/L with a 50% encapsulation efficiency. The nanoparticle was 192 nm size as determined by dynamic light scattering (DLS) (Fig. S38 in Supporting information), and the zeta potential was −36.9 mV [54,55]. Benefiting from the natural advantage of enzymatic reactions in terms of speed and HRP's excellent affinity for methylene blue precursors, the cleavage chemistry of compound 11 proceeds efficiently in tumor cells (4T1 and HepG2) by virtue of the H2O2 overgenerated by cells, as shown by the fluorescence of MB in cells (Fig. 4B and Fig. S39 in Supporting information) [56]. The fluorescence in cells increased over time and reached a maximum after 2 h, which is much faster than traditional H2O2-stimulated prodrug activation [5,57]. Importantly, the released MB shown high stability against HRP/H2O2 (Fig. S40 in Supporting information). In contrast, no detected fluorescence was observed in normal NIH-3T3 cells due to its low H2O2 concentration. In addition, all cells treated by compound 11 merely showed no detectable fluorescence due to its robustness toward endogenous H2O2 in the absence of exogenous HRP.
Then cooperatively activated phototoxicity toward both tumor and normal cells was evaluated. Guided by fluorescence of released MB, HRP/compound 11 were co-incubated with cells for 2 h to reach a maximum cellular uptake. The cells were subjected to irradiation (40 mW/cm2, 5 min) after being washed three times by PBS and replaced by fresh culture. The dark groups were treated with the same procedure without light irradiation.
In all tested cell lines, compound 11 showed excellent biosafety neither in the dark nor under irradiation (Fig. 4C). In the presence of HRP, compound 11 recovered its phototoxicity toward HepG2 cells (1.27 µmol/L) with close IC50 to free MB (0.70 µmol/L) (Fig. 4D). Similar result was observed in 4T1 cells (3.01 µmol/L vs. 1.19 µmol/L) (Fig. S41 in Supporting information). However, this enzyme/prodrug pair demonstrated much less phototoxicity toward normal NIH-3T3 cells compared to free MB (99.71 µmol/L vs. 3.09 µmol/L) (Fig. S42 in Supporting information). To evaluate the targeted effect of this enzyme/prodrug approach, the selectivity index (SI) was calculated by dividing the IC50 toward normal cell by IC50 toward the tumor cell. The SI was elevated significantly from 2.59 to 33.13 (NIH-3T3 vs. 4T1), and from 4.41 to 78.51 (NIH-3T3 vs. HepG2) separately (Fig. 4F). Both are much higher than the threshold of a selective drug (typical value = 10). Generally, this indicated that the cooperative-activated strategy showed much more specificity than free MB (~12-fold and 17-fold) and operate efficiently in living mammalian cells.
To intuitively visualize the cooperatively activated phototoxicity, calcein-AM and propidium iodide (PI) were used to distinguish live (green) and dead (red) HepG2 cells (Fig. S43 in Supporting information). Consistent with the above results, only cells co-treated with HRP/compound 11 show red fluorescence upon illumination, a marker of dead cells. In contrast, other groups (control, compound 11 in the dark, compound 11 upon irradiation, HRP/compound 11 under dark) showed strong green fluorescence, a marker of live cells, indicating the importance of enzyme and light. DCFH-DA assay further revealed that the cytotoxicity was initiated by ROS (Fig. S44 in Supporting information) [58,59].
In summary, we have firstly developed a cooperative bond cleavage strategy based on HRP and tumor abundant H2O2 for targeted and fast prodrugs activation. Urea-bond-containing precursors of MB could function as a prodrug platform for both MB and drugs containing primary or secondary amines, where urea bonds block both the functional phenothiazine structure in MB and the bioactive amine groups in drugs simultaneously. However, this kind of urea bond could be cooperatively cleaved by HRP and H2O2 to release MB and amine derivatives even with bulky hindrance. Cooperative cleavage reaction-mediated targeted phototherapy was demonstrated in HepG2,4T1, and NIH-3T3 cell lines. The selective indexes were significantly elevated (from 2.59 to 33.13 for NIH-3T3 vs. 4T1, from 4.41 to 78.51 for NIH-3T3 vs. HepG2). Mechanism study demonstrated that cancer cells were killed by 1O2 sensitized by released MB. This work demonstrated the superiority of bi-substrate enzyme reaction against traditional one-substrate enzyme reaction for prodrug activation. It may promote the development of both small-molecule prodrugs and enzymes in related fields.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank the National Natural Science Foundation of China (No. 22090011), the Fundamental Research Funds for China Central Universities (No. DUT22LAB608) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
H.H. Han, H.M. Wang, P. Jangili, et al., Chem. Soc. Rev. 52 (2023) 879–920. doi: 10.1039/D2CS00673A
Y. Xue, H. Bai, B. Peng, et al., Chem. Soc. Rev. 50 (2021) 4872–4931. doi: 10.1039/D0CS01061H
C. Choudhury, V. Kumar, R. Kumar, Eur. J. Med. Chem. 249 (2023) 115153. doi: 10.1016/j.ejmech.2023.115153
Y. Wang, D. Xiao, J. Li, et al., Signal Transduct. Target. Ther. 7 (2022) 20. doi: 10.1038/s41392-021-00833-8
J. Zhu, J. Chen, D. Song, et al., J. Mater. Chem. B 7 (2019) 7548–7557. doi: 10.1039/C9TB02041A
H. Ning, Y. Yang, C. Lv, et al., Nano Res. 16 (2023) 12294–12303. doi: 10.1007/s12274-023-5923-4
T. Xiong, Y. Chen, M. Li, et al., Sensors Actuators B: Chem. 352 (2022) 130990. doi: 10.1016/j.snb.2021.130990
J. Zheng, H. Ge, D. Zhou, et al., Adv. Mater. 35 (2023) 2308205. doi: 10.1002/adma.202308205
Y. Yang, Y. Zhang, R. Wang, et al., Chin. Chem. Lett. 33 (2022) 4583–4586. doi: 10.1016/j.cclet.2022.03.040
L. Zhou, C. Du, R. Zhang, C. Dong, Chin. Chem. Lett. 32 (2021) 561–564. doi: 10.1016/j.cclet.2020.02.043
Y. Yang, H. Ning, T. Xia, et al., Adv. Mater. 35 (2023) 2301409. doi: 10.1002/adma.202301409
T. Liu, H. Zou, J. Mu, et al., Chin. Chem. Lett. 32 (2021) 1751–1754. doi: 10.1016/j.cclet.2020.12.008
K.M. VanderMolen, W. McCulloch, C.J. Pearce, N.H. Oberlies, J. Antibiotics 64 (2011) 525–531. doi: 10.1038/ja.2011.35
G. Xu, H.L. McLeod, Clin. Cancer Res. 7 (2001) 3314–3324.
Y. Wu, W. Chen, C. Wang, D. Xing, Chin. Chem. Lett. 35 (2024) 109096. doi: 10.1016/j.cclet.2023.109096
A.B. Silver, E.K. Leonard, J.R. Gould, J.B. Spangler, Trends Pharmacol. Sci. 42 (2021) 1064–1081. doi: 10.1016/j.tips.2021.09.009
S.K. Sharma, K.D. Bagshawe, Adv. Drug Deliv. Rev. 118 (2017) 2–7. doi: 10.1016/j.addr.2017.09.009
B. Torres-Herrero, I. Armenia, M. Alleva, et al., ACS Nano 17 (2023) 12358–12373. doi: 10.1021/acsnano.3c01599
R.S. Greco O, C. Kanthou, L.K. Folkes, P. Wardman, G.M. Tozer, G.U. Dachs, Mol. Cancer Ther. 1 (2001) 151–160.
X. Zhang, Q. Yang, Y. Lang, et al., Anal. Chem. 92 (2020) 12400–12406. doi: 10.1021/acs.analchem.0c02149
A.K. Sangha, L. Petridis, X. Cheng, J.C. Smith, J. Phys. Chem. B 120 (2016) 7635–7640. doi: 10.1021/acs.jpcb.6b00789
P.P. Kelder, N.J. de Mol, M.J. Fischer, L.H. Janssen, Biochim. Biophys. Acta 1205 (1994) 230–238. doi: 10.1016/0167-4838(94)90238-0
C. Petit, K. Murakami, A. Erdem, et al., Electroanalysis 10 (1998) 1241–1248. doi: 10.1002/(SICI)1521-4109(199812)10:18<1241::AID-ELAN1241>3.0.CO;2-Q
Y. Tong, M. Li, H. Huang, et al., J. Am. Chem. Soc. 144 (2022) 16799–16807. doi: 10.1021/jacs.2c03669
S.M. Jo, H.S. Kim, M. Won, et al., Adv. Funct. Mater. 32 (2022) 2200791. doi: 10.1002/adfm.202200791
T. Meng, J. Han, P. Zhang, et al., Chem. Sci. 10 (2019) 7156–7162. doi: 10.1039/C9SC00910H
J. Peiró Cadahía, J. Bondebjerg, C.A. Hansen, et al., J. Med. Chem. 61 (2018) 3503–3515. doi: 10.1021/acs.jmedchem.7b01775
B. Yuan, H.L. Chou, Y.K. Peng, ACS Appl. Mater. Interfaces 14 (2022) 22728–22736. doi: 10.1021/acsami.1c13429
X. Zhao, S. Long, M. Li, et al., J. Am. Chem. Soc. 142 (2019) 1510–1517.
M.D. Toney, Arch. Biochem. Biophys. 544 (2014) 119–127. doi: 10.1016/j.abb.2013.10.002
M. Cui, J. Zhou, Y. Zhao, Q. Song, Sens. Actuator. B: Chem. 243 (2017) 203–210. doi: 10.1016/j.snb.2016.11.145
L. Gao, J. Zhuang, L. Nie, et al., Nat. Nanotechnol. 2 (2007) 577–583. doi: 10.1038/nnano.2007.260
M.B. Arnao, F. Garcia-Cánovas, M. Acosta, IUBMB Life 39 (1996) 97–107. doi: 10.1080/15216549600201101
P. George, Nature 169 (1952) 612–613. doi: 10.1038/169612a0
C.S. Chang, I. Yamazaki, R. Sinclair, et al., Biochemistry 32 (1993) 923–928. doi: 10.1021/bi00054a025
N. Wang, C.J. Miller, P. Wang, T.D. Waite, Anal. Chim. Acta 963 (2017) 61–67. doi: 10.1016/j.aca.2017.02.033
E. Niki, Am. J. Clin. Nutr. 54 (1991) S1119–S1124. doi: 10.1093/ajcn/54.6.1119s
T. Wang, K. Yang, Q. Tian, et al., Mol. Catal. 514 (2021) 111854. doi: 10.1016/j.mcat.2021.111854
N. Pessah, M. Reznik, M. Shamis, et al., Bioorg. Med. Chem. 12 (2004) 1859–1866. doi: 10.1016/j.bmc.2004.01.039
Y. Pommier, Nat. Rev. Cancer 6 (2006) 789–802. doi: 10.1038/nrc1977
Q. Miao, D.C. Yeo, C. Wiraja, et al., Angew. Chem. Int. Ed. 57 (2018) 1256–1260. doi: 10.1002/anie.201710727
R. Cao-Milán, S. Gopalakrishnan, L.D. He, et al., Chem 6 (2020) 1113–1124. doi: 10.1016/j.chempr.2020.01.015
Y. Li, M. Wang, F. Wang, et al., Smart Molecules 1 (2023) e20220003. doi: 10.1002/smo.20220003
M. Xiao, W. Sun, J. Fan, et al., Adv. Funct. Mater. 28 (2018) 1805128. doi: 10.1002/adfm.201805128
G. Varrassi, J.V. Pergolizzi, P. Dowling, A. Paladini, Adv. Ther. 37 (2020) 61–82. doi: 10.1007/s12325-019-01144-9
J.G. Mohanty, J.S. Jaffe, E.S. Schulman, D.G. Raible, J. Immunol. Methods 202 (1997) 133–141. doi: 10.1016/S0022-1759(96)00244-X
M. Yang, J. Fan, J. Zhang, et al., Chem. Sci. 9 (2018) 6758–6764. doi: 10.1039/C8SC02578A
A.P. Silva, C.L. Neves, E.D.A. Silva, et al., Photodiagnosis Photodyn. Ther. 23 (2018) 154–164. doi: 10.1016/j.pdpdt.2018.06.012
F. Xu, H. Li, Q. Yao, et al., Chem. Sci. 10 (2019) 10586–10594. doi: 10.1039/C9SC03355F
Y. Chen, T. Xiong, X. Zhao, et al., Adv. Healthc. Mater. 12 (2023) 2202085. doi: 10.1002/adhm.202202085
D. Huang, H. Huang, M. Li, et al., Adv. Funct. Mater. 32 (2022) 2208105. doi: 10.1002/adfm.202208105
L. Yu, Y. Xu, Z. Pu, et al., J. Am. Chem. Soc. 144 (2022) 11326–11337. doi: 10.1021/jacs.2c03256
X. Huang, R. Gu, J. Li, et al., Sci. China Chem. 63 (2019) 55–64.
H. Gu, W. Sun, J. Du, et al., Smart Molecules 2 (2024) e20230014. doi: 10.1002/smo.20230014
W. Liu, B. Wu, W. Sun, et al., Chem. Eng. J. 471 (2023) 144530. doi: 10.1016/j.cej.2023.144530
Q. Chen, C. Liang, X. Sun, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 5343–5348. doi: 10.1073/pnas.1701976114
E.J. Kim, S. Bhuniya, H. Lee, et al., J. Am. Chem. Soc. 136 (2014) 13888–13894. doi: 10.1021/ja5077684
H. Huang, D. Ma, Q. Liu, et al., CCS Chem. 4 (2022) 3627–3636. doi: 10.31635/ccschem.021.202101479
S. Luo, C. Liang, Q. Zhang, P. Zhang, Chin. Chem. Lett. 34 (2023) 107666. doi: 10.1016/j.cclet.2022.07.009

Scheme 1 Cooperative activation of prodrug by exogenous enzyme (HRP) and endogenous biomarker (H2O2) based on bi-substrate (prodrug and H2O2) enzyme reaction.

Figure 1 Cleavage of urea bond within compound 1 by HRP in the presence of H2O2. (A) Scheme illustration of decomposition of compound 1 by HRP/H2O2. (B) HPLC analysis of compound 1 with different treatments. Blue line: control. Green line: 5 nmol/L HRP, 50 µmol/L H2O2, 10 min. Yellow line: 5 nmol/L HRP, 3 h. Orange line: 50 µmol/L H2O2, 3 h. (C) HPLC analysis of compound 1 treated by different dose H2O2 with fixed HRP concentration. (D) Dynamic analysis of the cleavage reaction calculated by MB release rate. (E) Km for compound 1 and H2O2 calculated by Michaelis-Menten plots. Mean ± standard deviation (SD), n = 3.

Figure 2 Mechanism study of the reaction between HRP, H2O2, and compound 1. (A, B) Double-reciprocal plots of activity of HRP at a fixed concentration of one substrate versus varying concentration of the second substrate for H2O2 and compound 1. (C) The absorbance spectrum of HRP, HRP compound I, HRP compound I + compound 1 (1 equiv.). (D) The absorbance spectrum of compound 1, HRP, HRP + compound 1 (1 equiv.). (E) The absorbance spectrum of HRP compound I, HRP compound II (generated by self-decomposition of HRP compound I), HRP compound II + compound 1 (1 equiv.). (F) Cleavage of urea bond in compound 1 (5 µmol/L) by HRP/H2O2 (1 nmol/L + 10 µmol/L) in the presence of different dose of radical scavenger ascorbic acid (from 0 to 2 equiv. with an interval of 0.5 equiv.).

Figure 3 Cleavage of amide bond within compound 10 by HRP in the presence of H2O2. (A) Scheme illustration of decompostration of compound 10 by HRP/H2O2. (B) Fluorescence spectrum of 5 µmol/L compound 10 after coincubation with HRP/H2O2 (2 nmol/L, 10 µmol/L) for different times. (C) Fluorescence intensity change of compound 10 (5 µmol/L) at 585 nm in the presence of different amounts of HRP and a fixed amount of H2O2 (10 µmol/L). The red dots represent resorufin being oxidized to non-fluorescent resazurin. Mean ± SD, n = 3.

Figure 4 Cooperative cleavage strategy for targeted phototherapy. (A) Release of MB for 5 µmol/L compound 11 in the presence of 1 nmol/L HRP and different amounts of H2O2. (B) Fluorescence of MB in NIH-3T3 and HepG2 cells treated with 10 µmol/L compound 11 with/without 100 nmol/L HRP. Scale bar: 20 µm. (C) Photo and dark toxicities of compound 11 against NIH-3T3 (normal cell), 4T1 (cancer cell) and HepG2 (cancer cell). D: Dark, L: Light. (D) Photo and dark toxicities of HRP/compound 11 against HepG2 cells. (E) Photo and dark toxicities of MB against HepG2 cells. (F) Selectivity index of MB and HRP/compound 11 between different cell line. Mean ± SD, n = 3.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: