Citation:
Xinghong Cai, Qiang Yang, Yao Tong, Lanyin Liu, Wutang Zhang, Sam Zhang, Min Wang. AlO2: A novel two-dimensional material with a high negative Poisson's ratio for the adsorption of volatile organic compounds[J]. Chinese Chemical Letters,
2025, 36(2): 109586.
doi:
10.1016/j.cclet.2024.109586
AlO2: A novel two-dimensional material with a high negative Poisson's ratio for the adsorption of volatile organic compounds
English
AlO2: A novel two-dimensional material with a high negative Poisson's ratio for the adsorption of volatile organic compounds
Chongqing Key Laboratory for Advanced Materials and Technologies of Clean Energies, School of Materials and Energy, Southwest University, Chongqing 400715, China
b.
BatteroTech Co., Ltd., Shanghai 201417, China
c.
School of Astronautics, Harbin Institute of Technology, Harbin 150001, China
d.
Zhengzhou Research Institute, Harbin Institute of Technology, Zhengzhou 450000, China
Received Date:
31 December 2023 Accepted Date:
29 January 2024 Revised Date:
25 January 2024 Available Online:
15 February 2025
Abstract:
We propose and investigate a novel stable two-dimensional (2D) AlO2 with anomalous stoichiometric ratios based on first-principles calculation. 2D AlO2 has metallic properties. It possesses the rare in-plane and out-of-plane negative Poisson's ratio (NPR) phenomenon, originating from its special sawtooth-like structure. The absolute value of the NPR decreases as the number of layers increases. The adsorption of volatile organic compounds (VOCs) including CH2O, C2H3Cl and C6H6 by AlO2 exhibit small adsorption distance, large adsorption energy, large charge transfer and significant density of states (DOS) changes, indicating the presence of strong interactions. The desorption time of each gas molecule on the AlO2 surface is also evaluated, and the results further suggest that the desorption of VOCs can be controlled by changing the temperature to achieve the recycling of AlO2. These interesting properties make 2D AlO2 a promising material for electronic, mechanical and sensing applications for VOCs.
Volatile organic compounds (VOCs) are known as dominate precursors to urban haze and photochemical smog, mainly from processes such as coal chemical, petrochemical and fuel paint manufacturing and use [1,2]. Most VOCs are toxic, irritating, teratogenic, and carcinogenic, especially formaldehyde, vinyl chloride, and benzene, which seriously threaten the ecological environment and human health [3–5]. Studies have shown that regular exposure to formaldehyde increases the chance of developing cancers, such as blood and lymphoma [6]. Vinyl chloride enters the body and suppresses the central nervous system, damages the liver and spleen, and produces diseases such as hepatic angiosarcoma [7]. The intake of benzene paralyzes the central nervous system and affects the body's hematopoietic function, leading to aplastic anemia and leukemia in severe cases [8]. Therefore, it is urgent to eliminate VOC pollution. Adsorption technology has gained numerous attentions as an economical and environmentally friendly method in VOC reduction [1].
Because of the interesting physical and chemical performance of 2D metal oxides, they have recently been widely explored in transistors, inverters, photodetectors, memristors, sensors, etc. [9,10]. The developments of advanced synthesis methods such as mechanical exfoliation, vapor deposition, and solid solution methods have provided new technical supports for preparing novel layered 2D metal oxides [11,12]. Huang et al. [13] proposed that ultrathin 2D Al2O3 nanosheets designed by replicating graphene oxide from the bottom up exhibited excellent adsorption capacity for fluoride ions. In addition, HfO2 [14], In2O3 [15], ZnO [16], SnO2 [17], γ-Bi2O3 [18], etc., are also attracting attention as new 2D metal oxides.
Al2O3 is widely used in ultrafiltration membranes, catalysts, adsorbents, etc., due to its good electronic properties, mechanical properties and low cost [19–22]. Eunmi et al. [23] successfully synthesized porous Al2O3 with controlled mesoporous and macroporous characteristics using the solvent defect method to achieve efficient removal of the organic pollutant Congo red dye from water. Kumari et al. [24] used nitric-acid-activated Al2O3 to increase the efficiency of defluoridation in the industrial wastewater from 74.18% to 97.43%, and the adsorbent exhibited better regeneration and recyclability, which is beneficial for practical industrial applications. Recently, Liu et al. [25] found that CO2 can be effectively adsorbed and decomposed on the anoxic γ-Al2O3 (100) surface based on first-principles studies. Bai et al. [26] found good adsorption of both CS2 and COF2 on the surface of α-Al2O3 (0001) based on DFT studies. γ-Al2O3 showed good adsorption capacity for potentially carcinogenic chlorinated volatile organic compounds (Cl-VOC) such as benzyl chloride, chloroform, and tetrachlorine after 8 weeks of adsorption [27]. We assume and expect that the new AlO2 in this work can possess some potentials and possibilities for the adsorption of VOCs.
Besides, studying other 2D aluminides has also gained plenty of interests. Wang et al. [28] reported 2D AlN synthesized by metal-organic chemical vapor deposition between Si substrate and graphene with ultra-wide bandgap semiconductor (9.63–9.20 eV) showing great promise in deep-UV optoelectronic applications. The 2D Al2C is a medium bandgap semiconductor (1.02 eV) that maintains its structural integrity at 1500 K, indicating good thermal stability [29] and it was reported to have large adsorption energy (−1.972 eV ~ −2.244 eV) for four toxic VOCs (acetaldehyde, ethylene oxide, vinyl chloride and benzene) and can be used as an adsorbent and sensing medium for toxic VOCs [30]. Wang et al. [31] theoretically predicted that the novel monolayer AlP5 is promising for detecting toxic heavy metals (As, Hg and Cd) in the aqueous environment.
Recently, 2D CaCl and Na2Cl crystals with abnormal anion and cation ratios generated on graphene films have abnormal physical properties such as metallicity, ferromagnetism and piezoelectric behavior [32,33]. Therefore, the discovery of 2D AlO2 with abnormal stoichiometry compared with Al2O3 is expected to expand the development and application of novel electronic devices. Additionally, AlO2 molecule and thin film with anomalous stoichiometric ratios have been found experimentally. A new AlO2 thin film is formed at the interface of diffusion bonding between Pt and α-Al2O3 at 1200 ℃ in Ar atmosphere [34]. Lester et al. [35] reacted Al atoms evaporated by pulsed laser with O2 from a condensing Ar stream to obtain cyclic AlO2 molecule. The successful syntheses of AlO2 molecule and thin film promote more explorations of other new kinds of AlO2 including 2D materials.
Herein, a novel 2D AlO2 with anomalous stoichiometric ratios is designed based on DFT simulations, and its stability is determined by phonon band structure, calculation of elastic constants and ab initio molecular dynamics (AIMD) simulations. The mechanical and electronic performance of monolayer and multilayer AlO2 are investigated. Interestingly, AlO2 has a rare in-plane and out-of-plane NPR. In addition, the adsorption performance of AlO2 on three VOCs (CH2O, C2H3Cl and C6H6) is explored. The results show that monolayer AlO2 has good adsorption and desorption behaviors and is expected to be a potential gas sensor and a good adsorber for detecting and adsorbing VOCs.
The geometry optimization of pristine AlO2, the calculation of electronic properties and the simulation of adsorbed VOCs on AlO2 are implemented in the SIESTA code [36] based on DFT [37]. The exchange-correlation term of the generalized gradient approximation (GGA) [38] is performed under the Perdew-Burke-Ernzerhof (PBE) [39] scheme. In order to be closer to the theoretical limit, the double zeta polarization (DZP) [40] function is chosen for the basis set. A 9 × 12 × 1 sampling is used for Monkhorst-Pack grid point sampling, and the set plane wave cut-off energy is 300 Ry. All atoms during the geometry optimizations are completely relaxed until the maximum force is less than 0.02 eV/Å. Furthermore, the thermal and dynamic stability of AlO2 are confirmed by ab initio molecular dynamics (AIMD) [41] simulations based on the canonical ensemble (NVT) and phonon spectrum calculation, respectively. Herein, the canonical ensemble (NVT) refers to the simulated system in which the number of particles (N), volume (V), and temperature (T) remain constant.
To determine the stability of multilayer AlO2, we define the formation energy (Ef) as:
(1)
where n is the number of layers of AlO2; E(AlO2) and E(n-AlO2) denote the total energy of single-layer and n-layer AlO2, respectively.
In order to accurately describe the adsorption properties of VOCs on the AlO2 surface, we define the adsorption energy (Eads) as:
(2)
where E(AlO2) and E(VOCs) are the energy of the original AlO2 and independent VOCs gas molecules, respectively, and E(AlO2-VOCs) is the total energy of the adsorption system of VOCs molecules.
The differential charge density between AlO2 and VOCs molecules in the adsorption system is defined as:
(3)
where ρ(AlO2) and ρ(VOCs) denote the charge values of original AlO2 and independent VOCs gas molecules, respectively, and ρ(AlO2-VOCs) denotes the total charge value of the adsorption system of VOCs molecules.
The optimized 2D AlO2 structure is shown in Figs. 1a and b, which has a P-4m2 space group and belongs to the tetragonal crystal system (a = b = 3.27 Å, c = 15 Å, α = β = γ = 90°), and each cell includes one Al atom and two O atoms (see the light blue area in Fig. 1b). Section S7 (Supporting information) lists the specific position of each atom in the unit cell. The 2D AlO2 is a sawtooth-like structure with a monolayer height of d = 1.45 Å, as shown in the side view of Fig. 1b. The outer and inner atoms are O and Al ones, respectively. The Al-O bond has a bond length of 1.79 Å, which is close to the theoretical data of the Al-O bond length of α-Al2O3 (1.68–1.86 Å) [42] and γ-Al2O3 (1.75–1.77 Å) [43]. The bond angle of Al-O-Al is 132.14°. Additionally, AlO2 has anomalous stoichiometric ratios. The oxidation states may be abnormal. Lu et al. [34] reported that the possible form of AlO2 is (Al3+)2(O2)2−(O2−)2. Possible oxidation states of AlO2 may be Al3+, O2− and O−.
Figure 1
Figure 1.
(a) Geometrically optimized configuration of 2D AlO2, (b) top and lateral views. The unit cell is marked with a light blue rectangular area. Four possible adsorption sites (O1, O2, Al1 and I1) are marked. (c) Potential energy fluctuations of AlO2 in AIMD simulations at 300 and 1000 K, and the inset shows the structure after 5 ps of AIMD simulations at 1000 K. (d) Phonon band structure and phonon DOS of AlO2. (e) Band structure of AlO2 and the corresponding DOS.
The stability of AlO2 is evaluated by molecular dynamics (MD) simulations, phonon spectra and strain energy calculations. The results of MD simulation for AlO2 are plotted in Fig. 1c. It is obvious that no significant deformation and bond breaking occur in the structure of AlO2 heated at 300 K and 1000 K for 5 ps, and its potential energies at 300 K and 1000 K have few changes, indicating that AlO2 still has good thermal stability at 1000 K. The phonon spectrum of AlO2 is shown in Fig. 1d, and no imaginary frequencies are observed throughout the Brillouin zone, confirming the dynamic stability of AlO2. The maximum vibrational frequency of 1080.5 cm−1 is contributed by both Al and O atoms, which facilitates future experimental characterization. To further determine the mechanical stability of AlO2, the calculated elastic constants of C11 = 48.23 N/m, C12 = −12.32 N/m, C22 = 48.23 N/m and C44 = 1.25 N/m fully satisfy the Born-Huang criterion: C44 > 0 and C11C22-C122 > 0, indicating that AlO2 is mechanically stable.
In addition, the conductivity of AlO2 material is verified by calculating electronic properties. The valence band top of the band structure crosses the Fermi energy level (Fig. 1e), indicating that AlO2 is metallic. The contribution of O atoms near the Fermi level leads to a non-zero density of states (DOS), further proving the metallicity of AlO2. The electronic properties of AlO2 are similar to those of graphene oxide, which has a negative differential resistance (NDR) phenomenon [44], suggesting that AlO2 is promising for NDR nanodevices.
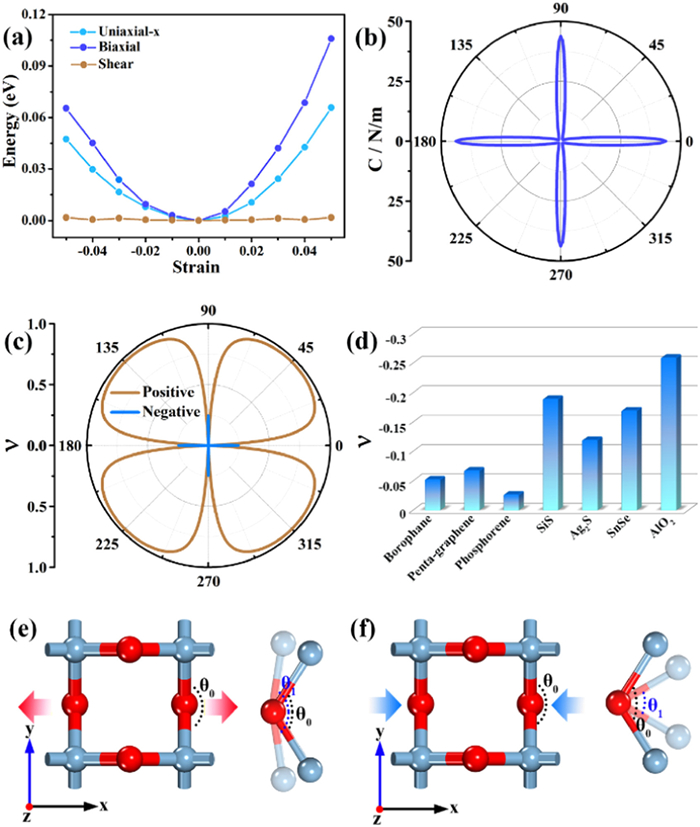
The flexibility or rigidity of the material is defined by the in-plane stiffness C, and flexible materials have less in-plane stiffness. The strain energy curves of AlO2 are shown in Fig. 2a. As shown in Fig. 2b, the in-plane stiffness along the x or y orientation is 48.23 N/m, which is considerably smaller than that of graphene (341 N/m) [45] and MoS2 (120.1 N/m) [46], but a little larger than those of g-C3N5 (37 N/m) [47] and g-ZnO (47.8 N/m) [48]. It is noted that when θ equals 45°, 135°, 225° and 315°, the corresponding in-plane stiffness is only 1.08 N/m. Such small in-plane stiffness along the diagonal direction indicates more flexibility and more softness than those along the x and y axes, which is attributed to its pleated structure. Poisson's ratio ν is defined as the ratio of lateral versus vertical strain of the material, and reflects the elastic constant of the lateral distortion of the material. As illustrated in Fig. 2c, AlO2 has a large ν of 0.97 along the diagonal direction. However, a rare negative Poisson's ratio (NPR) of −0.26 appears in the x and y orientations. Such NPR phenomenon mainly originates from its characteristic sawtooth configuration in the x and y directions, similar phenomena have been reported in structures such as black phosphorus [49], BP5 [50] and penta-graphene [51]. To further compare the NPR properties of AlO2, the Poisson's ratios of different two-dimensional materials with NPR properties predicted by recent theories are listed in Fig. 2d. The results show that the NPR performance of AlO2 materials is superior compared to the majority of 2D materials such as borophane (−0.053) [52], penta-graphene (−0.068) [51], phosphorene (−0.027) [53], SiS (−0.19) [54], Ag2S (−0.12) [55] and SnSe (−0.17) [56]. Due to the special mechanical properties of NPR materials, they have broad application prospects in aerospace, medical equipment, sensors, protective equipment and other fields [57].
Figure 2
Figure 2.
(a) AlO2 strain energy under various strains. (b) In-plane stiffness C and (c) Poisson's ratio ν of AlO2. (d) Comparison of in-plane Poisson's ratios of some 2D materials. The schematic images of in-plane NPR phenomenon caused by the angle ∠OAlO of AlO2 under transverse (e) stretching and (f) compression.
In order to explore the origination of the in-plane NPR of the single-layer AlO2 structure, we investigate the changes of the angle ∠OAlO under x-axis strain. As shown in Fig. S1, ∠OAlO increases by 0.88% and decreases by 1.27% under tensile and compressive strains, respectively. Therefore, the in-plane NPR of AlO2 mainly comes from the contribution of the angle ∠OAlO, as schematically shown in Figs. 2e and f.
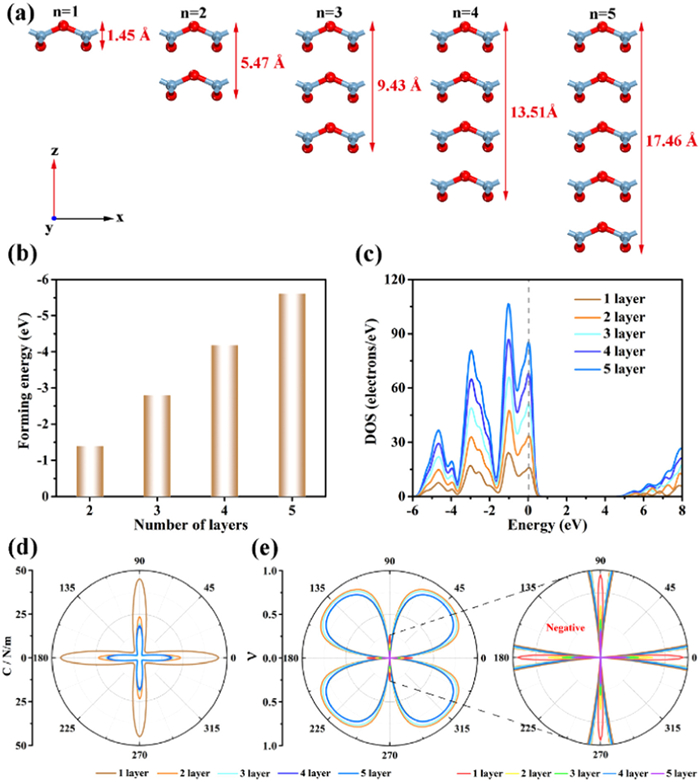
Since it is much easier to experimentally synthesize multilayer structures, the formation energy, DOS, C, ν and electronic properties of 1–5 layers of AlO2 are considered. The structures of 1–5 layers of AlO2 after geometry optimizations are shown in Fig. 3a, and the total layer thickness from 1.45 Å to 17.46 Å as the number of layers increases. In order to study the stability of multilayer AlO2, the calculation of formation energy is performed according to Eq. 1. Fig. 3b shows the variation of formation energy of 2–5 layers of AlO2 nanosheets, with the increase of the number of layers, the formation energy is more negative, which indicates the increased stability of the multilayer structure.
Figure 3
Figure 3.
(a) Side view of the geometry of 1–5 layers AlO2 with the intra-layer heights marked. (b) Forming energy of 2–5 layers of AlO2. (c) The DOS of 1–5 layers AlO2. (d) In-plane stiffness C and (e) Poisson's ratio ν of 1–5 layers AlO2. The inset shows an enlarged view of the NPR in (e).
The total DOS of 1–5 layers of AlO2 is shown in Fig. 3c. The DOS of all multilayer AlO2 crosses the Fermi level, confirming their conducting behavior, which is consistent with the band structures (Fig. S3 in Supporting information). As the number of layers increases, the DOS also increases, which is consistent with the increase in the number of energy bands. In addition, the DOS maps of 1–5 layers of AlO2 are similar, with all DOS peaks locate at nearly the same energy, suggesting that the multilayer AlO2 structures relies mainly on van der Waals interactions between neighboring layers.
The C and ν as a function of angle for 1–5 layers of AlO2 are shown in Figs. 3d and e. The in-plane stiffness and positive Poisson's ratio decrease as the layers increase. The NPR performance of the monolayer structure is still maintained for multilayer AlO2, however the NPR values along the x and y axes increase from −0.26 to −0.08 as the number of layers increases (Table S1 in Supporting information). Therefore, the tuning of mechanical properties such as in-plane stiffness and Poisson's ratio of AlO2 can be achieved by different numbers of layers.
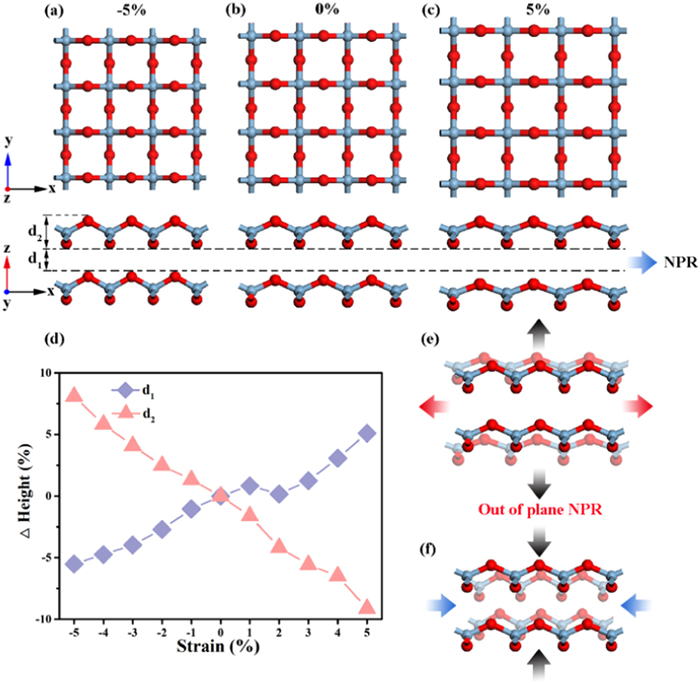
To explore the out-of-plane NPR of the bilayer structure, we compare the variation of interlayer height d1 and intra-layer height d2 in the bilayer AlO2 structure under different biaxial strains. The typical structures of the bilayer AlO2 after geometric optimization under the biaxial strains of −5%, 0% and 5% are shown in Figs. 4a–c. Obviously, with the applied compressive and tensile strains, the interlayer height d1 decreases and increases in the side view of Figs. 4a–c, respectively, revealing that the bilayer AlO2 has an out-of-plane NPR. Furthermore, the comprehensive variations of d1 and d2 with different biaxial strains are demonstrated in Fig. 4d. The results exhibit that by applying −5% compressive (or 5% tensile) strain in the transverse direction, d1 decreases by 5.5% (or increases by 5.1%); on the other hand, d2 increases by 8.1% (or reduces by 9.1%). Thus the out-of-plane NPR of bilayer AlO2 comes mainly from the contribution of the interlayer height d1. The schematic diagram of the out-of-plane NPR of bilayer AlO2 is shown in Figs. 4e and f. AlO2 becomes thicker or thinner vertically when it experiences transverse tensile or compressive strain respectively (Figs. 4e and f), similar to other 2D materials such as black phosphorene [58] and borophane [52] possessing out-of-plane NPR.
Figure 4
Figure 4.
Top and side views of the bilayer AlO2 structure under biaxial strain of (a) −5%, (b) 0%, and (c) 5%. d1 denotes the interlayer height of bilayer AlO2. d2 denotes the intra-layer height of single-layer AlO2. (d) Percentage change of interlayer height d1 and intra-layer height d2 with biaxial strain. (e) Longitudinal expansion phenomenon of bilayer AlO2 in the case of transverse stretching. (f) Longitudinal shrinkage phenomenon of bilayer AlO2 under lateral compression.
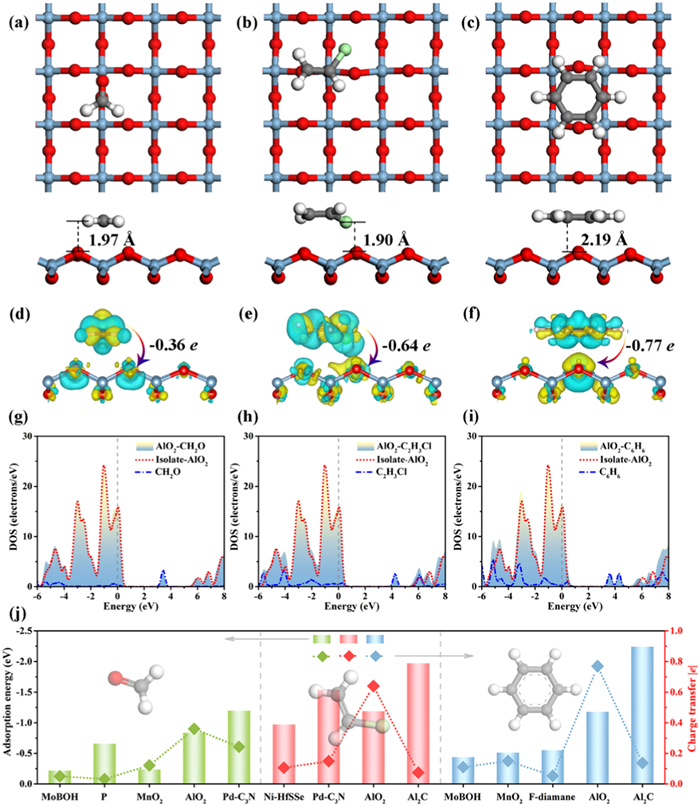
The adsorption performance is investigated to explore the potential application of AlO2 in VOC detection. As shown in Fig. 1b, there are three types of adsorption sites in the AlO2 structure: atomic tops (A1, O1, and O2), the middle of the bonds (Al-O1 and Al-O2), and hollow (I1). VOC molecules are placed in the positions mentioned above to find a suitable adsorption conformation, and then the geometry is optimized without imposing any constraints. The most negative adsorption energy is used as a criterion to find the most stable adsorption conformation. Figs. 5a–c show the most stable adsorption configurations of three VOCs adsorbed on AlO2. The adsorption energies of the AlO2-CH2O, AlO2-C2H3Cl and AlO2-C6H6 parallel (and vertical) adsorption systems are −0.84 eV (−0.51 eV), −1.19 eV (−0.72 eV) and −1.18 eV (−0.29 eV), respectively. The adsorption energies of the parallel systems are more negative than those of the vertical systems. CH2O, C2H3Cl and C6H6 are parallel to the AlO2 plane with adsorption distances of 1.97 Å, 1.90 Å and 2.19 Å, respectively. Mulliken charge analysis is performed for the most stable adsorption configurations, as shown in Figs. 5d–f, revealing that all VOC molecules act as charge donors and AlO2 acts as charge acceptors. The charge transfer amount of the adsorption system is defined as: ΔQ = Q(*VOCs) - Q(VOCs), where Q(*VOCs) denotes the charge of VOCs gas in the adsorption system, and Q(VOCs) denotes the charge values of independent VOCs gas molecules. The charge loss of CH2O, C2H3Cl and C6H6 molecules are −0.36 e, −0.64 e and −0.77 e, respectively.
Figure 5
Figure 5.
Stable configurations and charge density differences of (a, d) CH2O, (b, e) C2H3Cl and (c, f) C6H6 adsorbed on the surface of AlO2. The red, blue, gray, white and green balls are used to denote O, Al, C, H and Cl atoms. The value of 0.025 e/Å3 is determined as the isosurface. The amount and direction of charge transferred in the adsorption system are marked. The DOS before and after adsorption of (g) CH2O, (h) C2H3Cl and (i) C6H6 by monolayer AlO2. (j) Comparison of adsorption energies and charge transfers of some 2D materials for the adsorption of VOCs.
To further compare the adsorption stability of AlO2, the adsorption energies and charge transfer amounts of VOCs on the surfaces of different 2D materials (MoBOH [59], phospholene [60], MnO2 [61], Pd-C3N [62], Ni-HfSSe [63], Al2C [30] and F-diamane [64]) are presented in Fig. 5j. As a comparison, the adsorption energies of VOCs for the structures provided in previous works (MnO2 [61] and phospholene [60]) are calculated. The adsorption energies of MnO2-CH2O, MnO2-C6H6 and phospholene-CH2O are −0.52 eV, −0.24 eV and −0.56 eV, respectively. These data are close to those reported works [60,61]. The results show that the two-dimensional AlO2 material exhibits a better adsorption effect on VOCs and the adsorption energy is better than most 2D materials. The AlO2 surface has satisfactory VOCS desorption compared to Pd-C3N and Al2C, which have higher adsorption energy. It must be noted that too high adsorption energy is not conducive to VOCs desorption and material recovery. The charge transfer is much larger than that of several other 2D materials, confirming the strong interaction of VOCs with AlO2. The low adsorption distance, high adsorption energy and large charge transfer demonstrate the high efficiency of 2D AlO2 for removing these VOCs.
As shown in Figs. 5g–i, DOS analysis is also performed to examine the effect of the adsorption of VOCs on the electronic characteristics of AlO2. The total DOS before and after the adsorption of VOCs appear to be significantly different. After adsorption of VOCs molecules on AlO2, VOCs molecules not only have a significant contribution to the valence band but also have a major contribution to the conduction band, which is consistent with the band structures (Fig. S4 in Supporting information). Significant changes in the DOS before and after adsorption of VOCs molecules indicate the presence of strong interactions between AlO2 and VOCs, which is consistent with the results of large charge transfer amounts.
The effectiveness of VOCs desorption is mainly determined by the recovery time, which is the time the sensor needed to return to its original state after the gas is removed. Therefore, according to the transition state principle, the recovery time (τ) of VOCs molecules on the AlO2 surface is evaluated:
(4)
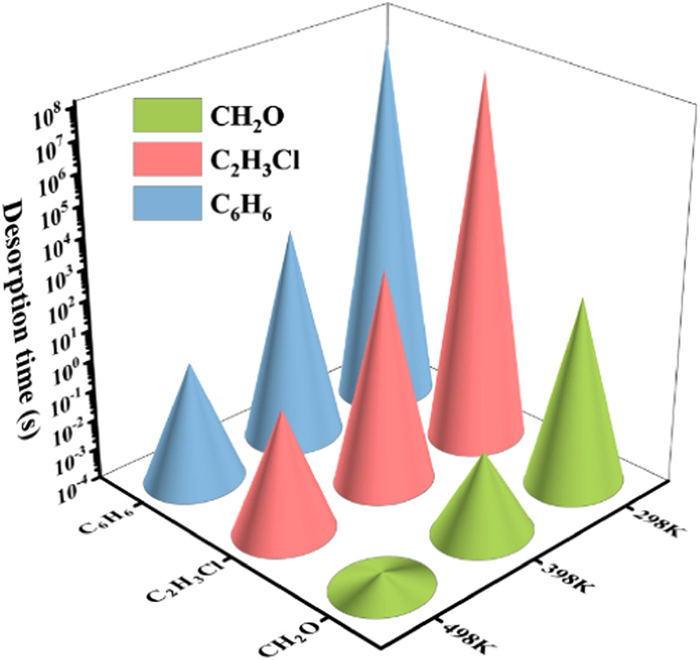
where A (1012 s−1) is the apparent frequency factor, kB (8.62 × 10−5 eV/K) is the Boltzmann constant, Eads (eV) is the adsorption energy which is the energy barrier of the desorption process, and T (K) is the system temperature. As shown in Fig. 6, the desorption times of VOCs molecules on the AlO2 surface at different temperatures are calculated. CH2O has a short recovery time (1.69 × 102 s) at room temperature, indicating that the AlO2 sensor is expected to detect CH2O at room temperature reversibly. The long desorption time of C2H3Cl (1.36 × 108 s) and C6H6 (9.93 × 107 s) implies that AlO2 is suitable as an adsorbent for C2H3Cl and C6H6 at room temperature. In addition, the desorption time decreases with increasing temperature. For C2H3Cl and C6H6 molecules, the desorption times at 498 K are 1.11 s and 0.92 s, respectively, indicating that the high temperature accelerates desorption. Therefore, at room temperature, AlO2 can be used as a gas sensor for the detection of CH2O and an adsorbent for C2H3Cl and C6H6; and at higher temperatures, it can also act as a gas sensor for the detection of C2H3Cl and C6H6.
Figure 6
Figure 6.
Desorption time of CH2O, C2H3Cl and C6H6 molecules on AlO2 surface at different temperatures.
For the future applications, the O2, N2 and water effects should be considered. Since AlO2 has the maximum adsorption energy for C2H3Cl, we continue to consider the effect of O2, N2 and H2O molecules on the adsorption of C2H3Cl. As shown in Figs. 7a–f, the adsorption energies (−0.18 eV, −0.45 eV and −0.37 eV) and the amounts of the charge transfer (−0.11 e, −0.26 e and −0.14 e) of N2, O2 and H2O on the AlO2 surface are much smaller than those of C2H3Cl on AlO2. Figs. 7g–l display the geometry, charge difference density distribution and DOS of C2H3Cl adsorbed on the AlO2 surface in O2, N2 and water environment, respectively. As shown in Fig. 7m, the adsorption energies (−1.06 eV, −1.01 eV and −1.04 eV) and the amounts of charge transfer (−0.56 e, −0.57 e and −0.57 e) of C2H3Cl on AlO2 in O2, N2 and water environment are slightly lower than those in the vacuum environment. Therefore, the effect of the presence of O2, N2 and H2O on the adsorption of C2H3Cl on the AlO2 surface is insignificant.
Figure 7
Figure 7.
Stable configurations, charge density differences and DOS of (a, d) N2, (b, e) O2, (c, f) H2O, (g, j) N2-C2H3Cl, (h, k) O2-C2H3Cl and (i, l) H2O-C2H3Cl adsorbed on the surface of AlO2. (m) Comparison of adsorption energies and charge transfers of monolayer AlO2 for the adsorption of gas molecular and C2H3Cl (in O2, N2 and water environment).
In summary, we propose a novel 2D AlO2 with anomalous stoichiometric ratios. The phonon spectra, AIMD simulations and elastic constants confirm its stability. Electronic calculations show that AlO2 is a conductor. Due to its special structure of serrated configuration, AlO2 offers low in-plane stiffness and a large negative Poisson's ratio (NPR). The Poisson's ratio of 1–5 layers of AlO2 is negative, and the absolute value of NPR decreases as the number of layers increases. The bilayer AlO2 exhibits a rare NPR in both in-plane and out-of-plane directions and reveals the underlying mechanism. The potential applications of AlO2 in the adsorption of CH2O, C2H3Cl and C6H6 detection are explored. The low adsorption distance (1.90 Å to 2.19 Å), high adsorption energy (−0.84 eV to −1.19 eV) and large charge transfer (−0.36 e to −0.77 e) indicate the high removal efficiency of 2D AlO2 for these VOCs. The DOS of AlO2 changed significantly before and after the adsorption of VOC molecules, indicating the presence of strong interactions between AlO2 and VOCs. The evaluation of the desorption time revealed that AlO2 could be used as an adsorbent or gas sensors for VOCs at room or higher temperatures. Besides adsorption of VOCs on the bare AlO2 situations, we also investigate the adsorption of C2H3Cl on the AlO2 surface in N2, O2, and water environment to support the possible applications of AlO2. Compared with normal Al2O3, 2D AlO2 with abnormal stoichiometric ratios can not only extend aluminum-oxide materials, but also provide some interesting properties including a high NPR and good adsorption of VOCs. Moreover, 2D AlO2 has potentials for the novel mechanical and electronic devices. Therefore, this research is expected to provide new insights into the development and application of AlO2 and other 2D materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (No. 22275149), Fundamental Research Funds for the Central Universities (No. SWU118105) and the Next-Generation Advanced Energy Materials Program of BatteroTech Co., Ltd.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109586.
Figure 1
(a) Geometrically optimized configuration of 2D AlO2, (b) top and lateral views. The unit cell is marked with a light blue rectangular area. Four possible adsorption sites (O1, O2, Al1 and I1) are marked. (c) Potential energy fluctuations of AlO2 in AIMD simulations at 300 and 1000 K, and the inset shows the structure after 5 ps of AIMD simulations at 1000 K. (d) Phonon band structure and phonon DOS of AlO2. (e) Band structure of AlO2 and the corresponding DOS.
Figure 2
(a) AlO2 strain energy under various strains. (b) In-plane stiffness C and (c) Poisson's ratio ν of AlO2. (d) Comparison of in-plane Poisson's ratios of some 2D materials. The schematic images of in-plane NPR phenomenon caused by the angle ∠OAlO of AlO2 under transverse (e) stretching and (f) compression.
Figure 3
(a) Side view of the geometry of 1–5 layers AlO2 with the intra-layer heights marked. (b) Forming energy of 2–5 layers of AlO2. (c) The DOS of 1–5 layers AlO2. (d) In-plane stiffness C and (e) Poisson's ratio ν of 1–5 layers AlO2. The inset shows an enlarged view of the NPR in (e).
Figure 4
Top and side views of the bilayer AlO2 structure under biaxial strain of (a) −5%, (b) 0%, and (c) 5%. d1 denotes the interlayer height of bilayer AlO2. d2 denotes the intra-layer height of single-layer AlO2. (d) Percentage change of interlayer height d1 and intra-layer height d2 with biaxial strain. (e) Longitudinal expansion phenomenon of bilayer AlO2 in the case of transverse stretching. (f) Longitudinal shrinkage phenomenon of bilayer AlO2 under lateral compression.
Figure 5
Stable configurations and charge density differences of (a, d) CH2O, (b, e) C2H3Cl and (c, f) C6H6 adsorbed on the surface of AlO2. The red, blue, gray, white and green balls are used to denote O, Al, C, H and Cl atoms. The value of 0.025 e/Å3 is determined as the isosurface. The amount and direction of charge transferred in the adsorption system are marked. The DOS before and after adsorption of (g) CH2O, (h) C2H3Cl and (i) C6H6 by monolayer AlO2. (j) Comparison of adsorption energies and charge transfers of some 2D materials for the adsorption of VOCs.
Figure 7
Stable configurations, charge density differences and DOS of (a, d) N2, (b, e) O2, (c, f) H2O, (g, j) N2-C2H3Cl, (h, k) O2-C2H3Cl and (i, l) H2O-C2H3Cl adsorbed on the surface of AlO2. (m) Comparison of adsorption energies and charge transfers of monolayer AlO2 for the adsorption of gas molecular and C2H3Cl (in O2, N2 and water environment).


 DownLoad:
DownLoad:


 下载:
下载:








 下载:
下载:
