State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body, College of Mechanical and Vehicle Engineering, Hunan University, Changsha 410082, China
b.
CAS key Laboratory of Design and Assembly of Functional Nanostructures, and Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
c.
College of Materials Science and Engineering, Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing Forestry University, Nanjing 210037, China
d.
Laboratory of Advanced Spectro-electrochemistry and Li-ion Batteries, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
e.
Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
f.
Xiamen Key Laboratory of Rare Earth Photoelectric Functional Materials, Xiamen Institute of Rare Earth Materials, Haixi institutes, Chinese Academy of Sciences, Xiamen 361021, China
Received Date:
21 January 2024 Accepted Date:
07 February 2024 Available Online:
15 January 2025
Abstract:
Phosphorus-based anode is a promising anode for sodium-ion batteries (SIBs) due to its high specific capacity, however, suffers from poor electronic conductivity and unfavorable electrochemical reversibility. Incorporating metals such as copper (Cu) into phosphorus has been demonstrated to not only improve the electronic conductivity but also accommodate the volume change during cycling, yet the underline sodiation mechanism is not clear. Herein, take a copper phosphide and reduced graphene oxide (CuP2/C) composite as an example, which delivers a high reversible capacity of > 900 mAh/g. Interestingly, it is revealed that the native oxidation PO components of the CuP2/C composite show higher electrochemical reversibility than the bulk CuP2, based on a quantitative analysis of high-resolution solid-state 31P NMR, ex-situ XPS and synchrotron X-ray diffraction characterization techniques. The sodiation products Na3PO4 and Na4P2O7 derived from PO could react with Na-P alloys and regenerate to PO during charge process, which probably accounts for the high reversible capacity of the CuP2/C anode. The findings also illustrate that the phosphorus transforms into nanocrystalline Na3P and NaP alloys, which laterally shows crystallization-amorphization evolution process during cycling.
In the past decade, lithium-ion batteries (LIBs) have been widely applied in portable electronics, electrical vehicles and other energy storage devices. To meet the demands with further increasement of low-cost and large-scale energy storage devices, sodium ion batteries (SIBs) or sodium-sulfur (Na-S) batteries are considered as promising candidate because of its abundant reserves and even distribution of sodium resource in earth crust [1–3]. Compared with Cu foil as anode current collectors for LIBs, Al foil that applied in SIBs is cost effectiveness. Although the fundamental mechanism is almost the same with LIBs, the molar mass of Na is heavier that is 23.00 compared to 6.94 (Li) and the atomic diameter is larger that is 1.02 Å regarded to 0.76 Å (Li-ion), which not only sacrifices in gravimetric and energy density of SIBs but also induces different sodium mechanism, such as complex electrochemical reactions and relatively low reaction kinetics [4,5].
To develop high-energy SIBs, it is significant to design high-capacity and appropriate-redox-potential electrode materials. However, suitable high-performance anode materials for SIBs are still challenging [6,7]. Recently, red phosphorus (rP) as anode has been paid much attention to because of its highest theoretical capacity of 2596 mAh/g (in view of reaction of P with three Na+ to form Na3P) [8,9]. But its low intrinsic electronic conductivity and large volume swelling during cycling are to the disadvantage of application [10]. Introducing metal to phosphors to form metal phosphides (MPx) is a proven method to mitigate the issues, for example Cu, Sn, and Ge [11–13]. Carbon coating, such as hard carbon, multilayer graphene (mG) and carbon nanotubes were also widely applied to enhance the conductivity and release the mechanical stress during cycling [14–17]. Additionally, the discharge product of MPx-Na3P has been investigated by X-ray diffraction (XRD), high resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) [18–22]. The deficiencies of above conventional diffraction measurements are difficult to obtain information about amorphous phase, thus the electrochemical sodium storage mechanisms during (dis)charging are not fully understood.
In this work, a copper phosphide and reduced graphene oxide (CuP2/C) composite material was synthesized by a simple ball milling method, which show a high reversible specific capacity of > 900 mAh/g. Taking this composite anode as a sample, the sodiation and desodiation mechanism and quantitative phase transformation CuP2/C were explored and revealed by the techniques of synchrotron XRD, ex-situ XPS and solid-state NMR. For the first time, it is found that the native POx on the surface of CuP2/C composite show higher electrochemical reversibility than the bulk CuP2, due to the existence of Cu catalyst. The POx firstly sodiates and forms Na3PO4 and Na4P2O7 during the first discharge process, after that the above sodiation products react with Na-P alloys and recover to POx during charge process. The reversible phase transformation of POx accounts for the high reversible capacity of the CuP2/C composite anode. The findings also illustrate that the phosphorus transforms into nanocrystalline NaP, Na3P and NaxP alloys, among which the size of nanocrystals becomes smaller along cycling and shows crystallization-amorphization evolution process during long-life test. Our work sheds light on the material design of phosphorus-based anode with high capacity and electrochemical reversibility toward high energy density SIBs.
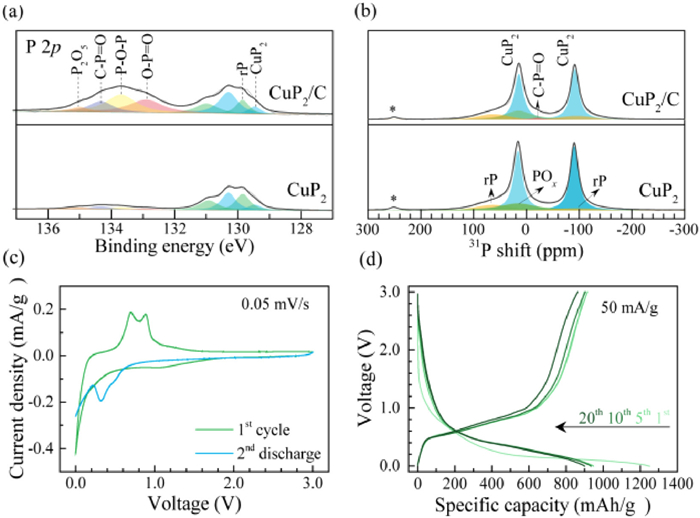
The synthesis details of CuP2/C are provided in Experimental Section in Supporting information. For simplicity, CuP2/C is firstly taken as an example for illustration. Fig. S1a (Supporting information) shows the XRD patterns of CuP2 and CuP2/C materials, all peaks can be assigned to CuP2 phase (JCPDS No. 076–1190) with a monoclinic lattice structure [23]. Compared with CuP2, the CuP2/C composite exhibits almost the same diffraction peaks, implying the ball milling process does not change the crystalline nature of CuP2. To investigate the chemical composition and bonding state of above materials, XPS was carried out. In P 2p spectra in Fig. 1a, there is a broad peak at around 130 eV for both samples, which is matched with the CuP2 and pristine rP. Additionally, broad peaks appear at 132.9, 133.7 and 135.0 eV due to the surface oxidation of rP, the first two peaks indicate the formation of P−O−P and O−P=O bonds [24], and the last one belongs to P2O5. The P2O5 was obtained during the ball milling process, which is inevitably incorporating some extent of air. At the same time, the size becomes smaller and the surface area increase, as a result, the reactivity between P and O increases and forms some native P2O5 on the surface. The peak at 134.3 eV corresponds to C−P=O bond, in which the C element is probably due to the inevitable C-containing contaminants in CuP2. Conspicuously, above four peaks all intensified in the CuP2/C composite. The XPS Cu 2p spectra in Fig. S1b (Supporting information) shows the two characteristic peaks, which are associated with Cu(Ⅰ) 2p3/2 (932.9 eV) and 2p1/2 (952.7 eV), respectively [25]. Solid-state 31P NMR spectra in Fig. 1b were collected to present a quantitative understanding about the materials. Two intense peaks located at 15 and −91 ppm is observed for both samples, corresponding to two P sites in the crystal structure of CuP2 as illustrated in Fig. S2 (Supporting information). Another two broad resonances at ~62.7 and −95.2 ppm is ascribed to the unreacted rP. A broad peak located at ~15 ppm attributed to the P−O bonding is distinguished. A weak peak at ~−20 ppm detected in CuP2/C composite is assigned to the C−P=O bond, which is produced during the ball-milling process by the reaction of reduced graphene oxide and POx. The morphologies of CuP2 and CuP2/C materials are characterized by SEM and TEM. In Fig. S1c (Supporting information), CuP2 is composed of irregular shape of particles in a range of 300−600 nm. While for CuP2/C composite in Fig. S1d (Supporting information), smaller particles with a diameter of 100−300 nm observed, which should be generated during ball milling process. High resolution TEM (HRTEM) image in Fig. S1e (Supporting information) manifests that CuP2/C composite has a crystal core with amorphous carbon shell. The marked parallel fringes have the space of ~0.289 nm, corresponding to the (112) plane of CuP2, which is in good agreement with the XRD patterns. Such a carbon layer will enhance the electronic conductivity and accommodate the volume variation during (dis)charge.
Figure 1
Figure 1.
The physical characterization of as-synthesized CuP2 and CuP2/C materials: (a) XPS P 2p spectra and (b) solid-state 31P NMR spectra. The asterisks indicate the spinning sidebands. The electrochemical profiles of CuP2/C material as anode for SIBs: (c) CV curves at a scan rate of 0.05 mV/s and (d) electrochemical profiles during cycling at 50 mA/g. The specific capacity and applied current densities are based on the mass of CuP2 material exclusive of carbon.
The electrochemical profiles were firstly evaluated by CV between 0 and 3 V at 0.05 mV/s versus Na+/Na and presented in Fig. 1c. During the initial cathodic sweep, there is a broad wave ranging from 1.6 V to 0.7 V, which is indicative of the decomposition of electrolyte and the formation of SEI (solid electrolyte interphase) layer on the fresh electrode. Upon further sodiation to 0 V, the current density increases continuously, stating that the occurrence of electrochemical sodiation on CuP2/C electrode. In the ensuing anodic sweep, two pronounced peaks centered at 0.70 and 0.89 V correspond to multi-step desodiation process from NaP and Na3P to P. By contrast, in the second discharge process, a peak located at 0.32 V appears contributed to the sodiation of amorphous rP, and the broad wave (1.6−0.7 V) disappears due to the absence of SEI contribution, which is different from the first cycle. The specific capacity is calculated based on the total mass of CuP2, POx and carbon, the mass loading is about 2 mg/cm2 on each of the discs for performance tests and 4 mg/cm2 for synchrotron XRD, ex-situ NMR characterization, respectively. Fig. 1d shows the voltage profiles of CuP2/C electrode at a current density of 50 mA/g, the specific capacity of the initial discharge can reach up to 1200 mAh/g (normalized to the mass of CuP2 exclusive of carbon), and roughly 900 mAh/g is recovered when the electrode is fully charged. Notably, the reversible capacity of ~900 mAh/g is lower than the theoretical value of 1281 mAh/g according to the formation of Na3P, which has been reported in the P-based sodium storage materials [26–31]. The rate performance and high rate cycling were also tested. As shown in Fig. S3 (Supporting information), the specific capacities of CuP2/C at 0.05, 0.2, 0.5, 1, and 2 A/g are 950, 880, 815, 708 and 595 mAh/g, respectively. Even at a high rate of 1 A/g, the CuP2/C anode could deliver a reversible capacity of 550 mAh/g after 50 cycles (Fig. S4 in Supporting information). With the purpose of elucidating the underlying sodiation/desodiation mechanism of CuP2/C composite, we disassemble the coin cells at certain (dis)charged states and track the phase evolution by ex-situ studies.
To investigate the reaction mechanism of CuP2/C composite as anode for SIBs, the crystal structure variation during (dis)charge process is researched by synchrotron XRD (Fig. S5 in Supporting information). The pattern of each state shows no pronounced peaks ascribed to other crystal phases as (dis)charge proceeds, indicating that amorphous phases present in addition to the residual CuP2 material.
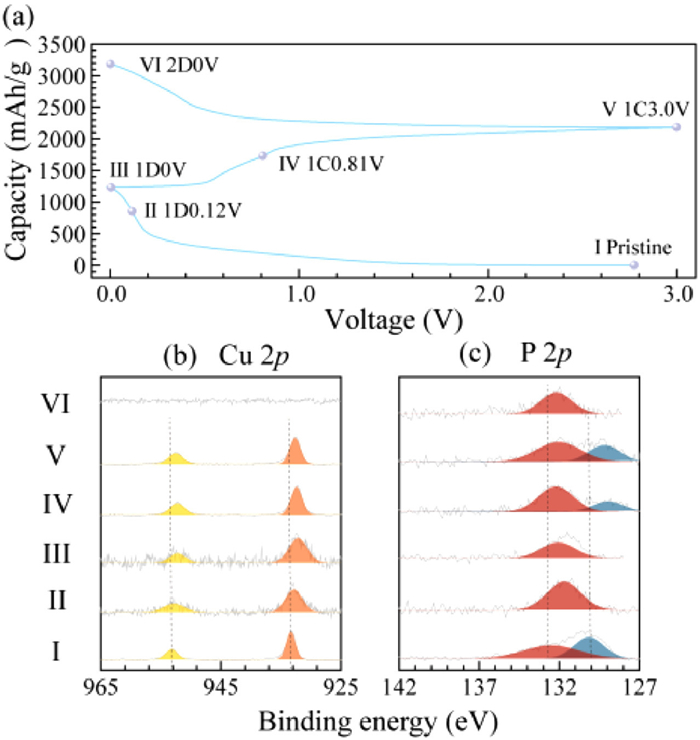
The surface oxidation states of the CuP2/C electrode at selected voltage points during cycling, as shown in Fig. 2a, were investigated by ex-situ XPS. Fig. 2b displays the XPS spectra of Cu 2p at (dis)charged states: the pristine electrode featuring Cu(Ⅰ) 2p1/2 (953.1 eV) and Cu 2p3/2 (933.2 eV) [25]. As discharge goes on, the peaks of Cu 2p all move to lower Eb. When fully charged to 3 V (Ⅵ), all of peaks shifted in the higher Eb direction, implying the occurrence of reversible electrochemical reaction. And at 2D0V (discharged to 0 V in the second discharge process, similarly hereinafter) state, it is distinct that no peaks of Cu(Ⅰ) are observed compared with 1D0V state, indicating possibly that the influence of high S/N (signal/noise) ratio. In Fig. 2c, the P 2p spectra at different voltages display a peak centered at ~132 eV, which is attributed to the sodium phosphorus intermediates, NaxP (x = 1–3) [32]. The peak at ~130 eV is corresponding to the rP and CuP2, which is consistent with the result in Fig. 1a.
Figure 2
Figure 2.
(a) The electrochemical profiles of CuP2/C electrodes for SIBs at 25 mA/g. (b) XPS Cu 2p spectra and (c) XPS P 2p spectra.
In addition, Cu-K edge XAS analysis was employed to further resolve the structural ambiguities of above weak XRD patterns and to probe the structural variations around Cu ions during electrochemical process of the CuP2/C electrode. In the X-ray absorption near-edge structure (XANES) region (Fig. S6a in Supporting information), the pristine electrode exhibited a CuI-like characteristic feature with a peak located at ~8986 eV assigned to a single bond of 1s → 4pπ*, the intensity of which suggests the fourfold coordination geometry of the Cu site. This hypothesis is evidenced by the structure presentation of CuP2 as illustrated in Fig. S2. For CuI, the transition shows highest intensity for the linear two-coordinated complexes and becomes less intense and broaden as the coordination number increases [33]. This peak gradually becomes weak with an increase of the depth of discharge. Upon subsequent charge, the XANES feature returned incompletely to its initial state. The second discharge process is similar with the first one. More interestingly, the radial distribution functions of the Fourier-transformed (FT) k2-weight extended X-ray absorption fine structure (EXAFS) spectra (Fig. S6b in Supporting information) present a distinct geometric structure variation around the central Cu ion. Yet surprisingly, an apparent peak at ~2 Å corresponding to the Cu-O shell is viewed. Upon discharge, this peak shifts to the higher r space region, indicating a distinct elongating of the bond length of the CuP2/C electrode. During the consecutive charge process, the structure is reversibly recovered. Ex-situ XRD was performed to further confirmed the crystalline phase transformation of CuP2/C electrode for the first cycle and second discharge processes (Fig. S7 in Supporting information). In Fig. S5a, the diffraction intensity of crystalline CuP2 decrease during the discharge process, implying the CuP2 transforms into sodiation products. Even after recharged to 3 V, no obvious diffraction peak was observed, demonstrating the CuP2 becomes amorphous after initial cycle. Expectedly, no crystalline peak was detected during the second discharge process (Fig. S5b). The ex-situ XRD results further confirms the phase transformation of CuP2 during charge and discharge. The crystalline phase transformation of CuP2/C during cycling was revealed while the amorphous phases needed to be further explored.
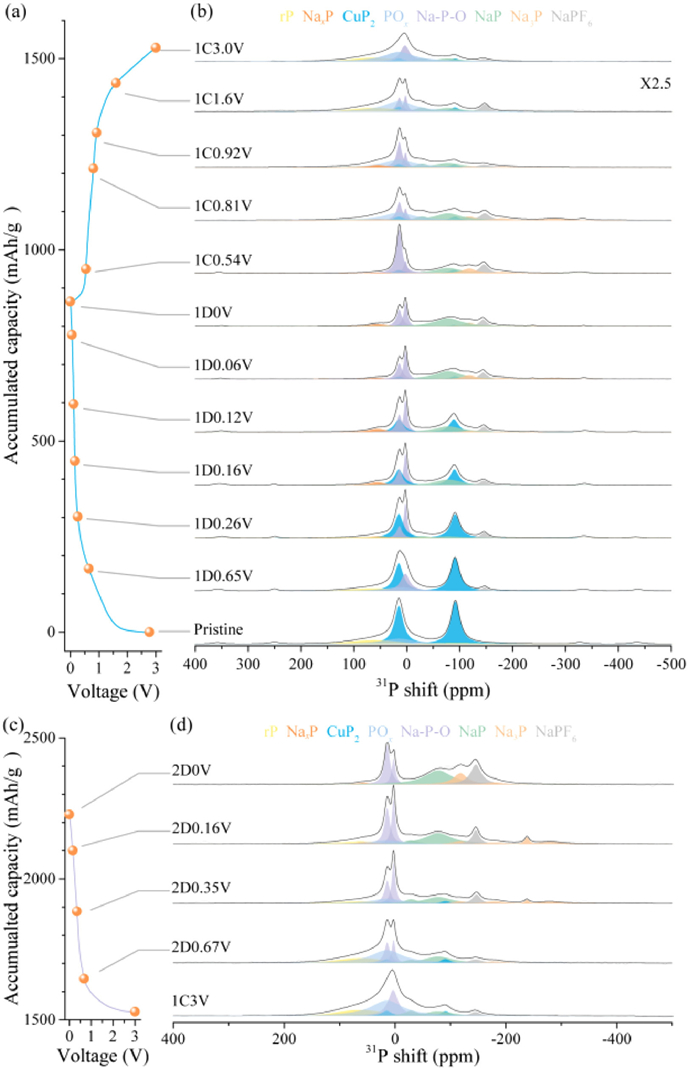
To elucidate the structural evolution including crystal and amorphous states of the CuP2/C composite during cycling, the usage of solid-state 31P NMR (Fig. 3) was conducive to circumvent the disadvantage of the traditional diffraction method, and quantify the variation of amorphous products. Accordingly, the peaks at −91 and 15 ppm are ascribed to CuP2 phase, the areal ratio of which is around 1:1. The peaks at 14.1 and 2.5 ppm are Na3PO4 and Na4P2O7, respectively [34,35]. In this case, with the Cu catalyst, the POx on the surface tends to be electrochemical active and transforms into Na-P-O at the early stage of the sodiation process, such as 1D0.65 V. With the sodiation process continues, the Na-P-O reacts with more Na atoms and forms Na3PO4 and other being Na4P2O7 products, as a result, the peak the transition state of phosphates is divided into two peaks. Red phosphorus shows two relatively broad peaks with FWHM of ~100 ppm, which are located at −95.2 and 62.7 ppm [36]. The presence of two environments of associated 31P shifts at −29 and −78 ppm that are correspond to amorphous NaP. While for Na3P, there are four environments of shifts at −118, −180, −238 and −280 ppm. Thereinto, the peak of −238 ppm with width of 10 ppm is probably originated in the NaxP species formed in side reactions, minor NaxP intermediates, or Na near P defects (such as the end of a P chain) [37]. Based on above discussion, we can analysis each ex-situ state during cycling.
Figure 3
Figure 3.
Tracking the structural evolution of CuP2/C electrodes for SIBs in the first cycle by solid-state 31P NMR spectroscopy: (a) The electrochemical profiles of the CuP2/C electrode at a current of 25 mA/g. (b) Solid-state 31P NMR spectra of the CuP2/C electrodes at selected states. Tracking the structural evolution of CuP2/C electrodes for SIBs in the second discharge process by solid-state 31P NMR spectroscopy: (c) The electrochemical profiles of the CuP2/C electrode at a current of 25 mA/g. (d) Solid-state 31P NMR spectra of the CuP2/C electrodes at selected states.
Solid-state 31P NMR spectra were further collected to gain a quantitative understanding on the prepared materials. The content of POx on the surface was determined by NMR fitting analysis. Two intense 31P NMR peaks located at 15 and −91 ppm, corresponding to two P sites in CuP2. Two other broad resonances at 62.7 and −95.2 ppm which are due to the unreacted rP as shown in the fitting plots. And a broad peak located at ~15 ppm is distinguished and attributed to the P–O bonding. It is noted that the fitting analysis indicates that the atomic ratio of P in CuP2, rP and P–O bonds (POx) is around 7:1.5:1.5. Also, the contents keep dropping as discharge proceeds on. Additionally, the peak of C−P=O bond shows the shift at −21 ppm with a width of 25 ppm, which is located at the same position of POx. During the first discharge, when the voltage is downs to 0.65 V (1D0.65 V, discharged to 0.65 V in the first discharge process, similarly hereafter), there is a peak with FWHM of ~20 ppm at 2.9 ppm accompanied with the vanishment of C−P=O bond, which is attributed to the transition state of phosphates. Then in the state of 1D0.26 V, the peak of mentioned transition state is divided into two peaks. One is the peak at 14.1 ppm with width of 12 ppm of Na3PO4, the other is the peak at 2.5 ppm with width of 8 ppm of Na4P2O7. It is worth mentioning that the content of Na3PO4 gradually increases but that of Na4P2O7 is almost unchanged. Meanwhile, the phases of NaxP and amorphous NaP (a-NaP) are observed along with the disappearance of POx. The peak of NaxP centers at 55 ppm with width of 40 ppm. In the next state (1D0.16 V), rP vanishes. When the voltage discharged to 0.12 V (1D0.12 V), the phase of Na3P generates, three of which could be viewed (shifts at −118, −180, −238 ppm with FWHM of 30, 40, 10 ppm, respectively). Above the contents of a-NaP and Na3P all increase with discharged depth raising. For 1D0.06 V, fourth peak of Na3P appears at −280 ppm with width of 50 ppm. In short, the discharge process is linked to a conversion of CuP2, rP and POx to Na-P alloys and phosphates. To demonstrate the role of Cu on the electrochemical reversibility of rP, the NMR data of P/C during charge and discharge was carried out. As shown in Fig. S8 (Supporting informatio), the POx peak remains almost the same during initial discharge and charge processes, indiacating that the POx is electrochemical inactive, which is totally different with the CuP2 results. Based on the above results, it is concluded that, the native oxide POx on the surface of CuP2/C becomes electrochemical active and reversible under the existence of Cu catalyst.
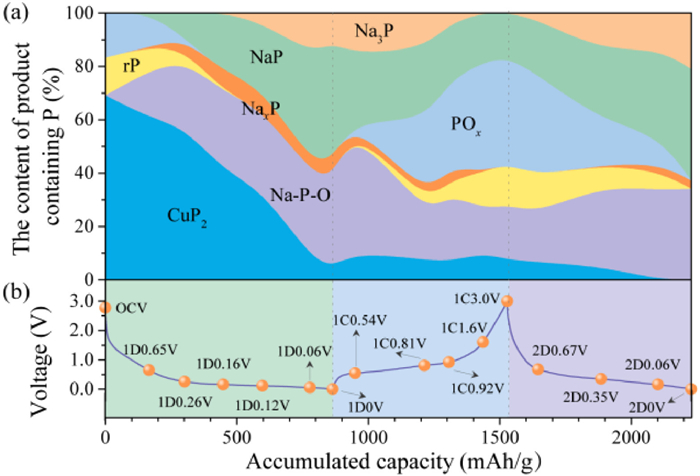
To better understand the sodiation/desodiation mechanism of CuP2/C, based on the 31P NMR spectra during the galvanostatic (dis)charge processes, the quantitative phase transformation diagram is analyzed (detailed in Table S1 in Supporting information) and shown in Fig. 4. Generally speaking, the diagram exhibits the reversibility of CuP2, rP and POx, especially for the high electrochemical reversibility of POx. After the first desodiation, only a small ratio of CuP2 convert to the initial state but POx shows high reversibility. It should be noted that the content of POx is higher than the pristine CuP2/C before cycling, which may be caused by atomic structure reconstruction between P and O atoms during cycling. Instead, the Na-O-P and Na-P alloy derived from CuP2 convert to POx, rP and CuP2, which should be the main reason responsible for the high reversibility of the unique CuP2/C anode. Besides, after the initial desodiation process, the residual Na-P alloy such as NaxP, Na3PO4 and Na4P2O7 may contribute to the irreversible capacity loss and low Coulombic efficiency. It is suggested that the mechanical stress caused by volume expansion could be regulated by controlling the content of POx in the phosphorus-based anodes. In addition, reducing the content of NaxP, Na3PO4 and Na4P2O7 is expected to improve the reversibility and Coulombic efficiency. For the second cycle, the contents of NaP and Na3P alloy increased compared to the initial cycle, which may be induced by the particles fracture and converting into nanosized P, thus enabling fast sodiation kinetics. During the sodiation process of rP, NaxP and NaP forms at the same time with the disappearance of POx. With the sodiation continues, more Na atoms insert into and forms more NaP and NaxP. At the end of sodiation process, some extent of NaxP trnaforms into Na3P. The NaP and Na3P tend to be amorphous and degree of disorder increase along repeated cycles.
Figure 4
Figure 4.
Phase transformation diagram for the CuP2/C composite with the galvanostatic (dis)charge processes: (a) The contents of produced Na-P alloys and Na-P-O compounds can be directly read in the Y axis, depending on the deconvolution results of ex-situ31P NMR spectra in Fig. 3. (b) The electrochemical profiles of the CuP2/C electrode cycling at a current of 25 mA/g.
In summary, combination of synchrotron XRD, ex-situ XPS and NMR, it is revealed that native oxidation components such as POx of the CuP2/C composite show better electrochemical reversibility even than the bulk CuP2. The surface POx firstly sodiate and generate Na3PO4 and Na4P2O7 during charge process, and substantially react with Na-P alloys and recover to the POx during the desodiation process. This is a critical factor that accounts for the high reversible capacity of 900 mAh/g for the CuP2/C composite. It is also demonstrated that the formation of amorphous Na3P and NaxP along repeated cycles. This work for the first time provides a quantitative understanding on the electrochemical sodiation mechanism of the CuP2/C composite, which shed light on a new road to design high-capacity P-based anode materials for sodium storage by manipulating the content and bonding nature between P and oxygen.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by National Nature Science Foundation of China (Nos. 21805278, 22272175 and 22209075), the Fujian Science and Technology Planning Projects of China (Nos. 2022T3067 and 2023H0045), the Self-deployment Project Research Programs of Haixi Institutes, Chinese Academy of Sciences (No. CXZX-2022-JQ12), the Self-deployment project of XIREM (No. 2023GG02).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109650.
Na-Ion batteries K. Kubota, S. Komaba, S. Passerini, D. Bresser, A. Moretti, A. Varzi, Encyclopedia of Electrochemistry Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, 2020, pp. 1–64.
[5]
N. Yabuuchi, K. Kubota, M. Dahbi, et al., Chem. Rev. 114 (2014) 11636–11682. doi: 10.1021/cr500192f
L.E. Marbella, M.L. Evans, M.F. Groh, et al., J. Am. Chem. Soc. 140 (2018) 7994–8004. doi: 10.1021/jacs.8b04183
Figure 1
The physical characterization of as-synthesized CuP2 and CuP2/C materials: (a) XPS P 2p spectra and (b) solid-state 31P NMR spectra. The asterisks indicate the spinning sidebands. The electrochemical profiles of CuP2/C material as anode for SIBs: (c) CV curves at a scan rate of 0.05 mV/s and (d) electrochemical profiles during cycling at 50 mA/g. The specific capacity and applied current densities are based on the mass of CuP2 material exclusive of carbon.
Figure 3
Tracking the structural evolution of CuP2/C electrodes for SIBs in the first cycle by solid-state 31P NMR spectroscopy: (a) The electrochemical profiles of the CuP2/C electrode at a current of 25 mA/g. (b) Solid-state 31P NMR spectra of the CuP2/C electrodes at selected states. Tracking the structural evolution of CuP2/C electrodes for SIBs in the second discharge process by solid-state 31P NMR spectroscopy: (c) The electrochemical profiles of the CuP2/C electrode at a current of 25 mA/g. (d) Solid-state 31P NMR spectra of the CuP2/C electrodes at selected states.
Figure 4
Phase transformation diagram for the CuP2/C composite with the galvanostatic (dis)charge processes: (a) The contents of produced Na-P alloys and Na-P-O compounds can be directly read in the Y axis, depending on the deconvolution results of ex-situ31P NMR spectra in Fig. 3. (b) The electrochemical profiles of the CuP2/C electrode cycling at a current of 25 mA/g.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
