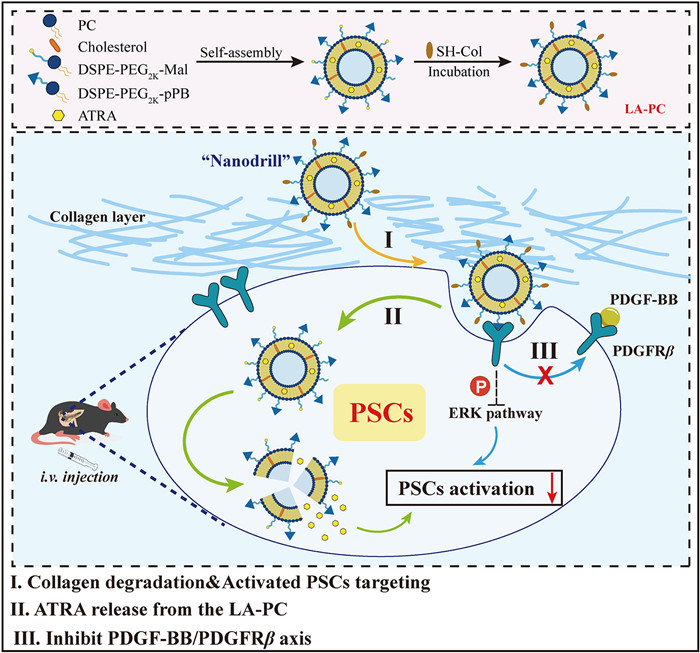
Scheme 1.
Schematic illustration of the construction, delivery and effect mechanism of PDGFRβ-targeting nano-system based on “nanodrill” strategy to induce PSCs quiescence from both intracellular and extracellular perspectives.
A PDGFRβ-targeting nanodrill system for pancreatic fibrosis therapy
Han Han , Bi-Te Chen , Jia-Rong Ding , Jin-Ming Si , Tian-Jiao Zhou , Yi Wang , Lei Xing , Hu-Lin Jiang

Chronic pancreatitis (CP) is a fibroinflammatory syndrome, hallmarked by enduring abdominal pain, diarrhea, and an augmented risk of pancreatic cancer [1]. The escalating global incidence of CP has elevated it to a pressing health issue of global concern [2]. Presently, the clinical management of CP is confined to palliative interventions aimed at symptom relief, with no effective treatment available [3].
Pancreatic stellate cells (PSCs), a kind of fibroblast-like cells, play a pivotal role in the pathogenesis of CP, making them a crucial therapeutic target [4,5]. Under chronic inflammatory conditions, PSCs become activated and secrete excessive extracellular matrix (ECM) proteins to form extensive pancreatic fibrosis in CP [6,7], thus severely impeding drug delivery [8,9]. To address this, we previously employed a nanodrill strategy based on collagen degradation, which effectively enhanced drug accumulation in fibrotic foci [10-12]. So, elevating the selectivity of nanodrill system on PSCs seems to be a crucial strategy for PSCs-quiescent therapy [13]. Further research has revealed the presence of certain receptors on fibroblast surfaces that can influence their activation [14,15]. Thus, it is imperative to design a nanodrill system with higher receptor selectivity specific to PSCs and regulate the signaling pathway for their activation.
Platelet-derived growth factor receptor beta (PDGFRβ) acts as a marker of activated fibroblasts in liver, pancreas, lung and so on [16-18]. When it binds to its ligand, such as platelet-derived growth factor subunit B (PDGF-BB), PDGFRβ can significantly activate fibroblasts, accelerating the fibrosis progression [19,20]. Current research has utilized PDGFRβ as a target for hepatic stellate cells (HSCs) [21], notably effective occupation of the site of PDGFRβ can block the downstream mitogen-activated protein kinase (MAPK) pathway and inhibit fibroblast proliferation and activation, thereby alleviating liver fibrosis [22]. However, no studies have yet explored the potential of PDGFRβ as a regulatory site for PSCs in CP treatment.
Herein, we constructed all-trans retinoic acid (ATRA)-loaded liposomes co-decorated with pPB (C*SRNLIDC*) and collagenase (Col) (LA-PC) for CP therapy. The pPB is a cyclic peptide that targets PDGFRβ [21], while ATRA is a pharmaceutical agent known to suppress PSCs activation [11,23]. The design of LA-PC aims to synergistically induce quiescence in PSCs from both intracellular and extracellular aspects (Scheme 1). Following intravenous administration, the surface-bound Col facilitates LA-PC penetration into the fibrotic pancreas. The surface-modified pPB peptide enables the LA-PC to selectively target activated PSCs, promoting drug uptake by PSCs and concurrently inhibiting the PDGF-BB/PDGFRβ axis in PSCs. This dual regulation strategy, targeting both intracellular and cell surface receptors, will offer new possibilities for CP treatment.
LA-PC used in this study was synthesized using the thin film dispersion and grafting methods [24]. ATRA was encapsulated within the lipid layer of liposomes, which were composed of soy lecithin, cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[pPB-(polyethylene glycol)-2000] (DSPE-PEG2K-pPB), and DSPE-PEG2K-maleimide (Mal), through a self-assembly process. Following this, thiolated type Ⅰ collagenase (SH-Col) was grafted onto the liposome surface via the Michael reaction [11]. The optimal modification ratio of Col and pPB was investigated by cell uptake assay and determined the ratio to be 2:3 (pPB:Col, w/w) (Fig. S1 in Supporting information). To evaluate the impact of different molecular modifications on both the mechanical properties and therapeutic effectiveness of liposomes, we procured unmodified liposomes (LA), liposomes modified with Col (LA-Col), and liposomes modified with pPB (LA-pPB) by substituting DSPE-PEG2K-pPB or DSPE-PEG2K-Mal with DSPE-PEG2K. The average particle sizes of the four liposomes (LA, LA-Col, LA-pPB, LA-PC) were 181.57 ± 1.22, 179.50 ± 1.44, 184.66 ± 2.49, and 182.10 ± 1.72 nm, respectively. Dynamic light scattering (DLS) measurements revealed that the polydispersity index (PDI) of the four preparations was less than 0.2, indicating a uniform size distribution [25,26]. Transmission electron microscopy (TEM) images confirmed that all four formulations exhibited uniform spherical shapes and core-shell structures (Figs. 1A–D).
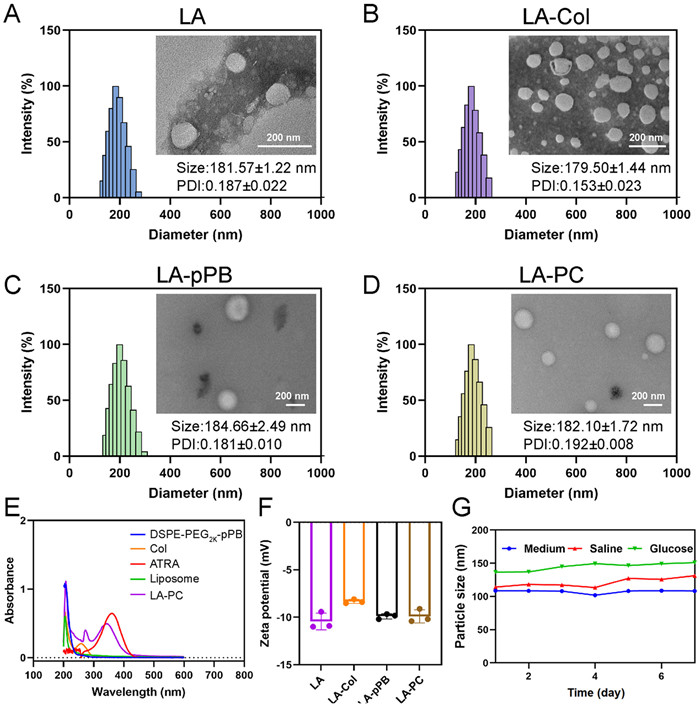
Next, we measured the UV absorption of blank liposomes, LA-PC, ATRA, and modified molecules. Within the 200–600 nm band, liposomes did not exhibit noticeable UV absorption. However, both ATRA and LA-PC demonstrated strong absorption in proximity to 340 nm (Fig. 1E). This result revealed that the characteristic peaks of LA-PC were synchronized with ATRA, confirming the successful fabrication of LA-PC.
The zeta potential values of LA, LA-Col, LA-pPB, and LA-PC were −10.44, −8.33, −9.89, and −9.90 mV, respectively (Fig. 1F). The negative surface potential ensures good biocompatibility [27]. Furthermore, throughout the 7-day stability experiment, the particle size of LA-PC remained unchanged in saline, 5% glucose solution (w/w), and DME/F12 medium. The average particle size consistently remained below 200 nm [28], demonstrating good stability, which ensures that subsequent in vitro/in vivo experiments can be conducted without hindrance (Fig. 1G and Fig. S2 in Supporting information).
The experimental results presented above have confirmed the successful synthesis of LA-PC. Following this, we first conducted biosafety tests of LA-PC on PSCs cell lines. Post 24 or 48 h of treatment with LA-PC, PSCs exhibited a survival rate exceeding 90%, even at an elevated dose of 3 µg/mL, indicating substantial safety of LA-PC (Fig. S3 in Supporting information). For subsequent in vivo/in vitro experiments, we utilized liposome solutions with a drug concentration of 1.5 µg/mL.
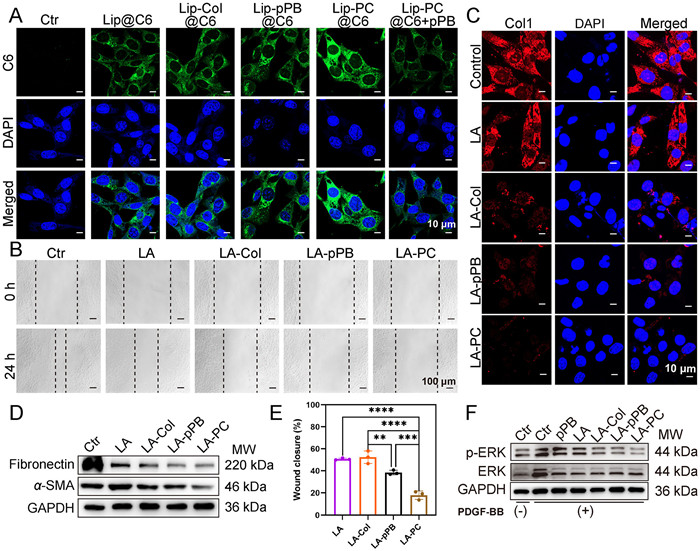
The successful accumulation of the drug within cells is a critical determinant of its therapeutic effectiveness. Consequently, we investigated the in vitro cellular uptake of liposomes. Adhering to the synthesis method of LA-PC, we prepared coumarin 6 (C6)-loaded liposomes (Lip@C6, Lip-Col@C6, Lip-pPB@C6 and Lip-PC@C6), which were examined by confocal laser scanning microscope (CLSM). Fluorescence images revealed that all four liposomes were effectively internalized by PSCs, with cells in the Lip-pPB@C6 and Lip-PC@C6 treated groups displaying higher fluorescence intensity than those in the Lip@C6 and Lip-Col@C6 treated groups. We hypothesized that the elevated uptake of Lip-pPB@C6 and Lip-PC@C6 is linked to the binding of the pPB peptide to PDGFRβ. Consequently, we assessed the expression of PDGFRβ on the surface of PSCs. Following the stimulation of PSCs with the PDGFRβ agonist PDGF-BB, there was an increase in the expression of PDGFRβ (Fig. S4 in Supporting information). Coinciding with this increase, the uptake of Lip-PC@C6 by PSCs also increased. However, after a period of incubation of the pPB peptide with PSCs, it was observed that the uptake of Lip-PC@C6 by PSCs was significantly diminished (Fig. 2A). This demonstrated that the cellular uptake of LA-PC is contingent on the recognition of pPB by PDGFRβ.
Next, we incubated PSCs with varying concentrations of PDGFRβ-antibody (Ab) for 12 h, then introduced Lip@C6 and Lip-PC@C6 to further assess the liposome uptake capability of PSCs. Flow cytometry results revealed that after treatment with high-concentration of PDGFRβ-Ab, the absorption of Lip@C6 remained relatively stable in comparison to treatment with a lower concentration. This suggests that the process of cellular uptake of Lip@C6 is independent of PDGFRβ. Conversely, treatment with high-concentration PGDFRβ-Ab markedly diminished the uptake of Lip-PC@C6. This provides evidence that the elevated cellular uptake instigated by pPB is reliant on PDGFRβ, a finding that aligns with the detection outcomes of CLSM (Fig. S5 in Supporting information).
After the successful confirmation of PSCs’ uptake of LA-PC, we investigated the in vitro quiescence ability of LA-PC on PSCs. Given the robust invasive capabilities of activated PSCs, we initially conducted a wound healing assay to evaluate the impact of different treatment regimens on PSCs migration and invasion. The findings revealed a significant reduction in the wound healing rate after drug-treated, particularly in the LA-pPB and LA-PC groups (Fig. 2B and Fig. S6 in Supporting information). Subsequently, we assessed the activation and collagen production capabilities of PSCs via immunofluorescence staining. The results demonstrated a significant reduction in both the secretion of collagen I (Col1) (Fig. 2C) and the expression of the fibroblast activation marker α-smooth muscle actin (α-SMA) (Fig. S7 in Supporting information) in PSCs within the treatment group, with LA-pPB and LA-PC exhibiting the most pronounced effects on collagen production inhibition. Western blot analysis also indicated a significant reduction in the expression of fibronectin and α-SMA in activated PSCs following LA-pPB and LA-PC treatment (Fig. 2D). In conjunction with the outcomes of the cellular uptake assay, we deduced that pPB could augment the cellular uptake of liposomes via its interaction with PDGFRβ. This facilitated the accumulation of ATRA within cells and enhanced the quiescence of PSCs.
It has been reported that PDGFRβ, a tyrosine kinase receptor, contains an intracellular tyrosine kinase domain [29]. PDGFRβ is predominantly expressed in fibroblasts, with its in vivo activator primarily being PDGF-BB from the PDGF family [30]. Upon binding to PDGF-BB, PDGFRβ undergoes dimerization, leading to autophosphorylation between the dimerized receptor molecules, thereby activating the kinase and its downstream pathways [31]. Concurrently, PDGF-BB can augment the expression of PDGFRβ and enhance their combination. Studies have shown that the use of the PDGFRβ antagonist GomisinD can effectively inhibit the activation of HSCs and mitigate liver fibrosis [22]. We hypothesize that the targeted binding of the pPB peptide to PDGFRβ may obstruct the receptor dimerization process by preventing the binding of PDGF-BB to PDGFRβ, thus inhibiting the activation of PSCs.
Therefore, we investigated whether the pPB peptide could suppress the activation of the PDGFRβ receptor and its subsequent signaling pathways. In the ensuing experiments, PDGF-BB was utilized to upregulate the expression of PDGFRβ on PSCs. Wound healing assays demonstrated that upon the addition of PDGF-BB, the wound healing rate of the group treated with LA-pPB was significantly divergent from that of the LA and LA-Col treated groups, a contrast to when it was not added (Fig. 2E and Fig. S8 in Supporting information). The outcomes of measuring Col1 expression in PSCs also revealed that following PDGF-BB stimulation, the pPB peptide alone could notably diminish the expression of Col1, but the pPB peptide failed to exhibit a similar effect when PSCs were not stimulated (Fig. S9 in Supporting information). These results suggest that the pPB peptide can induce the quiescence of PSCs via a PDGFRβ-dependent pathway.
The phosphorylation of the extracellular signal-regulated kinase (ERK) is a pivotal event post-activation of PDGFRβ [32]. The activation of the ERK pathway can bolster the proliferation and migration capabilities of PSCs [33]. Western blot analysis indicated that the phosphorylation of intracellular ERK was significantly augmented post-PDGF-BB stimulation. Following treatment with the pPB peptide, LA-pPB, and LA-PC, the intracellular level of phospho-ERK (p-ERK) was significantly diminished (Fig. 2F). These findings demonstrate that the pPB peptide can not only enhance the targeting of LA-PC to PSCs but also inhibit the activation of PDGFRβ and its downstream pathways, thereby promoting the quiescence of PSCs.
The data presented above substantiates that the pPB peptide-modified LA-PC not only enhances the cellular targeting and uptake of the ATRA but also inhibits the activation of PDGFRβ and its downstream signaling pathways, thereby suppressing the activation and proliferation of PSCs. Consequently, LA-PC is anticipated to play a pivotal role in the treatment of CP. Subsequently, we employed the cerulein-induced CP model in mice to evaluate the in vivo therapeutic efficacy of LA-PC. Animal experiments were conducted under protocols approved by the Ministry of Health of the People’s Republic of China and were approved by the Ethics Committee of China Pharmaceutical University (Acceptance number: 2023–08–014).
We confirmed the pancreatic-targeted aggregation abilities of LA-PC in CP mice firstly. We prepared 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR)-labeled liposomes (namely Lip@DiR, Lip-Col@DiR, Lip-pPB@DiR, and Lip-PC@DiR), and following the final injection of cerulein, free DiR and fluorescent dye-labeled preparations were administered (Fig. 3A). In vivo, near-infrared fluorescence images of mice were captured at 1, 2, 3, 5, 7, 9, 24, and 48 h post-injection (Figs. 3B and C). It was observed that Lip-PC@DiR exhibited stronger aggregation in the pancreas, displaying noticeable fluorescence from 3 h post-injection, and high fluorescence levels could still be detected up to 48 h. The in vivo fluorescence of mice treated with Lip-pPB@DiR peaked at about 7 h, followed by a gradual decrease. This could be attributed to the drug’s inability to penetrate the collagen barrier in the CP pancreas [34], resulting in the drug’s inability to accumulate over an extended period.
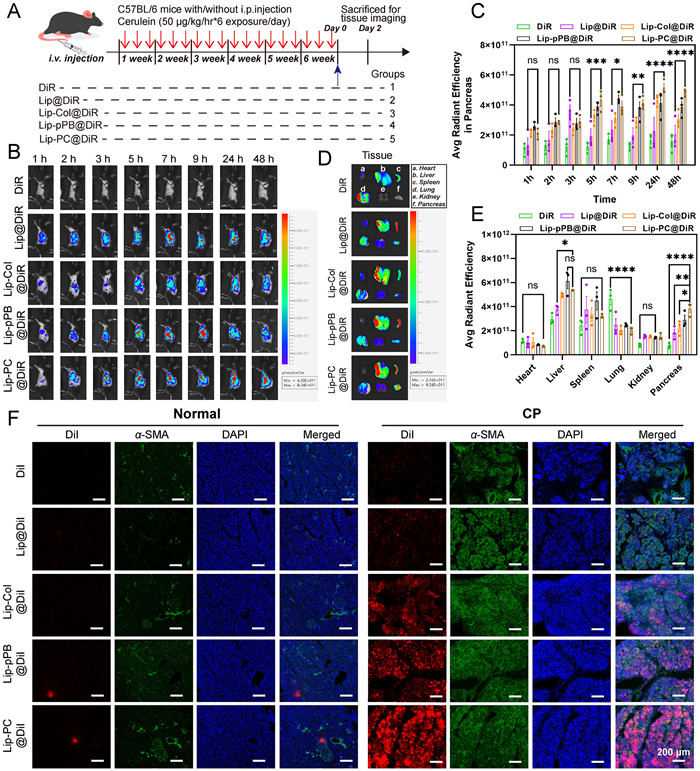
The 24 h post-treatment with different preparations and free drugs, mice were sacrificed to obtain the heart, liver, spleen, lung, kidney, and pancreas, and the fluorescence intensity was measured (Fig. 3D). The fluorescence intensity of the organs in each group was quantified (Fig. 3E). Notably, the accumulation of Lip-PC@DiR in the pancreas of the mice was found to be the most significant, with its fluorescence intensity increasing more than fivefold compared to free DiR. Subsequently, we applied staining the sections of pancreatic tissue to validate the drug’s distribution within the pancreas. Fluorescence images revealed that 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) and its preparations had negligible accumulation in the pancreas of normal mice. However, Lip-PC@DiI was extensively distributed in the pancreas of CP mice and exhibited conspicuous co-localization with α-SMA (Fig. 3F). This is partially attributed to the “nanodrill” effect generated post-Col modification, which facilitates the deep penetration of Lip-PC@DiI into fibrotic pancreatic tissue. Concurrently, the expression of PDGFRβ on activated PSCs in the CP pancreas was elevated, thereby enhancing the targeting of Lip-PC@DiI to PSCs. It was demonstrated that liposomes integrating Col and pPB serve as effective carriers for drug delivery to the fibrotic pancreas.
Following this, we assessed the capacity of LA-PC to modulate pancreatic fibrosis in vivo. Given the prolonged circulation of liposomes in the circulatory system, the drug release period is correspondingly extended [35]. Here, commencing from the 4th week of modeling, free drugs and preparations were administered to mice via intravenous injection thrice weekly for a total of three weeks (Fig. 4A). A day after the final injection, the mice were sacrificed, and the therapeutic effects of free drugs and the preparations on CP were evaluated through serum biochemical tests and tissue sample analyses.
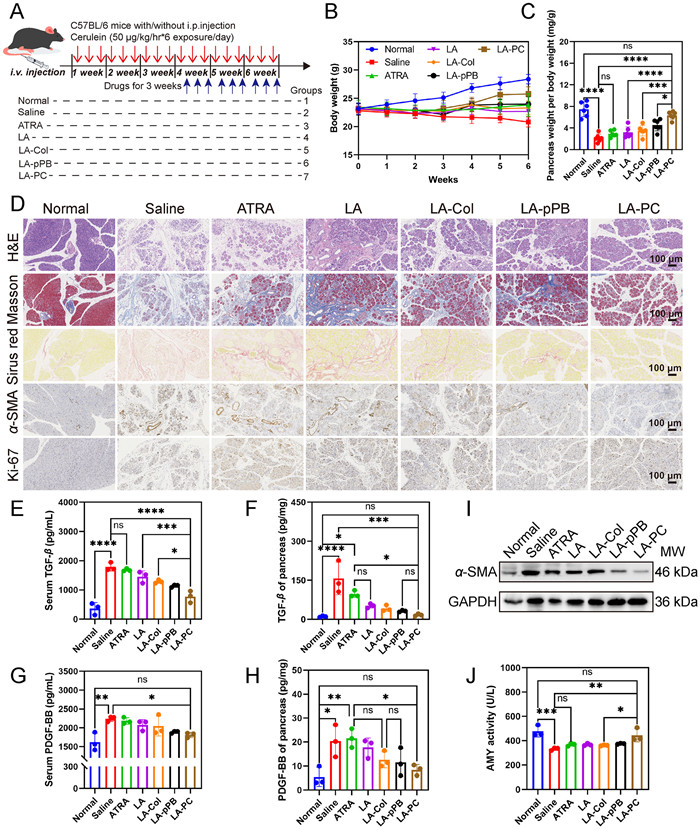
The body weight of mice was consistently monitored commencing from the initial modeling phase. Throughout the entire experimental duration, a continuous increase in weight of mice was observed in the normal group, while a consistent decrease was noted in the model group during the first 3 weeks, attributable to recurrent acute pancreatitis. Following the initiation of administration, the mice in the treatment group experienced varying degrees of weight gain, indicative of a relief in condition post-treatment (Fig. 4B). In untreated mice, the pancreas undergoes autodigestion and tissue atrophy due to damage to the acinar cells and leakage of digestive enzymes. However, mice in the treatment group exhibited significantly larger pancreas (Fig. S10 in Supporting information) and a higher ratio of pancreas weight to body weight (Fig. 4C). This substantiates the protective effect of the drug on the pancreas.
Hematoxylin and eosin (H&E) staining revealed that the pancreatic tissue of normal mice exhibited a preserved acinar structure. Conversely, the pancreas of fibrotic mice displayed severe acinar cell necrosis, extensive inflammatory cell infiltration, and collagen deposition. Masson staining and Sirius red staining were utilized for fibrosis analysis. In the normal pancreatic tissue of control mice, only a minor quantity of collagen fibers was present around the blood vessels. However, in fibrotic mice, blue or red-stained regions were observed throughout the pancreas, indicating extensive fibrosis. In contrast, the degree of fibrosis in mice in the treatment group (including mice treated with free drugs, LA, LA-Col, LA-pPB, and LA-PC) was reduced to varying extents. Following treatment with LA-PC, there was a marked reduction in the expression of the PSCs activation marker α-SMA. Simultaneously, there was a significant enhancement in acinar regeneration within the pancreas (Fig. 4D and Fig. S11 in Supporting information). These findings highlight the potent anti-fibrotic capability of LA-PC and its protective effect on the pancreas.
The serum biochemical indicator test results of mice serum demonstrated a significant increase in the concentration of inflammation-related cytokines such as tumor necrosis factor alpha (TNF-α) (Fig. S12A in Supporting information) and interleukin-1β (IL-1β) (Fig. S12B in Supporting information) in the serum of CP mice, which subsequently decreased following treatment. The levels of fibrosis-related cytokines, such as transforming growth factor beta (TGF-β) (Figs. 4E and F) and PDGF-BB (Figs. 4G and H), were elevated in the pancreatic tissue and serum of CP mice. This is believed to be a response from body cells like monocytes, macrophages, and endothelial cells to injury [36,37]. Post drug treatment, the concentrations of TGF-β and PDGF-BB in the pancreas and serum of mice decreased, correlating with the reduction of pancreatic tissue damage (Fig. 4I) and a decrease in the activation of PSCs (Fig. 4J). As previously mentioned, high concentrations of PDGF-BB can stimulate the expression of PDGFRβ on the surface of PSCs and promote the activation of PSCs. Therefore, the decrease in TGF-β and PDGF-BB content also aids in the quiescence of PSCs, thereby establishing a positive cycle of CP treatment.
Taken together, these findings suggest that LA-PC has the capacity to efficiently break down the collagen barrier, infiltrate profoundly into the pancreatic tissue, and provide space for pancreatic repair. The targeting attribute endowed by the pPB peptide on LA-PC facilitates the accumulation of ATRA within PSCs, thereby enhancing the therapeutic efficacy of ATRA. Concurrently, the suppression of PDGFRβ by the pPB peptide also fosters the quiescence of PSCs and effectively curtails the advancement of pancreatic fibrosis.
Lastly, we conducted an in vivo examination of LA-PC’s safety. Tissue sections from the mouse heart, liver, spleen, lung, and kidney exhibited no discernible pathological alterations (Fig. S13A in Supporting information). Furthermore, there was no significant disparity between the groups in the detection of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and blood urea nitrogen (BUN) levels (Figs. S13B–D in Supporting information). These findings suggest that LA-PC possesses excellent safety and biocompatibility within the body.
In conclusion, we have constructed PSCs-targeting liposomes LA-PC decorated by PDGFRβ-binding peptide and collagenase. After being injected into fibrotic mice via tail vein, the collagenase modified on LA-PC can remove the excess collagen layer, and the drug delivery efficiency through pPB targeting peptide was more than 5 times higher than that of free drug, as well as the degree of fibrosis significantly reduced. Notably, the LA-PC effectively inhibited PDGF-BB/PDGFRβ axis on activated PSCs via a down-regulated ERK pathway. Specially, PDGF-BB in fibrotic foci was also significantly down-regulated after LA-PC treatment in vivo, which reduces the PDGF-BB/PDGFRβ-induced PSCs activation. This strategy of synergistic resting PSCs has shown satisfactory anti-fibrotic effects in vivo, providing a promising therapeutic strategy for CP. In the future, owing to the collagen deposition and fibroblast activation are also a typical common feature of fibrosis in other organs, we believe that our nano-system LA-PC could be applied to more types of fibrosis, and other fibroblast-activated and collagen-rich diseases.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the National Natural Science Foundation of China (Nos. 82020108029, 82073398) and the National Key Research and Development Program of China (No. 2022YFE0198400). This work was also supported by Double First-Rate construction plan of China Pharmaceutical University (No. CPU2022QZ18).
Supplementary material associated with this article can be found, in the online version, at doi:
J. Kirkegård, F.V. Mortensen, D. Cronin-Fenton, Am. J. Gastroenterol. 112 (2017) 1366–1372. doi: 10.1038/ajg.2017.218
M.S. Petrov, D. Yadav, Nat. Rev. Gastroenterol. Hepatol. 16 (2019) 175–184. doi: 10.1038/s41575-018-0087-5
V.K. Singh, D. Yadav, P.K. Garg, J. Am. Med. Assoc. 322 (2019) 2422–2434. doi: 10.1001/jama.2019.19411
P. Mews, P. Phillips, R. Fahmy, et al., Gut 50 (2002) 535–541. doi: 10.1136/gut.50.4.535
R.G. Wells, J.M. Crawford, Gastroenterology 115 (1998) 491–493. doi: 10.1016/S0016-5085(98)70216-1
M. Erkan, G. Adler, M.V. Apte, et al., Gut 61 (2012) 172–178. doi: 10.1136/gutjnl-2011-301220
A. Masamune, T. Watanabe, K. Kikuta, T. Shimosegawa, Clin. Gastroenterol. Hepatol. 7 (2009) S48–S54. doi: 10.1016/j.cgh.2009.07.038
T. Ji, J. Lang, J. Wang, et al., ACS Nano 11 (2017) 8668–8678. doi: 10.1021/acsnano.7b01026
J.B. Qiao, Q.Q. Fan, C.L. Zhang, et al., J. Control. Release 321 (2020) 629–640. doi: 10.1016/j.jconrel.2020.02.049
M.M. Han, X.Y. He, L. Tang, et al., Sci. Adv. 9 (2023) eadg5358. doi: 10.1126/sciadv.adg5358
L. Qi, H. Han, M.M. Han, et al., Biomaterials 292 (2023) 121945. doi: 10.1016/j.biomaterials.2022.121945
L.F. Zhang, X.H. Wang, C.L. Zhang, et al., ACS Nano 16 (2022) 14029–14042. doi: 10.1021/acsnano.2c03858
Q.Q. Fan, C.L. Zhang, J.B. Qiao, et al., Biomaterials 230 (2020) 119616. doi: 10.1016/j.biomaterials.2019.119616
D. Chakraborty, H. Zhu, A. Jüngel, et al., Sci. Transl. Med. 12 (2020) eaaz5506. doi: 10.1126/scitranslmed.aaz5506
Z. Xu, W. Luo, L. Chen, et al., Hypertension 79 (2022) 2028–2041. doi: 10.1161/hypertensionaha.122.18657
C. Betsholtz, Cytokine Growth Factor Rev. 15 (2004) 215–228. doi: 10.1016/j.cytogfr.2004.03.005
J. Paulsson, T. Sjöblom, P. Micke, et al., Am. J. Pathol. 175 (2009) 334–341. doi: 10.2353/ajpath.2009.081030
Y.C. Wang, Q. Chen, J.M. Luo, et al., Exp. Mol. Med. 51 (2019) 1–11.
E. Patsenker, Y. Popov, M. Wiesner, et al., J. Hepatol. 46 (2007) 878–887. doi: 10.1016/j.jhep.2006.11.011
F. van Dijk, C.M. Hazelhoff, E. Post, et al., Pharmaceutics 12 (2020) 278. doi: 10.3390/pharmaceutics12030278
J. Zhang, H. Shen, J. Xu, et al., ACS Nano 14 (2020) 6305–6322. doi: 10.1021/acsnano.0c02633
R. Wang, F.B. Liu, P.P. Chen, et al., Int. J. Biol. Macromol. 235 (2023) 123639. doi: 10.1016/j.ijbiomac.2023.123639
L. Cai, Z. Gu, J. Zhong, et al., Drug. Discov. Today 23 (2018) 1126–1138. doi: 10.6023/cjoc201709046
M.Y. Yang, Y.J. Lin, M.M. Han, et al., J. Control. Release 351 (2022) 623–637. doi: 10.1016/j.jconrel.2022.09.054
X. Ji, Y. Meng, Q. Wang, et al., ACS Nano 17 (2023) 5421–5434. doi: 10.1021/acsnano.2c10042
Y. Yu, Y. Meng, X. Xu, et al., ACS Nano 17 (2023) 3334–3345. doi: 10.1021/acsnano.2c06313
L.W.C. Ho, Y. Liu, R. Han, et al., Acc. Chem. Res. 52 (2019) 1519–1530. doi: 10.1021/acs.accounts.9b00103
Y. Zhao, H. Chen, X. Chen, et al., Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 9 (2017) e1469. doi: 10.1002/wnan.1469
A.H. Shim, H. Liu, P.J. Focia, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 11307–11312. doi: 10.1073/pnas.1000806107
E.J. Battegay, J. Rupp, L. Iruela-Arispe, et al., J. Cell. Biol. 125 (1994) 917–928. doi: 10.1083/jcb.125.4.917
C.H. Heldin, Cell Commun. Signal. 11 (2013) 97. doi: 10.1186/1478-811X-11-97
X. Zou, X.Y. Tang, Z.Y. Qu, et al., Int. J. Biol. Macromol. 202 (2022) 539–557. doi: 10.1016/j.ijbiomac.2022.01.113
R. Jaster, G. Sparmann, J. Emmrich, S. Liebe, Gut 51 (2002) 579–584. doi: 10.1136/gut.51.4.579
H. Han, L. Xing, B.T. Chen, et al., Adv. Drug Deliv. Rev. 200 (2023) 115051. doi: 10.1016/j.addr.2023.115051
T.M. Allen, P.R. Cullis, Adv. Drug Deliv. Rev. 65 (2013) 36–48. doi: 10.1016/j.addr.2012.09.037
S.H. He, T.Y. Hou, J.L. Zhou, et al., Stem Cells Int. 2022 (2022) 2401693.
M. van der Kroef, T. Carvalheiro, M. Rossato, et al., J. Autoimmun. 111 (2020) 102444. doi: 10.1016/j.jaut.2020.102444

Scheme 1 Schematic illustration of the construction, delivery and effect mechanism of PDGFRβ-targeting nano-system based on “nanodrill” strategy to induce PSCs quiescence from both intracellular and extracellular perspectives.

Figure 1 (A–D) Size distribution and TEM image (inserted) of LA, LA-Col, LA-pPB, LA-PC. (E) Ultraviolet-visible spectra of DSPE-PEG2K-pPB, Col, free drugs, LA-PC and blank liposome. (F) Zeta potential of LA, LA-Col, LA-pPB, LA-PC (n = 3). (G) Stability of LA-PC in saline, glucose and 10% FBS-DME/F12 medium.

Figure 2 (A) Cellular uptake images of Lip@C6, Lip-Col@C6, Lip-pPB@C6, Lip-PC@C6 and pretreated PDGFR-β antibody Lip-PC@C6. C6 were shown in green and nucleus were shown in blue. Scale bar: 10 µm. (B) Wound-healing assay of PSCs, respectively. Scale bar: 100 µm. (C) Immunofluorescence staining of collagen I after treatment of activated PSCs with different preparations. Scale bar: 10 µm. (D) Western blot analysis of fibronectin, α-SMA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (E) Quantitative analysis of wound closure of LA, LA-Col, LA-pPB and LA-PC. (F) Western blot analysis of p-ERK, ERK and GAPDH. Values are expressed as means ± SD (n = 3). **P < 0.01, ***P < 0.001, ****P < 0.0001.

Figure 3 (A) Schematic summary of animal studies. (B, C) Representative in vivo near-infrared fluorescent images of mice conducted with caerulein for 6 weeks, acquired 1, 2, 3, 5, 7, 9, 24 and 48 h after intravenous injection of different preparations. (D, E) Fluorescence intensity in heart, liver, spleen, lung, kidney and pancreas of mice treated with caerulein for 6 weeks, expressed as average radiant efficiency units. (F) Colocalization of DiI and DiI-labeled liposomes with α-SMA in the pancreas of fibrotic mice treated with caerulein for 6 weeks. Pancreas tissue was sectioned and immunostained with α-SMA (green) for PSCs and counterstained with the nuclear dye, DAPI (blue). Fluorescence of DiI is shown in red. Scale bar: 200 µm. Values are expressed as means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, no significant difference.

Figure 4 (A) Schematic summary and dosing regimen of animal pharmacodynamic studies. (B) Body weight of both control and treatment groups was monitored over the 6 weeks’ experimental period. (C) Pancreatic tissues were weighed at the end of experiment and shown as pancreas weight per body weight. (D) Representative H&E, Masson, Sirius Red and IHC staining (α-SMA and Ki-67) of pancreas tissue sections. Scale bar: 100 µm. (E, F) Serum and tissue TGF-β in normal and fibrotic mice with or without different treatments. (G, H) Serum and tissue PDGF-BB in normal and fibrotic mice with or without different treatments. (I) Western blot analysis of α-SMA and GAPDH in pancreatic tissue. (J) Serum AMY activity in normal and fibrotic mice with or without different treatments. Values are expressed as means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: