Hunan Provincial Key Laboratory of Animal Models and Molecular Medicine, State Key Laboratory of Chemo/Bio-Sensing and Chemometrics, School of Biomedical Sciences, Hunan University, Changsha 410082, China
b.
Shenzhen Research Institute, Hunan University, Shenzhen 518000, China
yuandan@hnu.edu.cn (D. Yuan). 1 These authors contributed equally to this work.
Received Date:
14 December 2023 Accepted Date:
06 February 2024 Revised Date:
05 February 2024 Available Online:
15 December 2024
Abstract:
Achieving selectivity in cell penetrating peptide (CPP) design is crucial to mitigate systemic toxicity and enable precise targeting based on distinct cellular phenotypes. Herein, we designed an amphiphilic peptide, L17Yp, by incorporating phosphorylated tyrosine into natural occurring M-lycotoxin peptide, known for its potent membrane-lytic activity. This strategic modification induced a conformational shift, as confirmed by circular dichroism spectroscopy, transitioning it from its bioactive α-helix conformation to an inactive random coli configuration, effectively shielding its membrane-penetrating capacity. Upon exposure to alkaline phosphatase, L17Yp undergoes enzymatic dephosphorylation, prompting a conformational shift that restores its membrane-transduction capabilities. This unique property hold promises for selective drug delivery. This work introduces an enzymatic approach for targeted perturbation of the cell membrane, offering promising prospects for precise drug delivery applications.
Cell penetrating peptides (CPPs), also referred to as protein transduction domains (PTDs) or membrane translocating sequences (MTS), exhibit a remarkable capability to enter cells and facilitate the transport of diverse therapeutic agents [1]. These agents include small molecule drugs [2–6], proteins [7–9], nucleic acids [10] and nanoparticles [11–13], all of which traverse the cell membrane [14,15]. A majority of CPPs adopt α-helix [16] or β-hairpin [17–19] secondary structures, carefully balancing their hydrophobic and cationic charge properties [20]. These unique characteristics enable CPPs to efficiently traverse the cell membrane by interacting with its components (e.g., lipid) [21,22].
Despite their versatile utility, CPPs face a significant limitation- a lack of selectivity, leading to substantial systematic toxicities and hindering their widespread application [23]. CPPs, owing to their positive charge and amphiphilic properties, readily engage with a wide range of cell types, including both normal and tumorous cells [24]. However, this deficiency in selectivity often results in off-target effects and potential cytotoxicity when CPPs are employed for drug delivery or other therapeutic purposes [25]. Consequently, extensive research efforts have been dedicated to conferring CPPs with ability to specifically recognize and target particular cells types [26]. This endeavor involves various strategies, including the incorporation of pH-sensitive components into CPPs [27], creation of enzyme-responsive CPPs [28], or employing other innovative approaches tailored to precise targeting [29]. A widely employed approach for achieve selective delivery involves the utilization of enzyme-activatable CPPs. This approach leverages aberrant and elevated expression of specific enzymes, such as metalloproteinases [30], alkaline phosphatase (ALP) [31] and cathepsin B [32] within cancer cells. Notably, a significant portion of the research has focused on utilizing ALP to construct molecular assemblies for selectively inhibiting cancer cells [33–36]. Recently, Schneider group developed an ALP-activatable CPP that introduced a phosphorylated tyrosine to disturb hydrophilic-lipophilic balance, transforming the peptide’s conformation to β-hairpin [31]. This pioneering work has provided a valuable framework for designing ALP activated peptides. However, the exploration of ALP activated CPP remains relatively uncharted territory, especially in the context of α-helix CPPs (e.g., M-lycotoxin) [37]. α-Helix CPPs, while possessing formidable lytic properties, are often hindered by high cytotoxicity, limiting their extensive application [38]. Therefore, there is a pressing need to develop α-helix ALP-activable CPP capable of minimizing toxicity in normal cells while effectively targeting ALP-expressing cells.
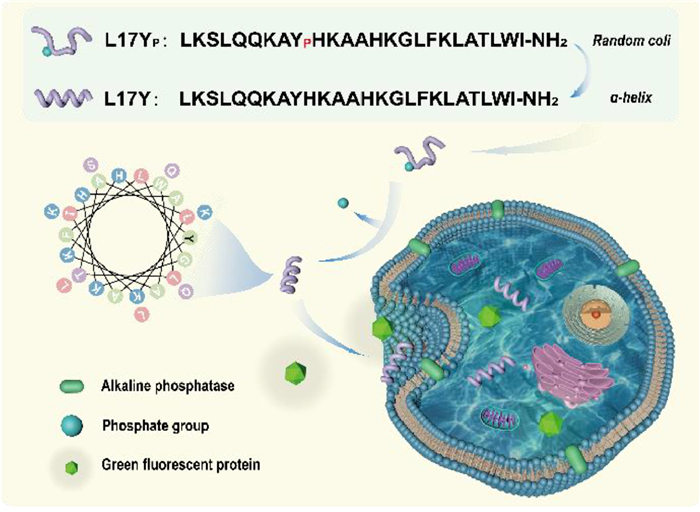
Building on previous work by Futaki et al., which demonstrated the reduction of the lytic activity of M-lycotoxin through introducing a glutamic acid (Glu) mutation [39], we designed an α-helix ALP-activated CPP (L17Yp) by incorporating a phosphorylated tyrosine (Yp, bearing two negative charges) to disrupt delicate amphiphilic balance of the peptide (Fig. 1). This strategic modification induced a transformation in peptide conformation, resulting in an inactive random coil structure that hindered its cell-penetrating ability. Upon encountering the cells with upregulated ALP expression, L17Yp underwent dephosphorylation by ALP, reverting to its active peptide conformation, denoted as L17Y. This process restored the peptide’s functional cell-penetrating properties and afforded its cell selective penetration toward high ALP-expressing cells. Leveraging this innovative strategy, we successfully achieved the delivery of green fluorescent protein (GFP) into ALP-overexpressing cell lines, exemplified by Saos-2. This approach holds significant potential for targeted delivery of drugs or bioactive molecules to specific cells and tissues, offering new avenues for precise therapeutic interventions.
Figure 1
Figure 1.
Conceptual illustration of functional mechanism of L17Yp. The inactive phosphorylated peptide L17Yp undergoes a conformational transition from a random coil to α-helix upon dephosphorylation catalyzed by ALP located on the cell surface. This transformation endows the dephosphorylated peptide L17Y with cell-penetrating ability, facilitating the efficiently delivery of GFP into cells. The sequences of both peptides are also shown.
Based on the rational above, we selected the natural α-helix peptide M-lycotoxin as a model peptide [39]. Herein, we designed a novel peptide L17Yp through altering leucine (Leu) to phosphorylated tyrosine (Yp) at position 17 of the M-lycotoxin peptide. As a control, we also prepared a dephosphorylated peptide, designated as L17Y. The synthetic characterization of these peptides is provided in Figs. S1–S4 (Supporting information). Subsequently, we investigated the influence of incorporated phosphorylated tyrosine on the peptide’s conformation. It was confirmed that phosphorylated polypeptides L17Yp undergo dephosphorylation after treatment with ALP (Fig. 2a) [40]. As expected, the incorporation of phosphorylated tyrosine maintained the peptide in an unfolded and inactive state until exposed to ALP. The removal of phosphate group restored its amphiphilic properties, and the converted L17Y adopted an α-helix conformation upon binding to a negative charged surface. This observation strongly indicates that the dephosphorylation event effectively restores the peptide conformation to its active forms (Fig. 2b).
Figure 2
Figure 2.
(a) Time-dependent dephosphorylation of L17Yp (200 µmol/L) in the presence of 1 U/mL ALP. (b) Circular dichroism (CD) spectra of 50 µmol/L L17Y and L17Yp incubated with negative charged large unilamellar vesicles (LUVs) (POPC/POPS = 1:1, 2.5 mmol/L) for 4 h. (c) Quantification of the ALP activity in various cell lines. (d) Determination of IC50 values for L17Yp towards Saos-2, NHDF and hFOB 1.19 cells lines. (e) Cell viability of L17Yp towards Saos-2 cells in the absence or presence of the ALP inhibitor (1.0 mmol/L levamisole), along with unphosphorylated peptide L17Y. Data were analyzed using ordinary one-way t-test in Excel. ****P < 0.0001. The results are presented as the mean ± standard deviation (SD) (n = 3).
The introduction of phosphate group was expected to reduce the membrane-perturbing activity of cationic amphiphilic peptide. To confirm this hypothesis, we utilized three cell lines with differential ALP activity: osteosarcoma (Saos-2) cells [41], normal human dermal fibroblasts (NHDF) cells and human fetal osteoblast (hFOB 1.19) cells (Fig. 2c). Here, our focus was on the contribution to cytotoxicity originating from ALP activity. The half-maximal inhibitory concentration (IC50) values of L17Yp against all types of cells exhibited substantial differences based on their ALP activity, approximately 10.3 µmol/L for Saos-2 cells, 52.5 µmol/L for NHDF cells, and 73 µmol/L for hFOB 1.19 cells. This nearly 5-fold and 7-fold contrast in IC50 values highlights the dependency of L17Yp cytotoxicity on the cell type under ALP action (Fig. 2d). Furthermore, Fig. 2e revealed that L17Yp and L17Y exhibited relatively similar high cytotoxicity in a dose-dependent manner, while the cytotoxicity of L17Yp could be significantly reduced by the addition of ALP inhibitor (levamisole) to Saos-2 cells. Additionally, the low cytotoxicity of L17Yp to NHDF cells and hFOB 1.19 cells could be reversed by the addition of exogenous ALP (Figs. S5 and S6 in Supporting information). These results indicate that phosphorylated peptides L17Yp maintained a random coli conformation and exhibited relatively low cytotoxicity until they were transformed into α-helix after dephosphorylating by ALP on the cell surface. This suggests L17Yp could function as an ALP-activated CPP at safe dosages, selectively killing cells in high concentrations.
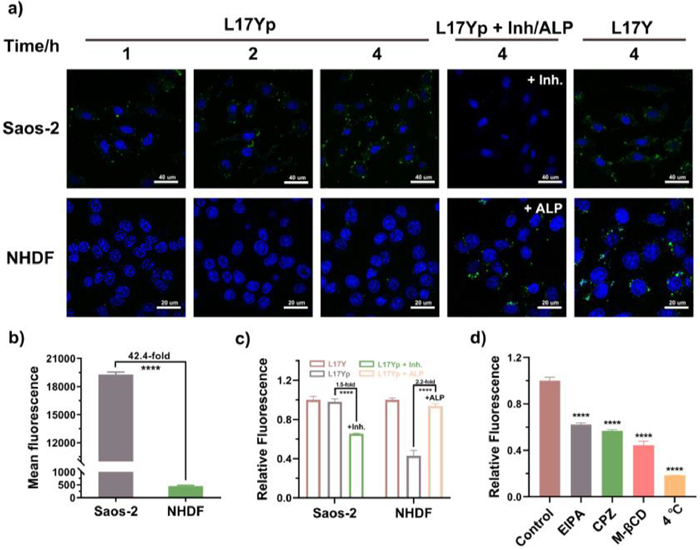
As shown in Fig. 1, the phosphorylated peptide L17Yp undergoes a conformational transformation facilitated by the action of the ALP enzyme on the cell surface, endowing it with membrane-penetrating ability. In this work, we chose Saos-2 cells and NHDF cells, which express varying levels of ALP and exhibit similar measured cell surface charges, to investigate the membrane penetration ability of L17Yp (Figs. S7 and S8 in Supporting information). The peptide concentration used (1 µmol/L) was below the IC50 value applied in the cell uptake study. Results obtained from confocal microscopy and flow cytometer unambiguously demonstrated that FITC-L17Yp exhibited a significantly higher fluorescent signal in Saos-2 cells compared to NHDF cells, as shown in Fig. 3a. The mean fluorescence intensity in Saos-2 was approximately 42.4 folds higher than that in NHDF cells (Fig. 3b). To further elucidate the role of the ALP, Saos-2 cells were treated with enzyme inhibitor (levamisole), while NHDF cells were supplemented with exogenous ALP. This differential treatment had a substantial impact on cell penetration of FITC-L17Yp. Incubation with levamisole significantly inhibited the entry of FITC-L17Yp, whereas the addition of exogenous ALP to NHDF cells facilitated the cellular uptake of FITC-L17Yp, as revealed in Fig. 3c. In contrast, the non-phosphorylated control peptide FITC-L17Y could enter both types of cells indiscriminately. Together, these results validate enzymatic dephosphorylation imparts selectivity to the precursor L17Yp, enabling it to enter cells at almost non-toxic concentrations in a time-dependent manner, as revealed in Fig. S9 (Supporting information).
Figure 3
Figure 3.
(a) Confocal images of Saos-2 and NHDF cells incubated with FITC-L17Yp (1 µmol/L) as a function of time under different conditions. Either ALP inhibitor (levamisole, 1 mmol/L) or exogenous ALP (5 U/mL) was added to Saos-2 or NHDF cells for comparative analysis. Scale bar: above: 40 µm; below: 20 µm. (b) Mean fluorescence intensity of cells incubated with 1 µmol/L L17Yp for 4 h. (c) Relative fluorescence of cells incubated with 1 µmol/L L17Yp in the absence or presence of either exogenous ALP (5 U/mL) or 1 mmol/L ALP inhibitor (levamisole) for 4 h. (d) Mechanisms underlying the cellular uptake of L17Yp towards Saos-2 cells under the treatment of micropinocytosis inhibitors (EIPA, 50 µmol/L), clathrin-mediated endocytosis inhibitors (CPZ, 15 µmol/L), lipid-raft inhibitors (M-β-CD, 2.5 mmol/L), and low temperature condition. Data were analyzed using ordinary one-way t-test in Excel. ****P < 0.0001. The results are presented as the mean ± SD (n = 3).
To investigate the internalization mechanism, we employed flow cytometry and confocal microscopy to monitor the cellular uptake of L17Yp under various conditions. Initially, Saos-2 cells pre-treated at 4 ℃ exhibited near-complete suppression of L17Yp internalization, implying that the entry of L17Yp into cells primarily occurs through an energy-dependent pathway. Further investigation into the specific cell entry mechanism involved pre-treating Saos-2 cells with three different inhibitors, 5-(N-ethyl-N-isopropyl)-amiloride (EIPA), chlorpromazine (CPZ) and methyl-β-cyclodextrin (M-β-CD), each at an effective concentration for 30 min. Among these inhibitors, M-β-CD, acting as a lipid-raft inhibitor, displayed the most notable reduction in penetration efficiency, resulting in a nearly 60% decrease in relative fluorescence. EIPA and CPZ both demonstrated comparable inhibitory effects, each reducing the fluorescence signal of L17Yp by around 40% (Fig. 3d). This tread was further confirmed in confocal images (Fig. S10 in Supporting information). These results collectively indicate that the internalization mechanism of L17Yp predominantly occurs through energy-dependent endocytosis, with lipid-raft-mediated endocytosis playing a pivotal role. Additionally, it is associated with clathrin and micropinocytosis pathways.
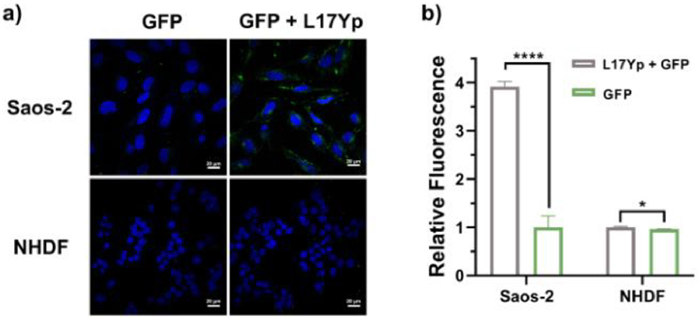
Protein-based therapeutics have emerged as a potent approach for treating various diseases [42]. However, intracellular protein delivery poses a significant challenge for clinical usage [43]. In this work, we chose GFP as a model cargo to investigate the delivery potential of L17Yp [44]. Through simple mixing, L17Yp and GFP bound together via electrostatic interaction, as confirmed by observed alternation in the zeta potential of the mixture (Fig. S11 in Supporting information). As shown in Fig. 4a, minimal GFP signals were detected when Saos-2 cells were exposed to GFP alone. In contrast, upon the introduction of L17Yp, a distinct and robust fluorescent signal was evident within the cells. This result indicated that L17Yp serves as an effective vehicle for delivering protein into cells at low concentrations. Unlike the phenomenon observed in Saos-2 cells, GFP signals in NHDF cells was virtually absent, regardless of co-incubation of L17Yp. Furthermore, the flow cytometry results in Fig. 4b confirm that following the introduction of L17Yp, the fluorescence value in Saos-2 cells increased by nearly four times, but remained almost unchanged in NHDF cells. Given the substantial impact of ALP presence on the cell surface in enhancing the cellular uptake of phosphorylated peptides, it is reasonable to infer that L17Yp holds promise as a candidate for selectively delivering cargoes to cells with high ALP expression levels.
Figure 4
Figure 4.
(a) Confocal images of Saos-2 cells co-incubated with L17Yp and GFP proteins to assess the delivery efficacy of L17Yp. Scale bar: 20 µm. (b) Quantification of the mean fluorescence signal. Data were analyzed using ordinary one-way t-test in Excel. *P < 0.05, ****P < 0.0001. The results are presented as the mean ± SD (n = 3).
In summary, we present a novel strategy involving the ALP-activated conformational transformation of peptide for the development of cell selective CPPs. Through the introduction of phosphorylated tyrosine to the hydrophobic face of M-lycotoxin, the inherent bioactive α-helix structure undergoes a transition into a random coil structure, resulting in the loss of membrane-perturbing activity. Subsequently, the restoration of its original conformation and transmembrane capability is initiated through the dephosphorylation process catalyzed by ALP. Therefore, the transmembrane process of L17Yp is exclusive to cells with elevated ALP expression, endowing the peptide with selectivity for specific cell types. This selectivity is exemplified by the pronounced internalization of L17Yp in high ALP-expressing cells, contrasting with limited uptake in ALP-deficient cells. Furthermore, our findings confirm that L17Yp serves as an efficient cytosolic delivery carrier by enhancing the cellular uptake of green fluorescent protein in ALP-overexpressing cells. In this work, ALP is responsible for the enzymatic-induced transmembrane process. Importantly, the concept of leveraging specific enzymes with high expression levels can be extended to other cell lines. This ALP-activated CPP represents a significant advancement in the design of selective molecule delivery systems and hold great promise for future applications in various biomedical contexts (e.g., drug delivery).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 21975068), Natural Science Foundation of Hunan (No. 2022JJ10008), Science and Technology and Development Foundation of Shenzhen (No. JCYJ20210324122403010), and Natural Science Foundation of Changsha (No. kq2202152). We would like to thank the Analytical Instrumentation Center of Hunan University for assistance in testing.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109628.
[1]
P. Lundberg, Ü. Langel, J. Mol. Recognit. 16 (2003) 227–233. doi: 10.1002/jmr.630
[2]
R. Tian, H. Wang, R. Niu, et al., Adv. Colloid Interface Sci. 453 (2015) 15–20. doi: 10.1016/j.jcis.2015.04.028
Figure 1
Conceptual illustration of functional mechanism of L17Yp. The inactive phosphorylated peptide L17Yp undergoes a conformational transition from a random coil to α-helix upon dephosphorylation catalyzed by ALP located on the cell surface. This transformation endows the dephosphorylated peptide L17Y with cell-penetrating ability, facilitating the efficiently delivery of GFP into cells. The sequences of both peptides are also shown.
Figure 2
(a) Time-dependent dephosphorylation of L17Yp (200 µmol/L) in the presence of 1 U/mL ALP. (b) Circular dichroism (CD) spectra of 50 µmol/L L17Y and L17Yp incubated with negative charged large unilamellar vesicles (LUVs) (POPC/POPS = 1:1, 2.5 mmol/L) for 4 h. (c) Quantification of the ALP activity in various cell lines. (d) Determination of IC50 values for L17Yp towards Saos-2, NHDF and hFOB 1.19 cells lines. (e) Cell viability of L17Yp towards Saos-2 cells in the absence or presence of the ALP inhibitor (1.0 mmol/L levamisole), along with unphosphorylated peptide L17Y. Data were analyzed using ordinary one-way t-test in Excel. ****P < 0.0001. The results are presented as the mean ± standard deviation (SD) (n = 3).
Figure 3
(a) Confocal images of Saos-2 and NHDF cells incubated with FITC-L17Yp (1 µmol/L) as a function of time under different conditions. Either ALP inhibitor (levamisole, 1 mmol/L) or exogenous ALP (5 U/mL) was added to Saos-2 or NHDF cells for comparative analysis. Scale bar: above: 40 µm; below: 20 µm. (b) Mean fluorescence intensity of cells incubated with 1 µmol/L L17Yp for 4 h. (c) Relative fluorescence of cells incubated with 1 µmol/L L17Yp in the absence or presence of either exogenous ALP (5 U/mL) or 1 mmol/L ALP inhibitor (levamisole) for 4 h. (d) Mechanisms underlying the cellular uptake of L17Yp towards Saos-2 cells under the treatment of micropinocytosis inhibitors (EIPA, 50 µmol/L), clathrin-mediated endocytosis inhibitors (CPZ, 15 µmol/L), lipid-raft inhibitors (M-β-CD, 2.5 mmol/L), and low temperature condition. Data were analyzed using ordinary one-way t-test in Excel. ****P < 0.0001. The results are presented as the mean ± SD (n = 3).
Figure 4
(a) Confocal images of Saos-2 cells co-incubated with L17Yp and GFP proteins to assess the delivery efficacy of L17Yp. Scale bar: 20 µm. (b) Quantification of the mean fluorescence signal. Data were analyzed using ordinary one-way t-test in Excel. *P < 0.05, ****P < 0.0001. The results are presented as the mean ± SD (n = 3).

 DownLoad:
DownLoad:


 下载:
下载:





 下载:
下载:
