State Key Laboratory of Medicinal Chemical Biology, College of Pharmacy and Tianjin Key Laboratory of Molecular Drug Research, Nankai University, Tianjin 300353, China
b.
College of Life Sciences, Nankai University, Tianjin 300071, China
Received Date:
14 December 2023 Accepted Date:
03 February 2024 Revised Date:
31 January 2024 Available Online:
15 December 2024
Abstract:
Multimodal bioorthogonal small molecule probes play a pivotal role in drug-focused biomedical research. However, existing drug tracking and imaging techniques face obstacles in living organisms, hindering precise drug localization and target protein capture. Herein, we introduced a multimodal probe named 1-(azidomethyl)pyrene-4,5–dione (AMPD). The probe incorporates adjacent dione structures at the pyrene core. AMPD selectively interacts with oxygen-rich alkene-labeled drug molecules under ice-blue LED light exposure, producing specific fluorescence emission and enabling in vivo tracking and flow cytometry sorting. A methyl azide group was also introduced at the pyrene core to help efficiently enrich target proteins via click chemistry with alkyne-functionalized beads. AMPD demonstrates exceptional biocompatibility, rendering it highly suitable for visual photo-triggered tracking studies. Combined with metabolic labeling using an oxygen-rich alkene-tagged drug molecule probe, AMPD is effective for live animal, tissue, cellular, and in-gel imaging, as well as target protein identification through magnetic capture. With its versatile capabilities, AMPD enhances our comprehension of drug-target interactions at the in vivo level and expedites the process of drug discovery.
The application of chemical biology techniques in drug discovery and development has been propelled by substantial advancements [1–4]. In particular, small molecule probes with multimodal functionalities serve as versatile tools, offering unique advantages in drug localization, target identification, and molecular imaging applications [5]. By integrating multiple functionalities into a single molecule, researchers gain comprehensive and detailed insights into the behavior of drugs within biological systems [6]. In recent years, bioorthogonal-triggered fluorescence probes and biologically specific multimodal probes have gained much attention [7,8]. These probes have expanded the possibilities for biomedical research and clinical applications by enabling molecular imaging and localization in various modes across different observation scales and conditions. Bioorthogonal chemistry tools have revolutionized numerous biological applications, greatly accelerating innovations in chemical biology [9]. Among established bioorthogonal or biocompatible reactions, cycloaddition reactions, such as copper-catalyzed azide-alkyne cycloaddition (CuAAC) [10], strain-promoted azide-alkyne cycloaddition (SPAAC) [11], and inverse-electron-demand Diels-Alder (IEDDA) [12] reactions, are the most widely utilized. Efforts have been made to enhance the reaction rates in biological applications, such as using metal catalysts in CuAAC reactions or employing strained alkyne/alkene substrates in SPAAC and IEDDA reactions [13]. However, certain challenges still exist with some bioorthogonal reactions, including cytotoxicity associated with metal catalysts, and accessibility and stability of strained alkynes/alkenes, among others. Therefore, efforts are continually being made to develop improved bioorthogonal crosslink methods.
Target discovery is an essential prerequisite for drug discovery and screening, enabling the precise design and validation of drug development [14–16]. Significant efforts have been made to enhance the specificity and efficacy of drug target identifications. These include covalently immobilizing drugs on specific matrices and selectively capturing potential targets through affinity-based methods without drug modification approaches such as drug affinity-responsive target stability (DARTS) [17], limited proteolysis-mass spectrometry (LiP-MS) [18,19], stability of proteins from rates of oxidation (SPROX) [20], and cellular thermal shift assay (CETSA) [21], among others. However, the effectiveness of unmodified target discovery methods heavily relies on alterations in protein abundance and physical and chemical properties, which may lead to false-negative outcomes if such alterations are absent. Traditional drug tracking and imaging techniques often face challenges in specificity and sensitivity. For instance, radioactive tracers such as 18F-fluorodeoxyglucose (FDG) used for tumor imaging may accumulate in other metabolically active tissues, leading to inaccurate imaging results [22]. Fluorescent dyes such as indocyanine green (ICG) are influenced by liver metabolism and excretion, limiting their applications [23]. Conventional methods often fall short in achieving precise in vivo imaging of drugs at the cellular level, and due to the heterogeneity of cells, effectively capturing proteins within specific target cells becomes challenging. One strategy to overcome these limitations is using light-triggered bioorthogonal cycloaddition reactions, generating highly reactive species, such as nitriles, quinone compounds, or substrates in an excited state, in situ [24,25]. However, the application of these photo-triggered bioorthogonal cycloaddition species is hindered by the limited penetration of ultraviolet (UV) or visible light into deep tissues and competing reactions involving highly reactive intermediates in complex biological systems [26]. Researchers continuously strive to develop improved versions of known bioorthogonal reactions to meet specific requirements, such as spatiotemporal resolution and metabolic transformation [27–31].
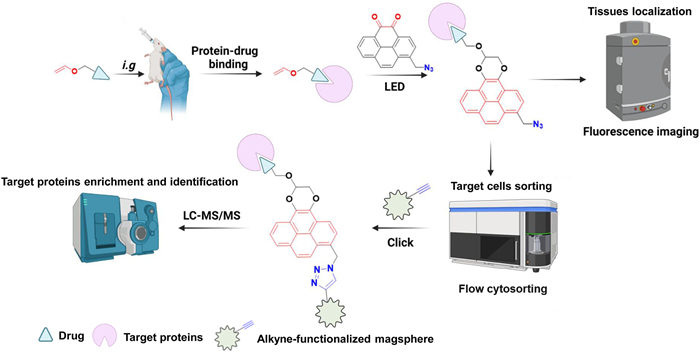
To address the limitations of traditional drug tracking and imaging techniques, innovative tracking technologies need to be developed, allowing for in situ localization, efficient capture, and enrichment of target proteins. Multimodal bioorthogonal 1-(azidomethyl)pyrene-4,5–dione (AMPD) probe enable simultaneous imaging and enrichment, allowing real-time visualization of target binding events in living animals. This facilitates a direct correlation between drug target identification and spatial distribution within tissues or cells, with applications in drug discovery, pharmacokinetics and pharmacodynamics studies, as well as personalized medicine. In this regard, we designed a promising pyrene-derived candidate, the AMPD multimodal molecule probe (Scheme 1). By incorporating adjacent dione structures at the C4 and C5 positions of a pyrene core, AMPD selectively reacts with oxygen-rich alkene-labeled drug molecules, generating specific fluorescence signals through light-triggered crosslinking. In addition, AMPD induces a methyl azide moiety at the C1 position of the core, facilitating specific binding and enrichment with target proteins through click chemistry with alkynes. Moreover, the introduction of the azide group elicits a redshift in the detection wavelength for in vivo imaging. Thus, due to its multimodal characteristics, AMPD holds great potential as a prospective tool for enhancing our comprehension of drug-target interactions and expediting the progress of drug discovery.
Scheme 1
Scheme 1.
Schematic illustration of the operational procedure for utilizing multimodal AMPD probe in drug target tissues localization, target cells sorting, target proteins enrichment and identification within living small animal studies.
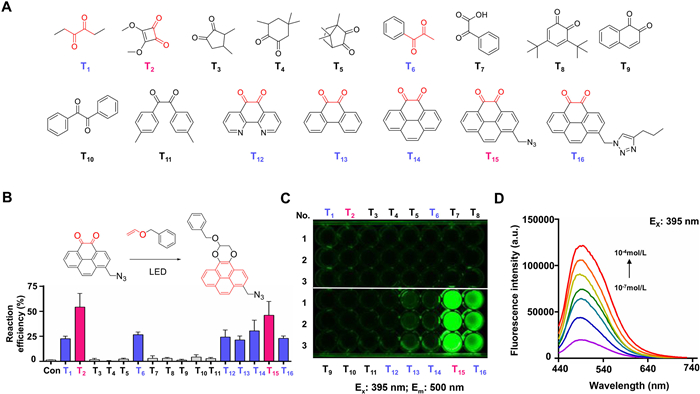
Screening substrate structure with ortho-dione scaffolds for visible light-triggered crosslinking. Light radiation to control chemical reactions is crucial in photo-biology [32]. This study examined the formation of fluorescent [4+2] cycloadducts between ortho-diones and electron-rich dienes under blue light emitting diode (LED) illumination. To investigate the chemical binding mode between electron-rich dienes and ortho-diones, we conducted a benzyl vinyl ether (BVE) reaction with various ortho-dione structures (Fig. 1A). Under physiological conditions, T1, T2, T6, and T12–T16 exhibited pronounced reactivity toward BVE, undergoing rapid reactions without any observed competing side reactions (Fig. 1B). It is worth noting that although T1 and T6 are not rigid ortho-diones, their highly twisted structures and the influence of neighboring alkyl groups retained their reactivity toward BVE under the same reaction conditions. Notably, other nonrigid ortho-diones did not react with BVE under the same reaction conditions (Figs. S1–S18 in Supporting information). Among them, only T15 exhibited significant fluorescence responsiveness under light-triggered conditions (Fig. 1C and Fig. S19 in Supporting information). The newly formed fluorescent moiety of T15 exhibited an optimal excitation wavelength of approximately 395 nm and a redshift emission wavelength of approximately 500 nm relative to the conjugation observed in T12, T13, and T14 (Fig. S20 in Supporting information). Moreover, the fluorescence intensity showed a concentration-dependent relationship, as shown in Fig. 1D, with T15 maintained at a constant level of 1 mmol/L and varying the concentration of BVE from 10−7 mol/L to 10−4 mol/L.
Figure 1
Figure 1.
Investigation and characterization of ortho-dione scaffold structures and fluorescence spectra. (A) Sixteen different ortho-dione structures. (B) Efficiency of the light-triggered bioorthogonal reaction between T1-T16 and BVE, which was detected by high performance liquid chromatography (HPLC). The data were shown as mean ± standard deviation (SD) (n = 3). (C) Fluorescence responsiveness after light-triggered crosslinking (Ex = 395 nm, Em = 500 nm). (D) Fluorescence spectrum of T15 at the optimal excitation wavelength (phosphate-buffered saline (PBS), Ex = 395 nm) in the experimental groups of different BVE concentrations.
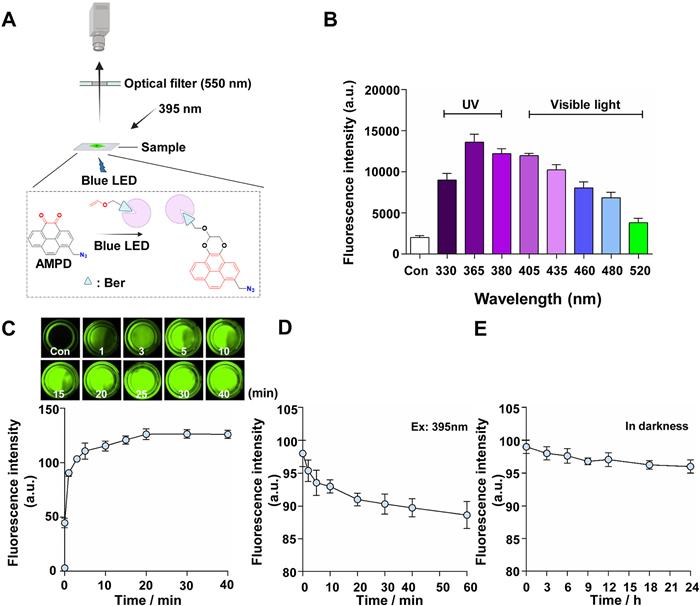
Optimal conditions for in vivo imaging and detection. Visible light is less harmful to biological systems than ultraviolet light, but it is still challenging to develop a bioorthogonal reaction that can effectively occur in complex biological environments. To assess the feasibility of imaging using photo-triggered orthogonal reactions, a multimodal AMPD probe (T15) was designed. By incorporating adjacent dione structures at the C4 and C5 positions of a pyrene core, AMPD selectively reacts with oxygen-rich alkene-labeled drug molecules, resulting in specific fluorescence signals via light-triggered crosslinking. Additionally, AMPD introduces a methyl azide moiety at the C1 position to facilitate specific binding and enrichment with target proteins through click chemistry with alkynes. Inspired by organic photo-reactions between electron-rich alkenes and ortho-dione substrates, the active natural compound bergenin (Ber), known for its anti-inflammatory and antioxidant properties [33], was modified into vinyl etherinbergenin (VE-Ber) (Figs. S21–S25 in Supporting information). Then, the VE-Ber probe was used as a model drug in photocatalytic research after confirming its bioequivalence with the prototype drug using a dual-fluorescence NF-κB reporting system (Fig. S26 in Supporting information). The schematic diagram is shown in Fig. 2A.
Figure 2
Figure 2.
Optimization conditions for photo-triggered orthogonal reaction and detection. (A) Schematic diagram of the photo-triggered crosslink and detection between AMPD and VE-Ber. (B) Investigation of the light source for photo-triggered crosslinking. (C) Investigation of the reaction efficiency for illumination time (Ex: 395 nm; Em: 550 nm). The changes in fluorescence intensity for the reaction mixtures were observed by an image reader. (D) Stability photo-triggered crosslinking between AMPD and VE-Ber (Ex: 395 nm). (E) Stability photo-triggered crosslinking between AMPD and VE-Ber (in darkness). The data were shown as mean ± SD (n = 3).
By conducting wavelength investigations using a microplate reader, it was found that orthogonal reactions can be effectively initiated in the range of 330–520 nm. However, considering that the maximum excitation of 365 nm does not apply to organisms, ice-blue LED visible light of nearly 430–460 nm was chosen as the best excitation light source (Fig. 2B). To mitigate background interference effectively, the existing gel imaging analyzer (HuaYue Enterprise Group Co., Ltd., China) was modified by implementing ice-blue LED light (50 W) for excitation and setting a 550 nm optical filter for detection. The efficiency and stability between VE-Ber and AMPD in a 1:1 CH3CN/PBS mixture were observed. The results showed that crosslinking could be achieved in 20 min (Fig. 2C). The stability investigation revealed that the fluorescence retention rate remained at 87% even after 40 min of irradiation, and it exhibited minimal decay in darkness through 24 h (Figs. 2D and E). The findings suggest that using ice-blue LED light to initiate the bioorthogonal reaction is more suitable for in vitro imaging.
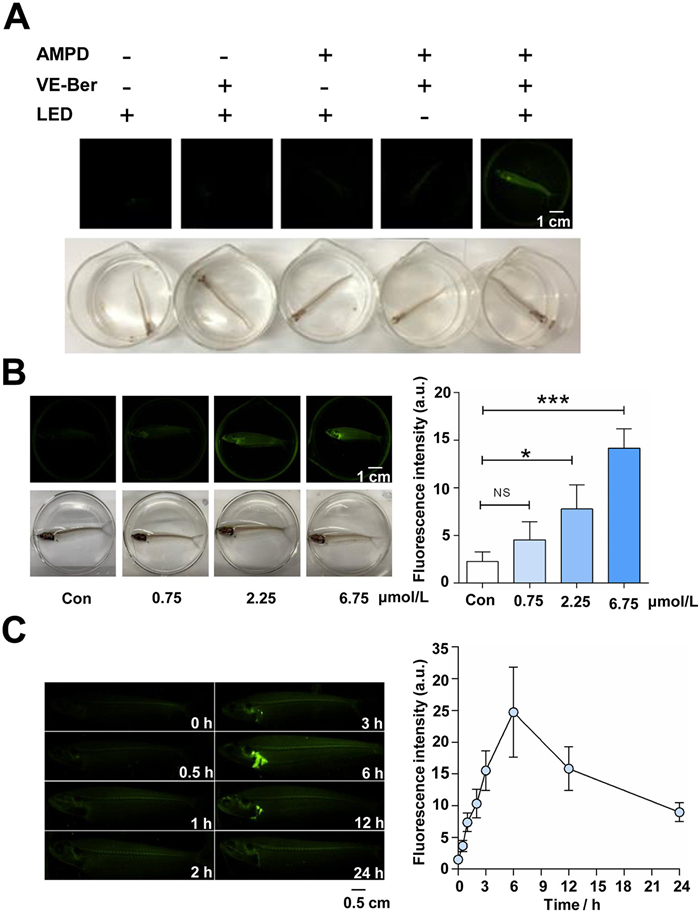
In vivo imaging with AMPD probe in model animals. In complex biological environments, intense illumination can trigger the reaction of nucleophilic thiols or highly reactive nitrile imine or quinonamine intermediates potentially competing with cycloaddition products [34,35]. Herein, we conducted a bioorthogonal reaction between AMPD and VE-Ber on glass catfish (Suxi Moon Biotechnology Co., Ltd., China) under ice-blue LED illumination to observe the reaction process and evaluate its efficiency. After incubation with VE-Ber (2.25 µmol/L) overnight, the glass catfish were placed in a 10 µmol/L AMPD probe solution and synchronously irradiated with ice-blue LED light for 20 min. The fluorescence signal was picked up by the above imaging analyzer with the optimum detection conditions. As depicted in Fig. 3A, AMPD can specifically recognize the oxygen-rich alkene probe on living glass catfish and initiate the cycloaddition reaction under ice-blue LED irradiation.
Figure 3
Figure 3.In vivo imaging of the AMPD probe on glass catfish. (A) Investigation of the bioorthogonal reaction conditions for in vivo fluorescence generation formed by the AMPD probe and VE-Ber (scale bar: 1 cm). (B) The in vivo imaging assay, generated by AMPD probe and serial-concentration VE-Ber. Scale bar: 1 cm. Con group (6.75 µmol/L Ber), low (0.75 µmol/L), medium (2.25 µmol/L), and high (6.75 µmol/L) dose of VE-Ber probe groups. *P < 0.1, ***P < 0.001 vs. Con group. NS: not significant. (C) Enrichment and tracer analysis of the VE-Ber probe in vivo with spatial and temporal resolution (scale bar: 0.5 cm). The data were shown as mean ± SD (n = 5).
To investigate the tissue distribution of Ber, more sophisticated in vivo tracing and metabolic labeling assays were carried out via VE-Ber. Twenty glass catfish were randomly divided into a control group (6.75 µmol/L, Ber) and different doses of VE-Ber groups (0.75, 2.25, 6.75 µmol/L). After overnight administration, the glass catfish were subjected to triple water soaking to ensure complete drug elimination. Subsequently, all groups were exposed to a 10 µmol/L AMPD probe solution and irradiated with ice-blue LED light for 20 min. As depicted in Fig. 3B, no fluorescence was observed in the prototype drug group. However, the photo-triggered orthogonal fluorescence signal was observed to be generated in the VE-Ber groups in a dose-dependent manner. Importantly, after metabolic labeling, the VE-Ber probes exhibited significant retention, specifically in hepatic tissue over time, demonstrating both spatial and temporal resolution (Fig. 3C). Additionally, the AMPD probe exhibits remarkable biocompatibility, as shown by the absence of any significant discomfort following overnight incubation on cells with 10 µmol/L AMPD (Fig. S27 in Supporting information) or administration of 10 µmol/L AMPD for 6 h on glass catfish (Supporting Video S1 in Supporting information). These findings suggest that the AMPD probe holds promising safety potential and can be effectively employed for in vivo animal imaging.
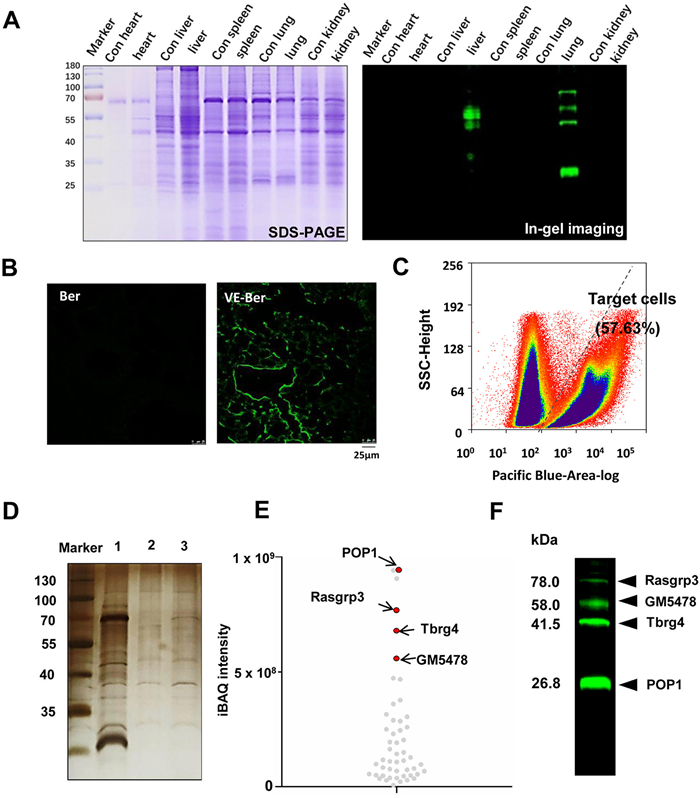
Target tissue localization, target cell sorting and target identification. Ultraviolet or visible light has limited penetration into deep tissues, which poses some limitations on the applications of photo-triggered crosslinks in vivo. To further investigate the feasibility of the drug on mouse target tissues, tissue homogenates or sections were used for photoinduced fluorescence imaging analysis. Protocols were reviewed and approved by the Nankai University of Chinese Medicine Institutional Animal Ethics and Welfare Committee (Certificate Number 2021-SYDWLL-000050; March 1st, 2021). The experiment was divided into a control group (50 mg kg−1 day−1 Ber) and an experimental group (50 mg kg−1 day−1 VE-Ber). After metabolic labeling for one week, the VE-Ber probes were considered to bind specifically with target proteins. Subsequently, mouse heart, liver, spleen, lung, and kidney tissues were individually collected and subjected to homogenization and centrifugation to obtain tissue protein lysates (1 mg/mL). These lysates were then combined with an AMPD probe (10−5 mol/L) and exposed to ice-blue LED light for 20 min to achieve in situ fluorescence labeling of the target proteins. The protein solutions were quantified using a BCA protein assay kit (Solaibao Science & Technology Co., Ltd., China), followed by equal loading of protein amounts onto SDS‒PAGE gels for subsequent in-gel imaging analysis using a fluorescence imaging analyzer (Ex: 395 nm, Em: 550 nm). Although SDS-PAGE is a denaturation technique, its purpose in this study was only to isolate and quantify the protein, and the in situ specific binding of the probe to the target protein was not affected by the denaturation properties of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. As shown in Fig. 4A, compared to Coomassie's bright blue staining (left image), the results showed pseudo-green signals only in the liver and lung tissues (right image). In the same way, specific fluorescence could also be observed in lung tissue sections by fluorescence confocal microscopy (Fig. 4B).
Figure 4
Figure 4.
Photo-triggered AMPD probe-guided target tissue tracing, target cell enrichment and target protein identification in mice. (A) Drug enrichment in the target tissue, investigated by the AMPD probe combined with oral administration of the VE-Ber probe. Coomassie brilliant blue staining (left panel) and in-gel imaging (right panel) for potential target proteins. (B) The localization of VE-Ber probe in lung tissue (scale bar: 25 µm). (C) Flow cytometry sorting for AMPD probe-labeled target cells from lung tissue. (D) The efficiency of the target protein by magnetic capture, evaluated by silver staining. Lane 1: Total protein lysate of AMPD probe-labeled lung cells. Lane 2: Captured proteins in the Ber control group. Lane 3: Captured proteins in the VE-Ber probe group. (E) Quantitative analysis of differential proteome content in lane 2 and lane 3, distinguished by protein scores. (F) In-gel imaging assay of potential target proteins in flow cytometric-sorted pulmonary cells, which were metabolically labeled with VE-Ber and detected with AMPD probes.
Due to the heterogeneity of physiological characteristics among different effector cell populations within biological systems, the precise identification of target cells for drugs has posed a significant benefit. To determine the appropriate dosage of the AMPD multimodal probe for cellular experiments, we conducted a 24 h investigation into the effects of different doses of AMPD administration on A549 cell viability. The results from the cell counting kit-8 (CCK-8) assay demonstrated that doses below 10−5 mol/L had no impact on cell viability (Fig. S27 in Supporting information). To achieve stringent capture of target proteins, cell sorting was conducted to obtain fluorescently labeled target cells. After administering VE-Ber (50 mg kg−1 day−1) for one week, the lung tissues of mice were collected, minced, and digested with collagenase to prepare single-cell suspensions. Subsequently, the cells were isolated using density gradient centrifugation and suspended in 1640 cell culture medium containing an AMPD probe (10−5 mol/L). Following irradiation with an ice-blue LED for 20 min while gently oscillating, the supernatant was discarded after centrifugation, and the labeled cells were resuspended for flow cytometry sorting using a FACSAria Fusion cell sorter (United States). As shown in Fig. 4C, approximately 57.63% of cells were labeled following one photocatalyzed fluorescent tracer sorting. The results indicate that the AMPD probe exhibits excellent recognition of oxygen-rich alkene-labeled drug molecules in living cells. At the same time, visible light-triggered cycloaddition enables precise temporal and spatial resolution labeling of cells.
Then, the captured cells were used to identify target proteins through the magnetic capture technique, while unlabeled cells served as the control group. The presence of an additional orthogonal handle in the multimodal AMPD probe enables a copper(I)-catalyzed bioorthogonal cycloaddition reaction to cooccur between the azido-alkynyl groups by incorporating an azido group at the C1 position. Consequently, acetylene-modified magnetic beads were utilized for target protein capture and enrichment, as previously reported [36]. The captured proteins were eluted and subjected to SDS-PAGE assay with silver staining (Fig. 4D). Subsequently, the captured proteins were identified using triple-quad ion trap and orbitrap fusion mass spectrometry systems (Thermo Scientific, USA). Compared to the control group, a total of 319 differential proteins were identified, among which processing of precursor 1 (POP1) (26.8 kDa), RAS guanyl-releasing protein 3 (Rasgrp3) (78.0 kDa), transforming growth factor beta regulator 4 (Tbrg4) (41.5 kDa), and Gene-Muridae 5478 (GM5478) (58.0 kDa) were determined as the most likely target proteins based on their abundance levels (Fig. 4E). These findings were consistent with those observed through in-gel imaging of flow cytometry-sorted cells (Fig. 4F). The inhibitory effect of POP1 on inflammasome activation has been reported [37], while Rasgrp3, acting as a guanine nucleotide exchange factor (GEF), restricts the Toll-like receptor-triggered inflammatory response and attenuates mitogen-activated protein kinase (MAPK) pathway activation [38,39]. These findings align with the documented anti-inflammatory properties of bergenin [40–42]. Additionally, Tbrg4 governs mitochondrial stress and the generation of reactive oxygen species (ROS) [43], while Ber has antioxidative properties [44], further substantiating our results.
This paper develops a multimodal AMPD probe, that exhibits selective reactivity toward the oxygen-rich alkene moiety on the drug molecule at the adjacent keto structures of the C4 and C5 positions under visible light catalysis. Simultaneously, the azido group located at the C1 position of AMPD can undergo a click reaction with an alkynyl group without any interference, enabling its capture and identification using acetylene-modified magnetic beads. The integration of a visual photo-triggered tracking system, encompassing tissue localization, cell sorting, and target fishing and identification processes, expands the repertoire of available bioorthogonal tools. The precise regulation of photo-triggered connections in biological systems by the multimodal AMPD probe, with high spatial and temporal resolution, will enable diverse applications and innovative avenues for non-invasive labeling and target identification in chemical biology research.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This research was supported by the National Key Research and Development Program of China (Nos. 2022YFC3500800 and 2022YFC3500805).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109646.
Scheme 1
Schematic illustration of the operational procedure for utilizing multimodal AMPD probe in drug target tissues localization, target cells sorting, target proteins enrichment and identification within living small animal studies.
Figure 1
Investigation and characterization of ortho-dione scaffold structures and fluorescence spectra. (A) Sixteen different ortho-dione structures. (B) Efficiency of the light-triggered bioorthogonal reaction between T1-T16 and BVE, which was detected by high performance liquid chromatography (HPLC). The data were shown as mean ± standard deviation (SD) (n = 3). (C) Fluorescence responsiveness after light-triggered crosslinking (Ex = 395 nm, Em = 500 nm). (D) Fluorescence spectrum of T15 at the optimal excitation wavelength (phosphate-buffered saline (PBS), Ex = 395 nm) in the experimental groups of different BVE concentrations.
Figure 2
Optimization conditions for photo-triggered orthogonal reaction and detection. (A) Schematic diagram of the photo-triggered crosslink and detection between AMPD and VE-Ber. (B) Investigation of the light source for photo-triggered crosslinking. (C) Investigation of the reaction efficiency for illumination time (Ex: 395 nm; Em: 550 nm). The changes in fluorescence intensity for the reaction mixtures were observed by an image reader. (D) Stability photo-triggered crosslinking between AMPD and VE-Ber (Ex: 395 nm). (E) Stability photo-triggered crosslinking between AMPD and VE-Ber (in darkness). The data were shown as mean ± SD (n = 3).
Figure 3In vivo imaging of the AMPD probe on glass catfish. (A) Investigation of the bioorthogonal reaction conditions for in vivo fluorescence generation formed by the AMPD probe and VE-Ber (scale bar: 1 cm). (B) The in vivo imaging assay, generated by AMPD probe and serial-concentration VE-Ber. Scale bar: 1 cm. Con group (6.75 µmol/L Ber), low (0.75 µmol/L), medium (2.25 µmol/L), and high (6.75 µmol/L) dose of VE-Ber probe groups. *P < 0.1, ***P < 0.001 vs. Con group. NS: not significant. (C) Enrichment and tracer analysis of the VE-Ber probe in vivo with spatial and temporal resolution (scale bar: 0.5 cm). The data were shown as mean ± SD (n = 5).
Figure 4
Photo-triggered AMPD probe-guided target tissue tracing, target cell enrichment and target protein identification in mice. (A) Drug enrichment in the target tissue, investigated by the AMPD probe combined with oral administration of the VE-Ber probe. Coomassie brilliant blue staining (left panel) and in-gel imaging (right panel) for potential target proteins. (B) The localization of VE-Ber probe in lung tissue (scale bar: 25 µm). (C) Flow cytometry sorting for AMPD probe-labeled target cells from lung tissue. (D) The efficiency of the target protein by magnetic capture, evaluated by silver staining. Lane 1: Total protein lysate of AMPD probe-labeled lung cells. Lane 2: Captured proteins in the Ber control group. Lane 3: Captured proteins in the VE-Ber probe group. (E) Quantitative analysis of differential proteome content in lane 2 and lane 3, distinguished by protein scores. (F) In-gel imaging assay of potential target proteins in flow cytometric-sorted pulmonary cells, which were metabolically labeled with VE-Ber and detected with AMPD probes.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
