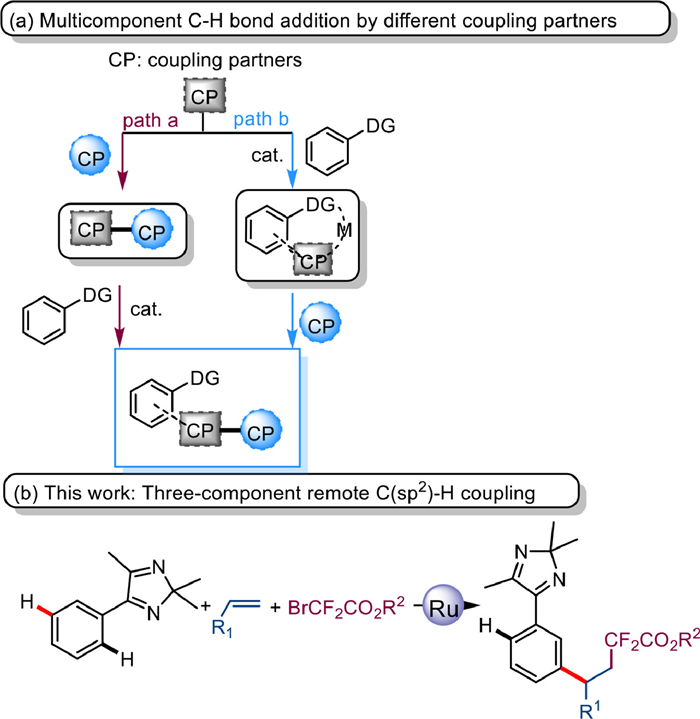
Scheme 1.
Multicomponent sequential C—H addition with two different coupling partners in C—H functionalization.

As a multipurpose synthetic method with high atom economy, C—H functionalization of the ubiquitous C—H bond enables various functional couplings catalyzed by transition metals, leading to its wide application in synthetic chemistry [1–5]. Considering the limitation of two-component C—H functionalization, an effective combination with a multicomponent strategy would be highly desirable. Multicomponent reactions (MCRs) provide convenient and comprehensive synthetic routes by assembling various heterocyclic scaffolds from simple starting materials, although they are trapped in the precise regulation of the reaction sequence [6–10].
The most recent primary approach to the modular construction of molecular complexity is the sequential addition of multiple components that forms potential stereo centers and performs various chemical combinations. This efficient transformation occurs in two ways: formation of a new coupling partner from two different coupling partners, followed by C—H functionalization (Scheme 1a, path a) [11–15]. The other way is to extend the reaction by introducing a second different coupling partner after activation of the C—H bond and addition to the first coupling partner (path b) [16–22].
In addition, the 2H-imidazole moiety embedded in drugs and active molecules is an important intermediate in synthesis [23–29]. Our group pioneered the use of 2H-imidazole as a lead group for performing various annulations in the field of C—H activation [30–32]. In view of this, the elaboration of a modular design approach to increase the complexity of 2H-imidazole derivatives is of great research value. Inspired by the above research progress, we have disclosed the Ru-catalyzed multicomponent addition reaction of 2H-imidazoles with bromodifluoroacetates via an olefin as a bridge with a long C(sp2)-H bond (Scheme 1b), which provides an efficient approach for the alkyl arylation of 2H-imidazoles with good substrate compatibility and tolerance to functional groups.
At the beginning of our studies, we investigated the coupling reaction of 2H-imidazoles 1a, 4-methoxystyrene 2a and bromodifluoroacetates 3a (Table 1). First, we investigated the ligands required for the reaction, and only in phosphine ligand systems containing strong electron-withdrawing groups could traces of coupling products 4 (entries 1–5) be detected. Considering the importance of ligands for metal catalysts, we prepared a series of ruthenium catalysts with different anionic ligands based on literature reports to screen the reaction conditions (entries 6–8) [33,34], and Ru(OAc)2(p-cymene) showed the optimal catalytic activity. Subsequent tests with different alkaline additives showed that potassium carbonate was best suited for this reaction (entries 9–13). Further heating was beneficial for the progress of the reaction, yielding 4aa and 4aa' in an overall yield of 76% (entry 14). Increasing the amount of catalyst led to a slight improvement in reaction efficiency (entry 15). In addition, ether solvent and toluene did not show any better effect compared to DCE (entries 16–18). Control experiments showed that ruthenium catalysts and phosphine ligands were essential for the reaction (entries 19 and 20).
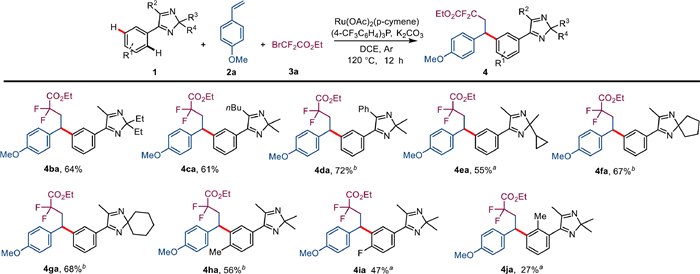
Under the best conditions, we investigated the possibilities of 2H-imidazoles for their suitability as three-component coupling products 4 (Scheme 2). The introduction of ethyl, butyl and phenyl substituents had no significant effect on the reaction efficiency and afforded 4ba-4da in 61%–72% yields. In addition, the cycloalkanes also participated in the reaction and afforded the compounds 4ea-4ga in 55%–68% yields. The incorporation of electron donating or withdrawing groups in the para position of the aromatic ring enabled the conversion of the substrates 1h-1i into the corresponding products 4ha-4ia in 47%–56% yields. Unfortunately, the introduction of substituents in the ortho position of the aromatic ring led to a significant decrease in reaction efficiency, resulting in the product 4ja in only 27% yield.
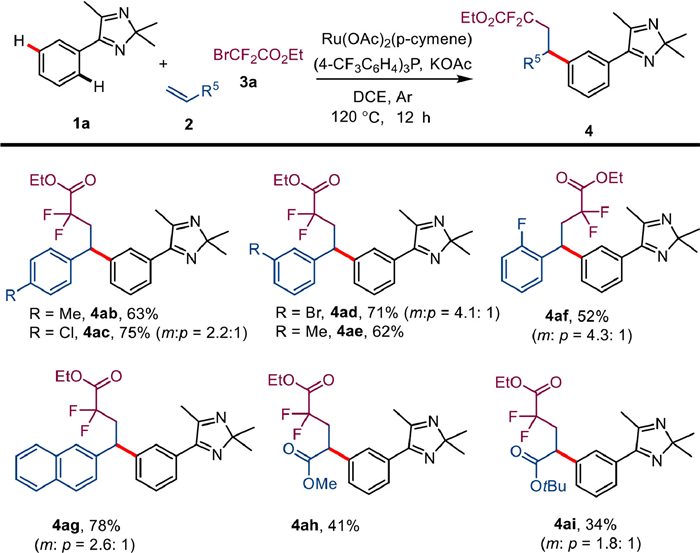
We then turned our attention to the substrate region of the olefins (Scheme 3). By introducing substituents in the para or meta positions of the aromatic styrene rings, the corresponding compounds 4ab-4ae can be obtained in good yield. However, the reaction activity of styrene containing fluorine in the ortho position was lower, so that the product 4af was obtained in only 52% yield due to steric hindrance. The positional selectivity of products 4ab and 4ae was consistent, as both afforded the single meta products, while substrates 2c, 2d, and 2f with electron-withdrawing groups afforded the regional isomers 4ac', 4ad', and 4af', which may be attributed to the influence of electronic effects. The 2-naphthalene-ethylene 2g was suitable for this transformation and formed the corresponding compounds 4ag in 78% yield. Taking into account the influence of electronic effects, methyl acrylate 2h and tert-butyl acrylate 2i showed lower activity in the reaction.
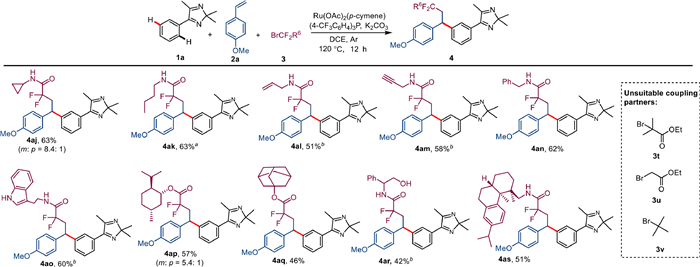
The next step was to test the applicability of the reaction to brominated difluoroalkanes (Scheme 4). Various bromodifluoroacetamides were suitable for this system. Cyclopropyl- and butyl-substituted difluoroacetamides could be readily used for this transformation and gave the compounds 4aj-4ak in 63% yield. The alkenyl and alkynyl substrates 3l-3m, which were suitable for further functionalization, were also successfully converted and afforded the corresponding compounds in 51%–58% yields. The benzyl- and tryptamine-substituted coupling partners showed good reactivity and afforded the products 4an and 4ao in 60%–62% yields. Inspired by the broad compatibility of the functional groups, the late functionalization of natural products was tested and the complex difluorinated products 4ap-4as were obtained in 42%–57% yield.
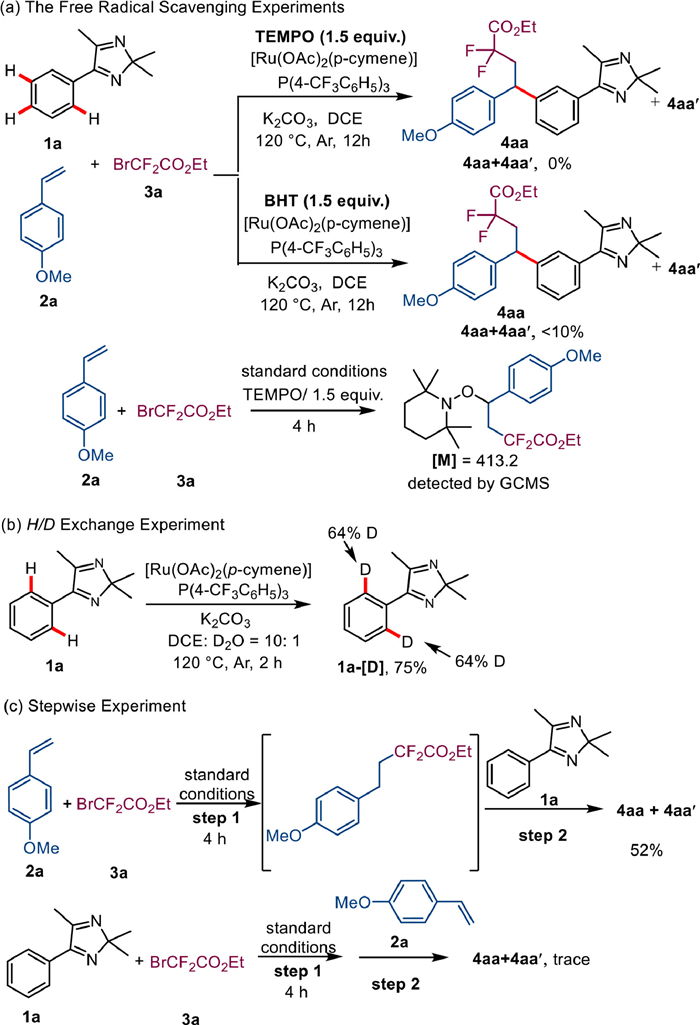
To investigate the reaction mechanism, the radical scavenging experiments were performed with 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT) (Scheme 5a), which suppressed this catalytic system. Meanwhile, without the presence of 1a, benzyl radicals were generated by the addition of 2a and 3a with TEMPO. Subsequently, deuterium-labeling was performed under standard conditions in the absence of 2a and 3a (Scheme 5b). Here, 64% deuterium was detected at the two ortho positions of the 2H-imidazole 1a. The stepwise experiment showed that styrene and bromodifluoroacetate reacted first in the system (Scheme 5c).
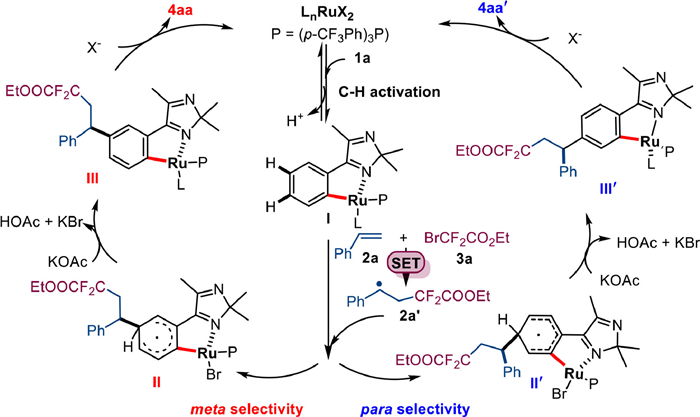
Based on previous studies on Ru metachemistry and experimental results [11–14], a possible mechanism was proposed in Scheme 6. The catalytic cycle starts with a reversible C—H cyclometalation between Ru(II) and 2H-imidazole 1a to form ruthenium complex I. Subsequently, benzyl radicals 2a' are formed by addition of 2a and 3a via a SET process. The newly formed radical 2a' completes the CAr—H bond addition at the para or meta position to the C—Ru bond and yields intermediates II or II', followed by rearomatization to ruthenacycles III or III'. Finally, the expected three-component coupling products 4aa and 4aa' are released by demetallation. At the same time, the active ruthenium(II) is regenerated.
In summary, we have successfully developed a Ru-catalyzed three-component remote C—H functionalization of 2H-imidazoles, which provides new ideas for the modular construction of complex 2H-imidazole scaffolds. Using radical scavenging experiments, we have shown that the reaction is based on a radical mechanism, and a possible mechanism is presented.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We highly appreciate the financial support from 111 Project B18035 and "the Fundamental Research Funds for the Central Universities".
J. Wencel-Delord, F. Glorius, Nat. Chem. 5 (2013) 369–375. doi: 10.1038/nchem.1607
T. Cernak, K.D. Dykstra, S. Tyagarajan, et al., Chem. Soc. Rev. 45 (2016) 546–576. doi: 10.1039/C5CS00628G
C. Sambiagio, D. Schönbauer, R. Blieck, et al., Chem. Soc. Rev. 47 (2018) 6603–6743. doi: 10.1039/C8CS00201K
P. Gandeepan, T. Müller, D. Zell, et al., Chem. Rev. 119 (2019) 2192–2452. doi: 10.1021/acs.chemrev.8b00507
X.L. Han, P.P. Lin, Q. Li, Chin. Chem. Lett. 30 (2019) 1495–1502. doi: 10.1016/j.cclet.2019.04.027
H.P. Wan, L. Gan, Y. Liu, Org. Biomol. Chem. 15 (2017) 9031–9043. doi: 10.1039/C7OB02011B
J.S. Zhang, L. Liu, T. Chen, et al., Chem. Asian J. 13 (2018) 227–229.
T. Shilpa, R. Dhanya, S. Saranya, et al., ChemistrySelect 5 (2020) 898–915. doi: 10.1002/slct.201904441
G.S. Susan Treesa, M. Neetha, S. Saranya, et al., ChemistrySelect 5 (2020) 7400–7416. doi: 10.1002/slct.202002021
D.S. Brandes, J.A. Ellman, Chem. Soc. Rev. 51 (2022) 6738–6756. doi: 10.1039/D2CS00012A
X.G. Wang, Y. Li, H.C. Liu, et al., J. Am. Chem. Soc. 141 (2019) 13914–13922. doi: 10.1021/jacs.9b06608
W.Y. Shi, Y.N. Ding, C. Liu, et al., Chem. Commun. 56 (2020) 12729–12732. doi: 10.1039/D0CC05491G
Y.Y. Luan, X.Y. Gou, W.Y. Shi, et al., Org. Lett. 24 (2022) 1136–1140. doi: 10.1021/acs.orglett.1c04182
H.C. Liu, X.P. Gong, Y.Z. Wang, et al., Org. Lett. 24 (2022) 3043–3047. doi: 10.1021/acs.orglett.2c00999
J. Wu, W. Wei, J. Pöhlmann, et al., Angew. Chem. Int. Ed. 62 (2023) 202219319. doi: 10.1002/anie.202219319
J.A. Boerth, S. Maity, S.K. Williams, et al., Nat. Catal. 1 (2018) 673–679. doi: 10.1038/s41929-018-0123-4
T. Pinkert, T. Wegner, S. Mondal, et al., Angew. Chem. Int. Ed. 58 (2019) 15041–15045. doi: 10.1002/anie.201907269
S. Maity, T.J. Potter, J.A. Ellman, Nat. Catal. 2 (2019) 756–762. doi: 10.1038/s41929-019-0330-7
A.G. Herraiz, N. Cramer, ACS Catal. 11 (2021) 11938–11944. doi: 10.1021/acscatal.1c03153
R. Mi, X. Zhang, J. Wang, et al., ACS Catal. 11 (2021) 6692–6697. doi: 10.1021/acscatal.1c01615
T. Pinkert, M. Das, M.L. Schrader, et al., J. Am. Chem. Soc. 143 (2021) 7648–7654. doi: 10.1021/jacs.1c03492
S. Dongbang, J.A. Ellman, Angew. Chem. Int. Ed. 60 (2021) 2135–2139. doi: 10.1002/anie.202010735
A. Mumtaz, A. Saeed, N. Fatima, et al., Bangladesh J. Pharmacol. 11 (2016) 756–764.
I. Ali, M.N. Lonea, H.Y. Aboul-Enein, Med. Chem. Commun. 8 (2017) 1742–1773. doi: 10.1039/C7MD00067G
N. Shalmali, M.R. Ali, S. Bawa, Mini-Rev. Med. Chem. 1 (2018) 142–163.
N. Rani, P. Kumar, R. Singh, et al., Curr Top. Med. Chem. 11 (2020) 1130–1155.
E.L. Gerasimova, E.R. Gazizullina, M.V. Borisova, et al., Molecules 26 (2021) 6534–6552. doi: 10.3390/molecules26216534
F. Hu, L. Zhang, K.S. Nandakumar, et al., Curr Top. Med. Chem. 21 (2021) 2514–2528. doi: 10.2174/1568026621666210527103225
S. Rulhaniaa, S. Kumar, B. Nehraa, et al., J. Mol. Struct. 1232 (2021) 129982. doi: 10.1016/j.molstruc.2021.129982
X.L. Wu, L. Dong, Org. Lett. 20 (2018) 6990–6993. doi: 10.1021/acs.orglett.8b02759
Y. Luo, H. Liu, J. Zhang, et al., Org. Lett. 22 (2020) 7604–7608. doi: 10.1021/acs.orglett.0c02805
Y. Luo, H.Y. Zhou, Y.C. Gang, et al., Org. Lett. 24 (2022) 6940–6944. doi: 10.1021/acs.orglett.2c02711
L. Ackermann, R. Vicente, H.K. Potukuchi, et al., Org. Lett. 12 (2010) 5032–5035. doi: 10.1021/ol102187e
J. Zhang, R. Shrestha, J.F. Hartwig, et al., Nat. Chem. 8 (2016) 1144–1151. doi: 10.1038/nchem.2602

Scheme 1 Multicomponent sequential C—H addition with two different coupling partners in C—H functionalization.

Scheme 2 Substrate scope of 2H-imidazoles 1. Unless otherwise stated, reaction conditions are as follows: Condition A: 1 (0.1 mmol), 2a (0.3 mmol), 3a (0.3 mmol), [Ru(OAc)2(p-cymene)] (5 mol%), P(4-CF3C6H5)3 (0.2 equiv.), K2CO3 (2.0 equiv.), 120 ℃, DCE (1 mL), Ar, 12 h, isolated total yield. Major isomer is shown. a [Ru(OAc)2(p-cymene)] (10 mol%). b Condition B: 1 (0.1 mmol), 2a (0.4 mmol), 3a (0.6 mmol), [Ru(OAc)2(p-cymene)] (5 mol%), P(4-CF3C6H5)3 (0.2 equiv.), KOAc (2.0 equiv.), 120 ℃, DCE (1 mL), Ar, 18 h, isolated total yield.

Scheme 3 Substrate scope of olefins 2. a Unless otherwise stated, reaction conditions are as follows: 1a (0.1 mmol), 2 (0.3 mmol), 3a (0.3 mmol), [Ru(OAc)2(p-cymene)] (5 mol%), P(4-CF3C6H5)3 (0.2 equiv.), KOAc (0.5 equiv.), 120 ℃, DCE (1 mL), Ar, 16 h, isolated total yield. Major isomer is shown.

Scheme 4 Substrate scope of brominated difluoroalkanes 3. Unless otherwise stated, reaction conditions are as follows: Condition A: 1a (0.1 mmol), 2a (0.3 mmol), 3 (0.3 mmol), [Ru(OAc)2(p-cymene)] (5 mol%), P(4-CF3C6H5)3 (0.2 equiv.), K2CO3 (2.0 equiv.), 120 ℃, DCE (1 mL), Ar, 12 h, isolated total yield. Major isomer is shown. a [Ru(OAc)2(p-cymene)] (10 mol%). b Condition B: 1 (0.1 mmol), 2a (0.4 mmol), 3a (0.6 mmol), [Ru(OAc)2(p-cymene)] (5 mol%), P(4-CF3C6H5)3 (0.2 equiv.), KOAc (2.0 equiv.), 120 ℃, DCE (1 mL), Ar, 18 h, isolated total yield.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: