Received Date:
07 December 2023 Accepted Date:
29 January 2024 Revised Date:
02 January 2024 Available Online:
15 December 2024
Abstract:
Highly active cathode catalysts for efficient formation/decomposition of Li2O2 are essential for the performance improvement of lithium-oxygen batteries (LOBs). In this study, a grain-refining Co0.85Se catalyst with a lattice spacing of 2.69 Å of (101) plane closely matching with the (100) plane (2.72 Å) of Li2O2 was applied for high-performance LOBs. Highly (101) plane exposed Co0.85Se@CNT was synthesized by a simple one-pot hydrothermal method. The Co0.85Se with the lattice matching effect not only led to the efficient conversion and polarized growth of Li2O2, but also prevented the formation of byproducts. Density functional theory (DFT) calculations reveal that Co0.85Se (101) plane has the intrinsic catalytic ability to generate/decompose Li2O2 during ORR/OER process, due to its homogeneous electron distribution, suitable adsorption energy, and promoted Li2O2 growth kinetics. As a consequence, the (101) plane highly exposed Co0.85Se@CNT-80 electrode exhibited remarkable cycle stability over 2400 h at 100 mA/g and 290 cycles at 500 mA/g, which is about 2 times longer than other electrodes.
Lithium oxygen batteries (LOBs) based on reversible reaction O2 + 2Li+ + 2e− ↔ Li2O2 are considered as one of the most promising electrochemical energy storage systems due to their high theoretical energy density (3500 Wh/kg), low cost and environmental friendliness [1–6]. Meanwhile, several challenges including poor circularity, low round-trip efficiency and serious polarization caused by sluggish kinetics of oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) need to be addressed before practical utilization [7–16]. Nevertheless, it is urgent to develop new bifunctional catalysts to improve OER and ORR kinetics. In order to improve the reaction rate, it is necessary to optimize the cathode catalyst, since cathode is the main place of electrochemical reaction [6,17–19].
The reversible formation and decomposition of discharge product Li2O2 on the cathode catalyst is closely related to the electrochemical performance of LOBs to a great extent. Most researchers postulated that the formation/decomposition process of Li2O2 undergoes through LiO2 intermediate, which is a strong oxidant and can lead to the passivation of the cathode catalyst and decomposition of electrolyte [4,20–22]. Furthermore, the accumulation of side products during the ORR/OER process also leads to the deterioration and capacity fading of LOBs cell [21,23–26]. It is reported that searching for a grain-refining agent catalyst with the lattice matching effect to discharge products, which can accelerate the reaction kinetics of discharge product for a controlled morphology and designed composition is a facile strategy to enhance the electrochemical performance of LOBs [20,27–29]. The grain-refining agents can serve as a grain promoter for the nucleation, initial growth and decomposition of discharge product through controlling the reaction kinetics based on lattice matching of the crystal structure between catalyst and discharge product. However, only few cathode catalysts have the lattice matching nature with Li2O2 or LiO2. For example, IrLi can serve as a template for crystal LiO2 growth because of the low interfacial energy (−0.36 eV/Å2) and good lattice matching between IrLi (111) plane and LiO2 (111), allowing the strong oxidant LiO2 to be generated and stabilized [30]. CeO2 effectively reduces overpotential during ORR and OER due to well matched interplanar spacing of CeO2 (200) facet (2.72 Å) and Li2O2 (100) facet (2.74 Å) [27]. Hexagonal MoSe2 with the same space group (P 63/mmc) and similar a, b-lattice parameters (3.288 Å) with Li2O2 can act as the grain promoter to induce initial nucleation and epitaxial growth of Li2O2 grains [20]. The similar d-spacing of Fe2O3 (104) and Li2O2 (100) plane induces epitaxial growth of Li2O2 on the Fe2O3 surface which enhances the nucleation of Li2O2 and suppresses the formation of amorphous Li2O2 [28]. Interestingly, (101) plane of Co0.85Se exhibits the lattice plane d-spacing closing to that of Li2O2 (100) and is expected to act as an effective grain refiner for the formation/decomposition of discharge products. In addition, as a promising transition metal selenide, Co0.85Se figures a vacancy defect structure and special electronic structure, which has been already applied in water splitting [31] and sodium-ion batteries [32].
In this work, Co0.85Se@CNT was applied as a grain-refining catalyst to investigate the lattice matching effect on the electrochemical performance of LOBs. The (101) plane highly exposed Co0.85Se nanocrystals with a size of 5–10 nm uniformly embedded on the CNT substrate. The (101) plane of Co0.85Se with a lattice spacing of 2.69 Å is closely matches with the (100) plane (2.72 Å) of Li2O2, which is favor to the conversion from LiO2 to Li2O2 and the epitaxial growth of Li2O2 crystal, reduce the overpotentials and extend the cycle life. It is identified that the (101) plane highly exposed Co0.85Se@CNT cathode exhibited low overpotentials and superior electrochemical performance. The effect of epitaxial growth of discharge product under the existence of Co0.85Se nanocrystals was observed. Density functional theory (DFT) calculations demonstrated that Co0.85Se (101) plane has the intrinsic catalytic ability to generate/decompose Li2O2 during ORR/OER process, with better conductivity, lower overpotential and higher catalytic activity. As a consequence, Co0.85Se@CNT-80 electrode exhibited remarkable cycle stability about 2448 h at 100 mA/g and 292 cycles at 500 mA/g, superior specific capacity of 9327.9 mAh/g.
The Co0.85Se@CNT samples were fabricated by the one-step hydrothermal method following a heat treatment as shown in Fig. 1a. Samples were denoted as Co0.85Se@CNT-80, Co0.85Se@CNT-40, and pure Co0.85Se, respectively, based on the addition of 80, 40, 0 mg of CNT in the synthesis process. The detailed morphologies of Co0.85Se@CNT-80, Co0.85Se@CNT-40, pure Co0.85Se nanoparticles and pure CNT captured by the field emission scanning electron microscope (SEM) were shown in Fig. 1b and Fig. S1 (Supporting information). The average diameter of pure CNT was about 30 nm with the length of several microns. After inlaying Co0.85Se nanocrystals, the surface of CNT became uneven with wrinkle-like protrusions. The pure Co0.85Se nanoparticles showed an irregular morphology with an average particle size of about 50 nm (Figs. S1b and c). Figs. 1c and d showed the transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images of Co0.85Se@CNT-80. Co0.85Se nanocrystals were dispersedly embedded on the surface of CNT. The diffraction ring in the selected area electron diffraction (SAED) pattern of Co0.85Se@CNT-80 (inset of Fig. 1c) could be perfectly indexed with the (101), (102) and (110) planes of Co0.85Se and the (100) plane of CNT, which further proved the formation of high crystalline Co0.85Se@CNT composite. The high-resolution TEM (HRTEM) image in Fig. 1d showed the size of the Co0.85Se nanocrystals was 5–10 nm with the lattice d-spacing of 0.27 nm matched well with the (101) plane of Co0.85Se, indicating the highly exposed (101) plane for Co0.85Se@CNT-80 sample. Figs. 1e–h showed the TEM and HRTEM images of Co0.85Se@CNT-40 and pure Co0.85Se samples. The Co0.85Se particles on Co0.85Se@CNT-40 exhibited a large particle size of about 20 nm and irregular morphology, which might be caused by the low content of CNT that decreased the surface area for the nucleation of Co0.85Se during synthesis process. The interlayer spacing of 0.18 nm and 0.27 nm corresponding to (110) plane and (101) plane of Co0.85Se were identified indicating the inhomogenous exposed crystal planes.
Figure 1
Figure 1.
(a) Schematic illustration of the synthetic process of Co0.85Se@CNT and the ORR/OER at Co0.85Se (101) plane. (b) SEM, (c) TEM and (d) HRTEM images with SAED pattern of Co0.85Se@CNT-80. (e) TEM and (f) HRTEM images with SAED pattern of Co0.85Se@CNT-40. (g) TEM and (h) HRTEM images with SAED pattern of pure Co0.85Se.
The Co0.85Se (101) plane has a surface spacing (2.69 Å) that closely matches with Li2O2 (100) plane (2.72 Å) with a mismatch degree smaller than 5%, which can act as a grain-refined agent to facilitate the nucleation and growth of peroxide on this plane (Fig. 1a), and in turn enhance the electrochemical performance of Co0.85Se cathode catalyst in LOBs. The lattice matching (101) plane can obtain superior adsorption capability for Li2O2 and Li4O4, optimized electronic state and catalytic capability for the formation/decomposition of Li2O2 (detail description see the following DFT calculation part).
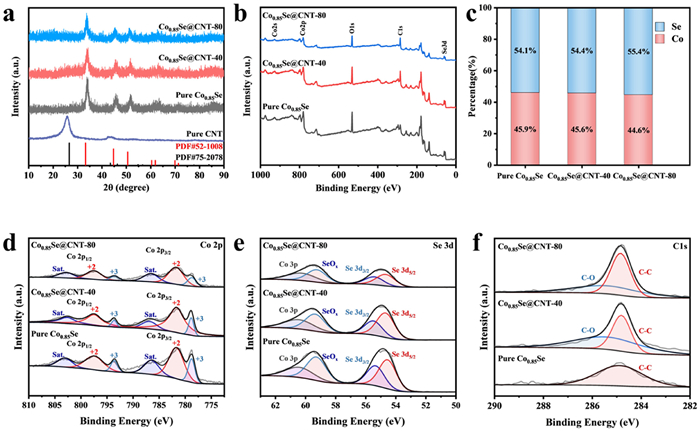
X-ray diffraction (XRD) patterns of Co0.85Se@CNT samples exhibited six diffraction peaks at 33.3°, 44.7°, 50.6°, 60.4°, 61.9° and 69.9°, corresponding to (101), (102), (110), (103), (112) and (202) planes of the hexagonal Co0.85Se (JCPDS No. 52–1008), respectively (Fig. 2a). The diffraction peak at 26.6° in pure CNT was matched to the (100) plane of CNT (JCPDS No. 75–2078). Therefore, the absence of secondary phase and other impurities confirmed the pure phase and high crystalline degree of the Co0.85Se@CNT samples.
Figure 2
Figure 2.
(a) XRD patterns of Co0.85Se@CNT and pure CNT. (b) XPS survey spectra, (c) Co: Se ratio and high-resolution of (d) Co 2p, (e) Se 3d, and (f) C 1s XPS spectra of Co0.85Se@CNT.
Fig. S2 (Supporting information) showed the N2 adsorption/desorption isotherms of the four samples. The specific surface area of the four samples calculated by the BET (Brunauer-Emmett-Teller) method was 45.132 (pure Co0.85Se), 66.561 (Co0.85Se@CNT-40), 62.243 (Co0.85Se@CNT-80), and 145.116 m2/g (CNT). The specific surface area of Co0.85Se@CNT-80 was smaller than that of CNT probably due to the incorporation of Co0.85Se particles and their dense coating. Nevertheless, the specific surface area of Co0.85Se@CNT-80 is approximately 1.4 times higher than that of pure Co0.85Se nanoparticles. The large specific surface area is beneficial to increase the active site and promote the wettability of electrolyte, thus improving the battery performance.
X-ray photoelectron spectroscopy (XPS) was applied to identify the surface electronic state and chemical composition of Co0.85Se@CNT composites. The survey spectrum in Fig. 2b confirmed the signals of the Co, Se and C elements, and their core energy level spectra were shown in Figs. 2d-f, respectively. In the Co 2p spectra, Co 2p1/2, Co 2p3/2 and two satellite peaks were deconvoluted by a Gaussian fitting method. These peaks fitted well with the two spin-orbit doublets characteristic of Co2+ and Co3+. The binding energies of Co3+ 2p3/2 and Co3+ 2p1/2 were 778.69 and 793.92 eV, respectively, which agreed well with the reported values [32]. The 781.56 and 797.42 eV peaks were attributed to Co2+ and the peaks observed at 786.80 and 802.59 eV were fitted with shakeup satellite peaks of Co 2p. The results indicated that cobalt mainly exists on the surface as Co2+ and the presence of Co3+ may be due to the Co vacancy [32,33]. Spectrum in Fig. 2e exhibited the electron-binding energies of 54.66 and 55.46 eV, corresponding to Se 3d5/2 and Se 3d3/2, respectively. On account of the surface oxidation of Se, the peak appeared at 59.35 eV was assigned to SeOx and the peak at 60.42 eV was attributed to Co 3p [34]. As illustrated in Fig. 2f, the high-resolution C 1s spectrum had two fitting peaks at 284.8 and 285.3 eV corresponding to C-C and C-O bonds, respectively. The contents of Co and Se were shown in Fig. 2c. The Co: Se ratios calculated from XPS spectra were 0.82 for Co0.85Se@CNT-80 and pure Co0.85Se, and 0.85 for Co0.85Se@CNT-40, which was close to the stoichiometry.
The electrochemical performance of Co0.85Se@CNT as the cathode catalyst of LOBs was carried out using the 2032-type coin cells with lithium sheet as the counter electrode. Fig. 3a exhibited the cyclic voltammetry (CV) curves of the four samples (Co0.85Se@CNT-80, Co0.85Se@CNT-40, pure Co0.85Se, and pure CNT) at a scanning speed of 0.15 mV/s over a voltage range of 2.35–4.35 V. During the cathodic scan, all four catalysts showed reduction peaks, corresponding to the ORR process related to the formation of discharge product Li2O2 (2Li+ + O2 + 2e− → Li2O2). Co0.85Se@CNT-80 had more positive ORR peak at 2.54 V. Meanwhile, during the anodic sweep, the oxidation peak of Co0.85Se@CNT-80 located at 3.88 V, lower than that of Co0.85Se-40 (4.24 V) and pure Co0.85Se, which is connected with the bulk decomposition process of the discharge product (Li2O2 → O2 + 2Li+ + 2e−) while only a weak peak was detected in pure CNT. In short, compared with other electrodes, much higher anodic peak current and large integration area were achieved for the Co0.85Se@CNT-80 electrode, implying that more discharge products can be reversibly decomposed and thus much ameliorated catalytic kinetics performance. The electrochemical impedance spectroscopy (EIS) spectra of these four samples were also collected before and after the CV test under open-circuit voltage in the frequency range of 105–0.01 Hz (Fig. S3 in Supporting information), and the charge transfer resistance (Rct) can be evaluated by the semicircles diameter in the middle frequency range. The Co0.85Se@CNT-80 electrode exhibited a Rct of 22.1 Ω before the CV test, which was lower than that of Co0.85Se@CNT-40 (74.8 Ω), pure Co0.85Se (41.5 Ω) and CNT (51.7 Ω). After the CV test, the charge transfer resistance of these four samples all showed a tendency to increase. Notably, the Rct of Co0.85Se@CNT-80 electrode (60.8 Ω) was also smaller than that of Co0.85Se@CNT-40 (81.7 Ω), pure Co0.85Se (61.1 Ω) and CNT (82.1 Ω) after the CV test. The low Rct observed in the Co0.85Se@CNT-80 electrode suggests a high efficiency of electron and ion transfer during ORR/OER. This characteristic facilitates the formation/decomposition of discharge products and contributes to an extended electrode lifespan.
Figure 3
Figure 3.
(a) CV curves at a scan rate of 0.15 mV/s at a voltage window of 2.35–4.35 V. (b) Initial discharge/charge profiles at a current density of 100 mA/g over a voltage range of 2.35–4.35 V of different electrodes. (c) The discharge/charge profiles at various current densities ranging from 100 mA/g to 500 mA/g of Co0.85Se@CNT-80. (d) The cycling performance of Co0.85Se@CNT-80 and (f) terminal discharge-charge voltages of different electrodes at 100 mA/g with a limited capacity of 600 mAh/g. (e) The cycling performance of Co0.85Se@CNT-80 and (f) terminal discharge-charge voltages of different electrodes at 500 mA/g with a limited capacity of 600 mAh/g.
The initial galvanostatic discharge-charge curves of four different cathodes were shown in Fig. 3b. It was noted that the Co0.85Se@CNT-80 cathode delivered a specific capacity of 9327.9 mAh/g at a current density of 100 mA/g when discharged to 2.35 V. However, Co0.85Se@CNT-40, pure Co0.85Se, and pure CNT cathodes could only exhibit 6264.6, 4911.0 and 2132.4 mAh/g, respectively. Furthermore, the LOBs with Co0.85Se@CNT-80 cathode could deliver a specific capacity of 7813.1 mAh/g when recharged to 4.35 V, which was much larger than that using Co0.85Se@CNT-40, pure Co0.85Se and pure CNT cathodes (5621.4, 3780.3 and 1550.9 mAh/g). More importantly, the initial overpotentials for ORR/OER process of the Co0.85Se@CNT-80 cathode were only 0.18/0.79 V with the voltage platforms of 2.73/4.19 V, which were remarkably lower than other cathodes, suggesting its superior electrocatalytic efficiency. The rate capability of the Co0.85Se@CNT-80 cathode was checked at different current densities from 100 mA/g to 500 mA/g (Fig. 3c). The initial discharge/charge capacity at current densities of 100, 300 and 500 mA/g were 9327.9/7813.1, 7081.8/5360.4 and 4655.3/3023.1 mAh/g, respectively. As the current density increased, the specific capacity did not fade quickly. The durability of the catalytic ability of Co0.85Se@CNT cathode in LOBs was evaluated by limiting the specific capacity at low and high current densities. Figs. 3d and e showed the selected discharge/charge profiles and terminal voltage of Co0.85Se@CNT-80, Co0.85Se@CNT-40, pure Co0.85Se, and pure CNT cathodes at a current density of 100 mA/g and limited capacity of 600 mAh/g. The (101) plane highly exposed Co0.85Se@CNT-80 cathode stably worked over 2400 h which was about 1.7 times longer than that of Co0.85Se@CNT-40 cathode embedded by irregular Co0.85Se particles. On the contrary, the batteries with pure Co0.85Se and pure CNT cathodes died after 600 and 288 h, respectively. It is worth noting that the Co0.85Se@CNT-80 cathode exhibited obviously lower ORR/OER overpotentials compared to other cathodes, in which the terminal discharge voltage was stable at around 2.62 V and the charge voltage stabilized at 4.36 V, indicating that the (101) plane highly exposed Co0.85Se@CNT-80 cathode can efficiently and sustainably maintain its catalytic ability for the formation and decomposition of discharge product Li2O2. When the current density increased to 500 mA/g (Figs. 3f and g), the Co0.85Se@CNT-80 electrode worked for 294 cycles with lower overpotentials compared to that of Co0.85Se@CNT-40 electrode (154 cycles), suggesting that the superior reversibility of Co0.85Se@CNT-80 cathode. For pure Co0.85Se and pure CNT, the voltage soon dropped to 2.0 V in the 91st, and 63rd cycle revealed in Fig. 3g and Figs. S5-S7 (Supporting information). These results demonstrated that Co0.85Se catalyst with highly exposed (101) plane exhibited superior cycle stability, large specific capacity and low overpotentials. It is thought that the (101) plane with the lattice matching effect with Li2O2 contributed significantly to the performance improvement.
To further investigate the underlying mechanism of the above-mentioned electrochemical performance improvements, the microstructures of discharged products and recharged electrodes during cycling were verified by ex-situ SEM, XRD spectroscopy, and XPS spectra, showing effective indicator of monitoring the oxygen reduction and evolution behaviors in LOBs. Figs. 4a-c showed the SEM images of pristine, full discharge, and recharged Co0.85Se@CNT-80 electrode. At the pristine state, Co0.85Se@CNT-80 presented a tubular structure with a diameter of about 30 nm, which was consistent with the previous characterization. After full discharge, the dendrite discharge products were observed with a size of 300 nm on the Co0.85Se@CNT-80. After charging, the discharge products decomposed completely and the nanotube structure was restored. These results indicated the polarized nucleation and growth of the discharge products on Co0.85Se@CNT-80 electrode, which would facilitate the infiltration of electrolyte, maintain the activity of tri-phase interface and accelerate the formation/decomposition kinetics of discharge products. It is deduced that the highly (101) plane is beneficial for the nucleation and growth of Li2O2. Figs. 4d-f showed the SEM images of Co0.85Se@CNT-40, pure CNT and pure Co0.85Se electrodes after full discharge. It can be seen that Co0.85Se@CNT-40 and CNT were still tubular structure after loading the products. The diameter of Co0.85Se@CNT-40 tube increased to about 400 nm, and the diameter of CNT increased to 100 nm, which indicated that the formation of film-like discharge products. Similarly, the film-like products can be still seen on pure Co0.85Se particles (Fig. 4f).
Figure 4
Figure 4.
SEM images of Co0.85Se@CNT-80 electrode at (a) pristine, (b) full discharge, and (c) 1st recharged at the current density of 100 mA/g. SEM images of (d) Co0.85Se@CNT-40, (e) pure CNT, and (f) pure Co0.85Se electrode at full discharge. (g) XRD patterns at different discharge/charge stages. (i) High-resolution XPS spectra of Li 1s at three selected states, corresponding to states (1–3) in (h), respectively.
Accordingly, as shown in ex-situ XRD pattern (Fig. 4g and Fig. S8 in Supporting information) of Co0.85Se@CNT-80, there were two characteristic peaks except for the peaks of carbon paper and Co0.85Se after discharge, which belonged to Li2O2 [20]. The peaks disappeared after 1st, 10th, and 100th recharging, demonstrating the good reversible cycling performance of Co0.85Se@CNT-80. Fig. 4i showed the ex-situ high-resolution XPS of Li 1s of Co0.85Se@CNT-80 electrode in different discharge states (corresponding to Fig. 4h) at a current density of 100 mA/g to identify the evolution of the chemical state of discharge products. At the initial discharge (100 mAh/g), Li2-xO2 (a mixture of LiO2 and Li2O2) with binding energies of about 56.10 eV [35–38] was the main discharge product. When discharged to 2000 mAh/g, the peak of Li2O2 with binding energies of about 55.07 eV [35,39] became stronger indicating the conversion from Li2-xO2 to Li2O2. After full discharge, Li2O2 became the dominant phase of the discharge products indicating the high-efficiency conversation from the intermediate to the final discharge products of the (101) plane highly exposed Co0.85Se cathode catalyst. For comparison, there was no obvious diffraction peaks of Li2O2 for the discharge product in the XRD pattern (Fig. S9 in Supporting information) of CNT electrodes after full discharge. The XPS spectra (Fig. S10 in Supporting information) showed that the products were mainly Li2-xO2 and a small amount of Li2CO3 and Li2O2. The existence of carbonate indicated that CNT was involved in the discharge reaction.
The polarized growth and composition evolution of discharge product demonstrated that the nucleation and growth of discharge products occurred directly on the surface of Co0.85Se@CNT-80, in which the (101) plane of Co0.85Se played an important role in promoting the formation, conversation and growth of Li2O2. The highly exposed Co0.85Se (101) plane matches well with Li2O2 (100) plane and exhibits superior catalytic capability, which can act as a grain refiner, promoting the formation of a dendrite-like Li2O2 structure instead of a closed membrane.
To further demonstrate the superior catalytic capability of lattice matching plane and the mechanism during ORR/OER process of the Co0.85Se catalyst, DFT calculations are conducted to provide reaction information at atomic scale. The (101) and (110) planes detected by HRTEM were selected for calculations. Fig. 5a shows the electron localization function (ELF) diagrams of (101) and (110) planes. The function ranges from 0 to 1, where values of 0, 0.5, and 1 represent very low charge density, completely delocalized electrons, and completely localized electrons, respectively [40–42]. It can be seen that the electrons are mainly concentrated on Se atom, while the charge density around Co is extremely low. The blue area in the (110) plane is deeper, which means that the stronger polarization of the Co-Se bond on the (110) plane. Compared with (110) plane, the green area (where electrons are fully delocalized) of (101) plane is arranged more closely and uniformly, which is conducive to the efficient transfer of electrons during the catalytic reaction process. Figs. 5b and c show the charge density difference diagrams and Bader charge of bonded Co and O atoms of (101) and (110) planes adsorbed with Li2O2 and Li4O4. An evident charge transfer can be observed between the adsorbents and the substrate (yellow and blue represent the increase and decrease in charge density, respectively). On both planes, charge transfer paths are established between the substrate and the adsorbents via Co–O bonds, with Co acting as charge transfer center. The difference is that the adsorption site of O atom on (101) plane is the Co top site, while on (110) plane is the Co-Co bridge bond. Additionally, one oxygen atom of Li2O2 after adsorption on (110) plane is almost detached and bonded to the Co atom on the surface (Fig. 5c), which could lead to instability of the peroxide on the surface and be detrimental to the OER process. On the contrary, the structure of Li2O2 on (101) plane remains basically unchanged. Compared to the (101) plane, the O atoms on (110) plane acquire more charge and form stronger Co-O bonds after the adsorption of Li2O2 and Li4O4. Fig. 5d and Table S1 (Supporting information) depict the Bader charge transfer (Q), adsorption energy (Eads), and distance (Å) between the different adsorbents and substrates on the (101) and (110) planes. It can be seen that the (101) plane shows low adsorption energy and considerable distance between the adsorbent and the substrate, which is conducive to the ORR/OER reaction. Especially for Li2O2, the adsorption energy on the (110) plane (−4.08 eV) significantly exceeds that on the (101) plane (−1.85 eV). The large adsorption energy may be attributed to the large change in the Li2O2 structure on the (110) plane. In addition, the Bader charge of (110) plane adsorbed Li2O2 is negative while that of (101) is positive. The negative Bader charge of (110) plane signifies a greater charge exchange between O atoms of Li2O2 and Co atoms on (110) plane, indicating stronger Co-O bonds. This is consistent with the large adsorption energy. Large adsorption energy and increased charge exchange will hinder the decomposition of Li2O2 during the OER reaction.
Figure 5
Figure 5.
(a) Electron localization function (ELF) diagrams of (101) plane and (110) plane. Charge density difference plots of Li2O2* and Li4O4* on (b) (101) plane and (c) (110) plane. (d) Bader charge transfer (Q), adsorption energy (Eads), and distance (Å) between different adsorbates and substrates on Co0.85Se (101) and (110) planes. DOS map before and after binding of Li2O2 and Li4O4 for the Co0.85Se (e) (101) plane and (f) (110) plane. Calculated energy diagram of Co0.85Se (g) (101) plane and (h) (110) plane in the ORR and OER processes. (i) Phase diagrams of the cathode reaction on Co0.85Se (101) and (110) planes.
Furthermore, the electron density of state (DOS) diagram of Co0.85Se (101) and (110) plane before and after adsorption of Li2O2 and Li4O4 obtained by DFT calculations are shown in Figs. 5e and f. The electronic state distributions near the Fermi energy level exist in both planes, where the DOS value of the (101) plane is larger than that of the (110) plane, indicating the excellent conductivity and charge transfer efficiency of the former. After loading Li2O2 and Li4O4, the total DOS of (101) plane near the Fermi level decreases slightly because of the poor conductivity of the adsorbents. In addition, the obvious overlap between the O and Co 3d orbitals observed in the DOS signifies their interaction, which provides evidence for the existence of Co-O bond. For (110) plane, the total DOS near the Fermi level increases slightly after loading Li2O2. After loading Li4O4, the total DOS increases significantly and the O and Co 3d orbitals contributed to the increase, which is consistent with the previously mentioned strong Co-O binding on (110) plane. It is unfavorable for the desorption of reaction products during OER process. The instability of the Co electronic structure is detrimental to the maintenance of catalyst stability. In contrast, stable DOS of (101) plane indicates the excellent charge transfer ability and bifunctional catalytic capability for LOBs.
Figs. 5g and h record the calculated energy diagrams for ORR and OER procedures on the (101) and (110) plane. Four elementary reaction steps for the generation of Li4O4 are considered as following: (Ⅰ) (Li+ + e−) + O2 ↔ LiO2*, (Ⅱ) LiO2 + (Li+ + e−) ↔ Li2O2*, (Ⅲ) Li2O2 + (Li+ + e−) + O2 ↔ Li3O4*, and (Ⅳ) Li3O4 + (Li+ + e−) ↔ Li4O4*. Where the symbol * indicates that the species are adsorbed on the surface. Compared with (110) plane (ηORR/ηOER = 1.04/0.86 V; ηTOT = 1.90 V), (101) plane exhibits lower overpotentials (ηORR/ηOER = 0.61/0.64 V; ηTOT = 1.25 V) during discharge/charge processes. Fig. 5i depicts the reaction phase diagram for the generation of LiO2 and Li2O2 on (101) and (110) planes. On (101) plane, LiO2 is spontaneously formed initially when the discharge voltage is between 2.27 V and 3.15 V due to lower ΔG of LiO2. Meanwhile, Li2O2 also formed when the discharge voltage is lower than 2.71 V and the conversion from LiO2 to Li2O2 occurs at 2.27 V. The result is consistent with ex-situ XPS that the initial product of the (101) plane highly exposed Co0.85Se@CNT-80 is LiO2. For (110) plane, Li2O2 is preferentially generated below 3.82 V, while LiO2 is also produced below 3.62 V, so the product on (110) is mainly Li2O2 and LiO2, which is consistent with the observation of the ex-situ XPS on the pure Co0.85Se electrode (Fig. S9 in Supporting information). DFT calculations reveal that the lattice matching (101) plane of Co0.85Se exhibits excellent bifunctional catalytic performance with uniform electron distribution, efficient charge transfer, and lower overpotentials. The (101) plane acts as a grain refiner and forms dendritic Li2O2 instead of membranous Li2O2 by a single-electron reaction.
In conclusion, Co0.85Se@CNT catalysts with a well-designed structure were successfully prepared by a simple one-pot hydrothermal method and used as a cathode catalyst for lithium-oxygen batteries. Co0.85Se can effectively reduce the overpotential due to its intrinsic metal conductivity and the lattice matching effect with Li2O2. The Co0.85Se@CNT-80 electrodes exhibited superior cycle stability, cycling over 2448 h at 100 mA/g and lower discharge/charge overpotentials due to smaller particle size and large exposed lattice matching (101) plane, which serves as a template for growth of Li2O2 and promotes epitaxial growth of Li2O2. The ex-situ analysis showed that crystalline Li2O2 accumulated as dendrites on the Co0.85Se@CNT-80 under the assistant of Co0.85Se without other by-products. DFT calculations revealed that the Co acts as the adsorption site and charge transfer center, and the lattice matching (101) plane exhibits excellent catalytic ability for ORR and OER due to its homogeneous electron distribution, suitable adsorption energy and promoted Li2O2 growth kinetics. The present work not only demonstrates the electrocatalytic performance of Co0.85Se@CNT, but also expands the selection and design of efficient catalysts by lattice matching.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Nos. 52173286, 52207249), Major basic research project of Natural Science Foundation of Shandong Province (No. ZR2023ZD12), the State Key Laboratory of Marine Resource Utilization in South China Sea (Hainan University) (No. MRUKF2023013) and Open Program of Guangxi Key Laboratory of Information Materials (No. 221024-K).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109595.
[1]
M. Armand, J.M. Tarascon, Nature 451 (2008) 652–657. doi: 10.1038/451652a
[2]
D. Aurbach, B.D. McCloskey, L.F. Nazar, P.G. Bruce, Nat. Energy 1 (2016) 16128. doi: 10.1038/nenergy.2016.128
H. Huang, C. Cheng, G. Zhang, et al., Adv. Funct. Mater. 32 (2022) 2111546. doi: 10.1002/adfm.202111546
[41]
G. Zhang, G. Li, J. Wang, et al., Adv. Energy Mater. 12 (2022) 2103910. doi: 10.1002/aenm.202103910
[42]
A.D. Becke, K.E. Edgecombe, J. Chem. Phys. 92 (1990) 5397–5403. doi: 10.1063/1.458517
Figure 1
(a) Schematic illustration of the synthetic process of Co0.85Se@CNT and the ORR/OER at Co0.85Se (101) plane. (b) SEM, (c) TEM and (d) HRTEM images with SAED pattern of Co0.85Se@CNT-80. (e) TEM and (f) HRTEM images with SAED pattern of Co0.85Se@CNT-40. (g) TEM and (h) HRTEM images with SAED pattern of pure Co0.85Se.
Figure 2
(a) XRD patterns of Co0.85Se@CNT and pure CNT. (b) XPS survey spectra, (c) Co: Se ratio and high-resolution of (d) Co 2p, (e) Se 3d, and (f) C 1s XPS spectra of Co0.85Se@CNT.
Figure 3
(a) CV curves at a scan rate of 0.15 mV/s at a voltage window of 2.35–4.35 V. (b) Initial discharge/charge profiles at a current density of 100 mA/g over a voltage range of 2.35–4.35 V of different electrodes. (c) The discharge/charge profiles at various current densities ranging from 100 mA/g to 500 mA/g of Co0.85Se@CNT-80. (d) The cycling performance of Co0.85Se@CNT-80 and (f) terminal discharge-charge voltages of different electrodes at 100 mA/g with a limited capacity of 600 mAh/g. (e) The cycling performance of Co0.85Se@CNT-80 and (f) terminal discharge-charge voltages of different electrodes at 500 mA/g with a limited capacity of 600 mAh/g.
Figure 4
SEM images of Co0.85Se@CNT-80 electrode at (a) pristine, (b) full discharge, and (c) 1st recharged at the current density of 100 mA/g. SEM images of (d) Co0.85Se@CNT-40, (e) pure CNT, and (f) pure Co0.85Se electrode at full discharge. (g) XRD patterns at different discharge/charge stages. (i) High-resolution XPS spectra of Li 1s at three selected states, corresponding to states (1–3) in (h), respectively.
Figure 5
(a) Electron localization function (ELF) diagrams of (101) plane and (110) plane. Charge density difference plots of Li2O2* and Li4O4* on (b) (101) plane and (c) (110) plane. (d) Bader charge transfer (Q), adsorption energy (Eads), and distance (Å) between different adsorbates and substrates on Co0.85Se (101) and (110) planes. DOS map before and after binding of Li2O2 and Li4O4 for the Co0.85Se (e) (101) plane and (f) (110) plane. Calculated energy diagram of Co0.85Se (g) (101) plane and (h) (110) plane in the ORR and OER processes. (i) Phase diagrams of the cathode reaction on Co0.85Se (101) and (110) planes.


 DownLoad:
DownLoad:




 下载:
下载:






 下载:
下载:
