lq580202@sdu.edu.cn (Q. Li). 1 These authors contributed equally to this work.
Received Date:
10 January 2024 Accepted Date:
07 February 2024 Revised Date:
04 February 2024 Available Online:
15 December 2024
Abstract:
Patients with oral squamous cell carcinoma (OSCC) encounter challenges in achieving efficient antitumor immunity, primarily due to the inherent pathophysiological characteristics of solid tumors affecting drug accumulation and penetration. Insufficient T-cells and immune escape induced by tumor-associated macrophages (TAMs) further exacerbate these issues. This study utilized M1 macrophage membrane-modified spatial dimension conversion drug delivery systems (SDDDSs) and introduced photosensitizers chlorophyll Pyro and the immune agonist R848. This innovative approach enhanced tumor targeting and accumulation by transforming stimulus-responsive size-reductive SDDDSs into smaller-sized iRGD-Pyro and R848 within the extracellular tumor microenvironment (TME). This facilitated effective drug penetration into deep tumor regions and cellular uptake. The synergistic treatment strategy for OSCC, combining photodynamic therapy (PDT) and tumor immunotherapy, induced tumor cell apoptosis, triggered immunogenic cell death (ICD), polarized TAMs towards the M1 phenotype, promoted sufficient T-cell infiltration, and resulted in significant therapeutic outcomes. This approach offers a promising avenue for future OSCC therapeutic interventions.
Oral squamous cell carcinoma (OSCC) typically originates from the epithelium of the oral mucosa and has a relatively shallow location. Its unique anatomic structure makes tumors susceptible to invasion and metastasis, making complete surgical removal difficult, increasing the risk of recurrence, and driving rapid progression to advanced stages [1]. Despite the impressive clinical efficacy of immune checkpoint inhibitors, such as programmed cell death-ligand 1/programmed cell death-1 (PD-L1/PD-1) and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), in activating anti-tumor immune responses, preventing invasion, metastasis, and recurrence, and prolonging survival in OSCC patients with advanced cancer, a significant challenge remains [2]. The inherent pathophysiological characteristics of solid tumors such as OSCC affect drug accumulation and penetration, limiting the bioavailability of therapeutic agents. This impact is a critical determinant of the efficacy of immunomodulatory drugs, acting synergistically with T cell deficiency [3] and tumor-associated macrophages (TAMs)-induced immune escape to collectively impede effective immunotherapy [4]. These challenges contribute to the difficulty in achieving efficient antitumor immunity in patients with OSCC. Therefore, developing strategies to effectively achieve drug accumulation at the tumor site, enable deep tumor penetration, and induce anti-tumor immunity holds great promise for treating OSCC. Fortunately, drug delivery systems (DDSs) are a new perspective for the realization of this treatment strategy.
Research suggests that the intricacies of the tumor microenvironment (TME) present challenges to DDSs in achieving effective accumulation at the tumor site and facilitating deep tumor penetration and cellular uptake [5]. Yet, larger DDSs favor prolonged in vivo retention, while smaller DDSs, especially small molecule drugs, facilitate deep tumor penetration [6]. To overcome this contradiction, developing size-variable DDSs is essential. Significantly DDSs can change size in response to specific TME characteristics, thereby achieving tumor-adaptive transformation for precise spatial and temporal delivery and releasing therapeutic components as stimulus-responsive size-reductive spatial dimension conversion drug delivery systems (SDDDSs). However, existing SDDDSs face challenges such as complex preparation, poor biocompatibility, and low drug loading, which necessitate the design of efficient SDDDSs [7].
Almost all extracellular TME contain high levels of matrix metalloproteinase-2 (MMP2) and reactive oxygen species (ROS) [8]. MMP2- and ROS-cleavable peptide-modified DDSs are an innovative approach for developing stimulus-responsive size-reductive SDDDSs to enhance drug accumulation, penetration and cellular uptake in tumors [9,10]. In addition, full utilization of macrophage membrane-modified SDDDSs can prolong their in vivo circulation time, improve active targeting effects, and reduce side effects [11]. Recent studies suggest that M1 macrophage membrane-modified SDDDSs exhibit higher efficacy in targeting, deep tumor accumulation [12,13].
Photodynamic therapy (PDT) has emerged as a promising modality for treating OSCC with a relatively shallow location due to its specificity and non-invasive nature [14]. In addition, insufficient immunogenicity is observed in deep tumor tissues due to inadequate penetration of photothermal agents, resulting in localized tumor regions remaining non-responsive to treatment [15]. Additionally, TME poses a challenging problem for the efficacy of PDT-induced immunogenic cell death (ICD) in activating tumor immunity [16]. Therefore, the synergistic potential of combining photodynamic therapy with immunotherapy holds great promise for achieving remarkable clinical outcomes in OSCC [17]. SDDDSs play a critical role in integrating PDT and immunotherapy by overcoming challenges such as insufficient drug accumulation in tumors, limited permeability and uptake, and significant systemic toxicities.
The present study exploited MMP2- and ROS-responsive peptides to construct SDDDSs. Combining the photosensitizers chlorophyll Pyro and the Food and Drug Administration (FDA)-approved immune agonist R848 with responsive, functional peptides by self-assembly exploits the masking and targeting effects of M1 macrophage membranes. Consequently, it enables more selective targeting and accumulation of SDDDSs at tumor sites. Within the extracellular TME, elevated levels of MMP2 and ROS facilitate the rapid degradation of SDDDSs into smaller-sized tumor-targeting iRGD-Pyro and R848. This process achieves deep tumor penetration and efficient uptake, leading to synergistic treatment of OSCC by PDT and tumor immunotherapy. The synergistic approach induces apoptosis in tumor cells, triggering ICD, while polarizing TAMs toward the M1 phenotype and promoting adequate T-cell infiltration. These combined effects result in remarkable therapeutic outcomes, highlighting the significant clinical translational potential for the treatment of OSCC using highly synergistic PDT and tumor immunotherapy (Scheme 1).
Scheme 1
Scheme 1.
Diagram depicting the design of SDDDSs (R-PD-Pyro NPs@M) of synergistic PDT and enhance immunotherapy for OSCC. (ⅰ) ROS cleavage site. (ⅱ) MMP2 cleavage site. (a) Augmented tumor targeting and (b) accumulation. (c) Transformation of stimulus-responsive size-reductive SDDDSs into smaller-sized tumor targeted iRGD-Pyro and R848. (d) Deep tumor penetration and cellular uptake.
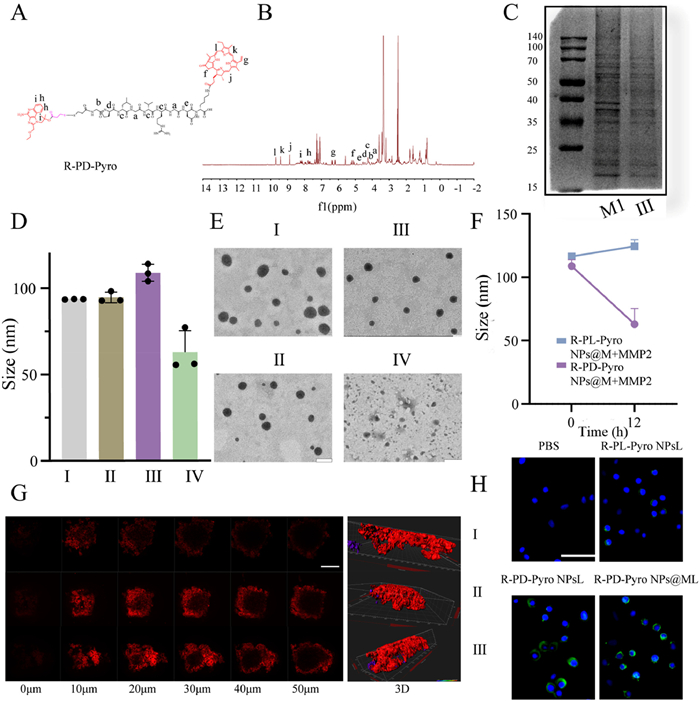
In this investigation, all in vivo experiments adhered to the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Shandong University School of Stomatology (No. 20220503) and the regulations for the administration of laboratory animals. We synthesized R-PD-Pyro (Fig. S1 in Supporting information) and R-PL-Pyro (Fig. S2 in Supporting information) by solid-phase peptide synthesis using amide condensation and thiol-disulfide exchange reactions. 1H NMR analysis (Figs. 1A and B, Fig. S3 in Supporting information) validated the structural integrity of the target products. In addition, ultraviolet–visible spectroscopy (UV–vis) detection (Fig. S4 in Supporting information) confirmed the compositional structures of R-PD-Pyro and R-PL-Pyro. R848 has a robust ultraviolet absorption peak at 274 nm, while Pyro exhibits a pronounced ultraviolet absorption peak at 675 nm. To improve in vivo circulation time, active targeting, tumor accumulation, and penetration of SDDDSs, we functionalized the surface with M1 macrophage membranes, resulting in R-PD-Pyro NPs@M. Gel electrophoresis analysis (Fig. 1C) indicates that R-PD-Pyro NPs@M experiences minimal protein loss. This not only imparts functional similarity to M1 macrophages but also confers the R-PD-Pyro NPs@M with the ability to evade the mononuclear phagocyte system. Dynamic light scattering (DLS) analysis revealed that R-PD-Pyro NPs have a diameter of 94 ± 2.5 nm, while R-PD-Pyro NPs@M exhibits only a minimal increase in size (Fig. 1D). This aligns with prior studies, affirming the successful camouflage with sizes well-tolerated by the mononuclear phagocyte system. The size is 108 ± 4 nm, with a favorable polydispersity index (PDI). Subsequent tests assessed R-PD-Pyro NPs@M’s size and PDI variations of R-PD-Pyro NPs@M in phosphate buffered solution (PBS) and 10% fetal bovine serum (FBS) (Fig. S5 in Supporting information). It demonstrated stability in PBS and remarkably maintained stability in 10% FBS. This robust stability is credited to the M1 camouflage, enabling R-PD-Pyro NPs@M to resist protein adsorption. Coating with the M1 macrophage membrane minimally altered the size of the R-PD-Pyro NPs@M, which was confirmed by transmission electron microscopy (TEM) results revealing the core-shell structure (Fig. 1E).
Figure 1
Figure 1.
Characterization of SDDDSs. (A) Structural formula and (B) 1H NMR structure of R-PD-Pyro. (C) Gel electrophoresis analysis protein expression of R-PD-Pyro NPs@M. (D) DLS analysis of R-PL-Pyro NPs, R-PD-Pyro NPs, R-PD-Pyro NPs@M, R-PD-Pyro NPs@M+MMP2. (E) TEM images of R-PL-Pyro NPs, R-PD-Pyro NPs, R-PD-Pyro NPs@M, R-PD-Pyro NPs@M+MMP2 (scale bar: 100 nm). (F) Changes in diameter of R-PD-Pyro NPs@, R-PL-Pyro NPs@M incubated with MMP2 for 12 h. (G) Confocal images of SCC7 cells at different depths after 12 h incubation with R-PL-Pyro NPs, R-PD-Pyro NPs, R-PD-Pyro NPs@M (scale bar: 250 µm). (H) Fluorescence images of CRT exposure on the surface of SCC7 cells (scale bar: 100 µm). M1: macrophage membranes; Ⅰ: R-PL-Pyro NPs; Ⅱ: R-PD-Pyro NPs; Ⅲ: R-PL-Pyro NPs@M; Ⅳ: R-PD-Pyro NPs@M+MMP2. Data are represented as mean ± SD (n = 3).
The expression of MMP2 has been reported to be elevated in nearly all malignant tumors and is closely associated with tumor progression. Therefore, utilizing MMP2 to develop SDDDSs that specifically respond to the tumor microenvironment, particularly by achieving cleavage outside tumor cells, appears to be a favorable choice. The adaptive stimulus-responsive effects of SDDDSs were characterized by co-cultivation with MMP2 enzyme using DLS and TEM (Figs. 1E and F). After 12 h of co-cultivation with MMP2, the size of R-PD-Pyro NPs@M significantly decreased to 62.97 ± 10 nm. TEM also revealed a reduction in size and even the loss of the stable structure of R-PD-Pyro NPs@M, indicating its adaptive response within TME. The release of R848, linked to the peptide chain by disulfide bonds, was evaluated under different concentrations of ROS (Fig. S6 in Supporting information). Under high ROS stimulation, R-PD-Pyro NPs@M showed an increase in R848 release within 12 h, demonstrating rapid and stable release in the TME at abnormal ROS levels. This smart, stimulus-triggered drug release ensures the accurate release of the drug at the tumor site, thereby avoiding toxic side effects such as fever, reduced lymphocytes, and severe cytokine storms caused by R848.
Tumor penetration of SDDDSs was evaluated using multicellular spheroids as a simulated SCC7 tumor model in vitro. The fluorescence intensity of the R-PD-Pyro NPs@M group was significantly higher than that of the control groups (R-PL-Pyro NPs and R-PD-Pyro NPs) (Fig. 1G), which was attributed to the adaptive response capability that allowed iRGD-Pyro to penetrate the tumor more effectively, facilitated by the M1 macrophage membrane. These results validate the successful preparation of stable R-PD-Pyro NPs@M, demonstrating its capability to respond to MMP2 and ROS in TME, leading to the cleavage into iRGD-Pyro and R848, and facilitating enhanced deep tumor penetration.
The uptake of SDDDSs by tumor cells and the resulting ROS generation are critical factors for PDT efficacy [18]. SDDDSs co-cultured with SCC7 cells for 4 h at the same concentration (Fig. S7 in Supporting information) showed Pyro fluorescence intensity in both R-PD-Pyro NPs and R-PD-Pyro NPs@M groups, with the latter showing the most robust fluorescence intensity. The enhanced cellular uptake in the R-PD-Pyro NPs@M group was attributed to an adaptive response to the high concentration of MMP2, which promoted effective uptake by tumor cells. ROS generation induced by PDT with laser irradiation for different SDDDSs groups showed that the R-PD-Pyro NPs@ML group exhibited the most vigorous green fluorescence intensity, indicating effective Pyro uptake and ROS generation (Fig. S8 in Supporting information). Cell counting kit-8 (CCK8) assay results (Fig. S9 in Supporting information) confirmed the robust in vitro anti-tumor activity of the R-PD-Pyro NPs@ML group induced by PDT with laser irradiation. This is primarily attributed to the ability of PDT to inhibit tumors by generating ROS.
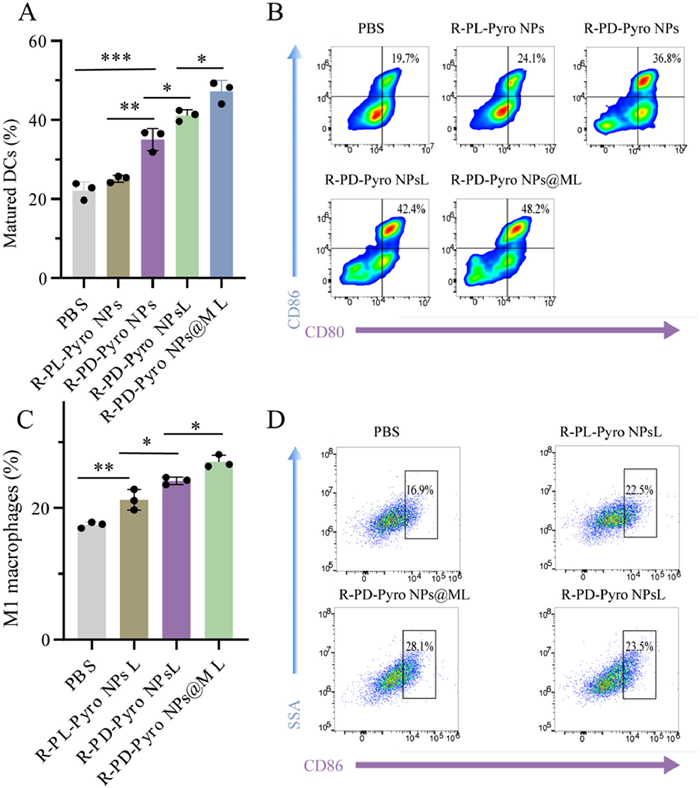
As is well known, calreticulin (CRT) serves as the primary marker of ICD induction, and PDT can induce ICD, promoting the maturation of dendritic cells (DCs) and antigen presentation. CRT expression in SCC7 cells induced by PDT when the laser is on (Fig. 1H) showed the strongest significantly increased exposure of CRT in the R-PD-Pyro NPs@ML group, indicating a potent ICD. DCs are crucial antigen-presenting cells (APCs). The complex interactions within the TME lead to their functional impairment, significantly diminishing the effectiveness of tumor immunotherapy. This has laid the foundation for a series of immune restoration approaches targeting DCs, aiming to improve the immunosuppressive microenvironment and enhance tumor immunotherapy [19]. Maturation of DCs after co-culture with SCC7 cells and SDDDSs showed approximately 2.1-fold higher DCs maturation in the R-PD-Pyro NPs@ML group compared to the PBS control, suggesting synergistic promotion of DCs maturation by PDT and R848 (Figs. 2A and B). M1 macrophages play a critical role in the immune system [20]. And their abundance is closely linked to the low survival rates observed in various cancers, especially when TAMs lean toward the M2-polarized state [21]. M2 polarization tends to promote tumor growth and immune evasion. Consequently, interventions aimed at modulating macrophage polarization, particularly inducing a shift towards the M1 phenotype, emerge as a promising avenue in cancer therapy. The expression of CD86, a surface marker for M1 macrophages, increased by 11% in the R-PD-Pyro NPs@ML group compared to the PBS group (Figs. 2C and D). This finding suggests that R-PD-Pyro NPs@ML exhibits potential efficacy in promoting TAMs’ polarization towards the M1 phenotype.
Figure 2
Figure 2.
(A, B) Flow cytometric quantification of DCs maturation. (C, D) Flow cytometric quantification of M1 macrophages. Data are represented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Tumor accumulation and organ distribution of SDDDSs were investigated using an in vivo imaging system. Over time, the fluorescence intensity of R-PD-Pyro NPs@M exceeded that of the R-PD-Pyro group, highlighting the evaded surveillance active targeting capability of the M1 macrophage membrane (Fig. 3A). After 36 h, R-PD-Pyro NPs@M fluorescence intensity decreased significantly, indicating easy metabolism, clearance, and good biological safety. Mouse necropsy confirmed the successful evasion of the liver’s phagocytic system by R-PD-Pyro NPs@M, demonstrating its enhanced capability for tumor accumulation (Fig. 3B). Tumor sections showed enhanced fluorescence images in the R-PD-Pyro NPs@M group, confirming effective accumulation and penetration (Fig. 3C).
Figure 3
Figure 3.
(A) Fluorescence images of mice in (a) R-PD-Pyro NPs@M, (b) R-PD-Pyro NPs-injected groups. (B) Ex vivo fluorescence imaging of tumors and main organs at 36 h post intravenous injection. (C) Fluorescence images of tumor frozen sections at 36 h post intravenous injection. Treatment groups: (a) R-PD-Pyro NPs@M, (b) R-PD-Pyro NPs (scale bar: 200 µm). (D) Changes of Tumor volume during the treatment. (E) Changes of Tumor weight from each group at the end of the treatment. (F) The tumor sections were stained with KI67, TUNEL and H&E (scale bar: 100 µm). Treatment groups: (a) PBS, (b) R-PD-Pyro NPs, (c) R-PL-Pyro NPsL, (d) R-PD-Pyro NPsL, (e) R-PD-Pyro NPs@ML. Data are represented as mean ± SD (n = 4). *P < 0.05, **P < 0.01, ****P < 0.0001.
Tumor suppression by SDDDSs was evaluated by comparing relative tumor volume and weight (Figs. 3D and E, Fig. S11 in Supporting information). R-PD-Pyro NPsL and R-PL-Pyro NPsL groups exhibited superior inhibitory effects compared to the PBS control, attributed to the high ROS concentration, which promoted R848 release and inhibited OSCC growth R-PD-Pyro NPsL showed superior inhibition to R-PL-Pyro NPsL due to high MMP2 levels, promoting iRGD-Pyro release and enhanced penetration. R-PD-Pyro NPs@ML showed the best tumor suppression, maximizing efficacy through efficient M1 macrophage membrane-mediated accumulation.
Validation by KI67, terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL), and haematoxylin and eosin (H&E) staining further supported tumor suppression by R-PD-Pyro NPs@ML, demonstrating reduced proliferation, increased apoptosis, and consistent histological changes (Fig. 3F). These results highlight R-PD-Pyro NPs@ML’s efficient accumulation, adaptive response-induced release of iRGD-Pyro, and deep penetration into the TME, thereby realizing synergistic PDT and tumor immunotherapy against OSCC.
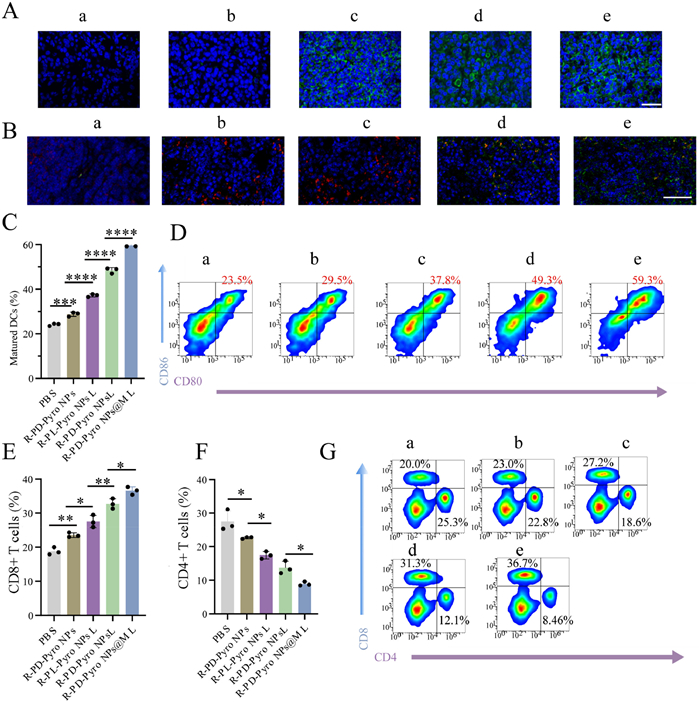
The preceding findings have validated the efficacy of R-PD-Pyro NPs@M in inhibiting tumor proliferation. A subsequent in-depth exploration of its mechanism uncovered that PDT induces ICD reactions, activating the immune microenvironment, while R848 promotes DCs maturation and repolarizes TAMs toward the M1 phenotype. CRT expression in different tissues was examined to gauge the extent of ICD reactions post-nanoparticle action (Fig. 4A). The R-PD-Pyro NPs@M group exhibited a robust ICD reaction under laser stimulation, a factor conducive to subsequent DCs maturation. Furthermore, macrophages within tumors exhibit notable plasticity, with a shift toward the M1 phenotype demonstrating an anti-tumor effect. Following treatment with R-PD-Pyro NPs@M, M1 macrophage expression markedly increased, while TAMs decreased (Fig. 4B). Subsequent evaluation focused on the maturation of DCs within lymph nodes. Mature DCs migrating from tumors to lymph nodes contribute to effective antigen presentation (Figs. 4C and D) [19]. In comparison to other groups, the R-PD-Pyro NPs@M group demonstrated outstanding anti-tumor immunity (Figs. 4E–G). This success was attributed not only to the efficient tumor site accumulation and deep tumor penetration but also to PDT’s indirect induction of DCs maturation through ICD and the direct benefits derived from the action of R848. The subsequent examination of T cell activation in tumor tissue revealed, as anticipated that the R-PD-Pyro NPs@M group exhibited the highest proportion of CD8+ T cells (Figs. 4E and G). Based on the above, it is inferred that the outstanding intratumoral immunotherapeutic effect of R-PD-Pyro NPs@M is not only due to the synergistic immune action of PDT and R848, but also inseparably linked to the deep tumor infiltration by Pyro and R848, enabling them to function more effectively. Therefore, R-PD-Pyro NPs@M possesses the potential to reshape the immune dynamics within the tumor microenvironment, enhancing the body’s ability to resist tumor progression.
Figure 4
Figure 4.
(A) Fluorescence images of CRT exposure in the tumor sections from each treatment group (scale bar: 50 µm). (B) Fluorescence images of M2 macrophages marked as CD206+ (red) and M1 macrophages marked as CD80+ (green) from each treatment group (scale bar: 200 µm). (C, D) Flow cytometric quantification of DCs maturation from each treatment group. (E–G) Flow cytometric quantification of CD8+, CD4+ T cells from each treatment group. a: PBS, b: R-PD-Pyro NPs, c: R-PL-Pyro NPsL, d: R-PD-Pyro NPsL, e: R-PD-Pyro NPs@ML. Data are represented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Concerns about the immunotoxicity of R848 were addressed by prioritizing the biosafety of the SDDDSs during OSCC inhibition using PDT and an immunostimulatory agent. Blood samples, liver function, and blood biochemical indicators in SDDDSs-treated mice showed no significant abnormalities (Fig. S12 in Supporting information). Consistent body weight (Fig. S13 in Supporting information) and H&E staining of major organs (Fig. S14 in Supporting information) further confirmed the reliable safety profile of STDDS.
In conclusion, our study successfully developed SDDDSs (R-PD-Pyro NPs@M) with stimuli-triggered downsized functionality specifically tailored to the extracellular TME. This innovative design effectively exploits the synergistic therapeutic potential of PDT and enhanced tumor immunotherapy. Our treatment strategy skillfully addresses the challenges of precise tumor targeting and accumulation, deep tumor penetration and efficient cellular uptake. In particular, it significantly overcomes the limitations associated with conventional PDT and immunotherapy approaches. Our therapeutic approach demonstrates the ability to induce apoptosis in tumor cells, induce ICD, polarize TAMs toward the M1 phenotype, and promote substantial T-cell infiltration into tumors. Consequently, these results culminate in exceptional therapeutic efficacy for OSCC.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81802709); the Shandong Provincial Natural Science Foundation, China (Nos. ZR2023MH230, ZR2023MH096); the Shandong Provincial Postdoctoral Innovative Talents Funded Scheme; Plan of Young Scholars of Shandong University.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109619.
[1]
G. Barillari, O. Melaiu, M. Gargari, et al., Int. J. Mol. Sci. 23 (2022) 8336. doi: 10.3390/ijms23158336
[2]
J. Michot, C. Bigenwald, S. Champiat, et al., Eur. J. Cancer 54 (2016) 139–148. doi: 10.1016/j.ejca.2015.11.016
[3]
S. Han, K. Huang, Z. Gu, et al., Nanoscale 12 (2020) 413–436. doi: 10.1039/C9NR08086D
N.R. Anderson, N.G. Minutolo, S. Gill, et al., Cancer Res. 81 (2021) 1201–1208.
[21]
A. Mantovani, F. Marchesi, A. Malesci, et al., Nat. Rev. Clin. Oncol. 14 (2017) 399–416. doi: 10.1038/nrclinonc.2016.217
Scheme 1
Diagram depicting the design of SDDDSs (R-PD-Pyro NPs@M) of synergistic PDT and enhance immunotherapy for OSCC. (ⅰ) ROS cleavage site. (ⅱ) MMP2 cleavage site. (a) Augmented tumor targeting and (b) accumulation. (c) Transformation of stimulus-responsive size-reductive SDDDSs into smaller-sized tumor targeted iRGD-Pyro and R848. (d) Deep tumor penetration and cellular uptake.
Figure 1
Characterization of SDDDSs. (A) Structural formula and (B) 1H NMR structure of R-PD-Pyro. (C) Gel electrophoresis analysis protein expression of R-PD-Pyro NPs@M. (D) DLS analysis of R-PL-Pyro NPs, R-PD-Pyro NPs, R-PD-Pyro NPs@M, R-PD-Pyro NPs@M+MMP2. (E) TEM images of R-PL-Pyro NPs, R-PD-Pyro NPs, R-PD-Pyro NPs@M, R-PD-Pyro NPs@M+MMP2 (scale bar: 100 nm). (F) Changes in diameter of R-PD-Pyro NPs@, R-PL-Pyro NPs@M incubated with MMP2 for 12 h. (G) Confocal images of SCC7 cells at different depths after 12 h incubation with R-PL-Pyro NPs, R-PD-Pyro NPs, R-PD-Pyro NPs@M (scale bar: 250 µm). (H) Fluorescence images of CRT exposure on the surface of SCC7 cells (scale bar: 100 µm). M1: macrophage membranes; Ⅰ: R-PL-Pyro NPs; Ⅱ: R-PD-Pyro NPs; Ⅲ: R-PL-Pyro NPs@M; Ⅳ: R-PD-Pyro NPs@M+MMP2. Data are represented as mean ± SD (n = 3).
Figure 3
(A) Fluorescence images of mice in (a) R-PD-Pyro NPs@M, (b) R-PD-Pyro NPs-injected groups. (B) Ex vivo fluorescence imaging of tumors and main organs at 36 h post intravenous injection. (C) Fluorescence images of tumor frozen sections at 36 h post intravenous injection. Treatment groups: (a) R-PD-Pyro NPs@M, (b) R-PD-Pyro NPs (scale bar: 200 µm). (D) Changes of Tumor volume during the treatment. (E) Changes of Tumor weight from each group at the end of the treatment. (F) The tumor sections were stained with KI67, TUNEL and H&E (scale bar: 100 µm). Treatment groups: (a) PBS, (b) R-PD-Pyro NPs, (c) R-PL-Pyro NPsL, (d) R-PD-Pyro NPsL, (e) R-PD-Pyro NPs@ML. Data are represented as mean ± SD (n = 4). *P < 0.05, **P < 0.01, ****P < 0.0001.
Figure 4
(A) Fluorescence images of CRT exposure in the tumor sections from each treatment group (scale bar: 50 µm). (B) Fluorescence images of M2 macrophages marked as CD206+ (red) and M1 macrophages marked as CD80+ (green) from each treatment group (scale bar: 200 µm). (C, D) Flow cytometric quantification of DCs maturation from each treatment group. (E–G) Flow cytometric quantification of CD8+, CD4+ T cells from each treatment group. a: PBS, b: R-PD-Pyro NPs, c: R-PL-Pyro NPsL, d: R-PD-Pyro NPsL, e: R-PD-Pyro NPs@ML. Data are represented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.


 DownLoad:
DownLoad:


 下载:
下载:






 下载:
下载:
