Received Date:
01 December 2023 Accepted Date:
23 January 2024 Revised Date:
08 January 2024 Available Online:
15 January 2025
Abstract:
Customized design of well-defined cathode structures with abundant adsorption sites and rapid diffusion dynamics, holds great promise in filling capacity gap of carbonaceous cathodes towards high-performance Zn-ion hybrid supercapacitors (ZHC). Herein, we fabricate a series of dynamics-oriented hierarchical porous carbons derived from the unique organic-inorganic interpenetrating polymer networks. The interpenetrating polymer networks are obtained through physically knitting polyferric chloride (PFC) network into the highly crosslinked resorcinol-formaldehyde (RF) network. Instead of covalent bonding, physical interpenetrating force in such RF-PFC networks efficiently relieves the RF skeleton shrinkage upon pyrolysis. Meanwhile, the in-situ PFC network sacrifices as a structure-directing agent to suppress the macrophase separation, and correspondingly 3D hierarchical porous structure with plentiful ion-diffusion channels (pore volume of 1.35 cm3/g) is generated in the representative HPC4via nanospace occupation and swelling effect. Further removal of Fe fillers leaves behind a large accessible specific surface area of 1550 m2/g for enhanced Zn-ion adsorption. When used as the cathode for ZHC, HPC4 demonstrates a remarkable electrochemical performance with a specific capacity of 215.1 mAh/g at 0.5 A/g and a high Zn2+ ion diffusion coefficient of 11.1 × 10−18 cm2/s. The ZHC device yields 117.0 Wh/kg energy output at a power density of 272.1 W/kg, coupled with good cycle lifespan (100,000 cycles@10 A/g). This work inspires innovative insights to accelerate Zn diffusion dynamics by structure elaboration towards high-capacity cathode materials.
With the depletion of limited fossil energy reserves and the rapid advancement of renewable energy sources in recent years, the urgent demand for high-performance electric energy storage devices comes to the fore [1–7]. Aqueous metal hybrid supercapacitors have drawn significant attention for their eco-friendliness, high abundance, and excellent safety performance [8,9]. Aqueous metal hybrid supercapacitors integrate the strengths of metal-ion batteries and electric double layer capacitors, which adopt metal as the anode and capacitive electrode material as the cathode, thereby enabling respective contributions in power density and energy density [10–13]. Among the various metal hybrid supercapacitors, zinc metal stands out as a highly promising candidate for practical implementation due to its inherent advantages, including exceptional chemical stability, a high theoretical capacity of 820 mAh/g, and a low redox potential of −0.76 V vs. standard hydrogen electrode [14–19]. Cathode material serves a vital function in determining the performance of zinc hybrid supercapacitors (ZHCs) to a large extent [20]. Carbon materials are one of the most commonly used cathodes because of the tunable pore structure, large specific surface area, and modifiable chemical composition [21–26]. Nevertheless, it is still facing many challenges at present, such as sluggish reaction kinetics, unstable carbon framework, and mismatched pore structure, resulting in unsatisfactory power, capacity, and cycling performance of ZHCs [27–29]. Hence, it is imperative to engineer porous carbon materials that possess abundant adsorption sites and rapid diffusion dynamics to optimize the electrochemical performance of ZHCs.
The pore structure is the pivotal element that affects the electrochemical performance of porous carbon electrodes [30–32]. Hierarchical porous carbons (HPCs) consisting of micropores, mesopores and macropores, present substantial advantages in the field of high efficiency energy storage [33]. Currently, the approaches for fabricating HPCs primarily involve activation process and template methods [34]. The activation process typically yields more micropores, but sometimes lead to non-interconnected or dead pores. In contrast, the template methods offer greater ease in regulating the pore size of carbon materials, which are categorized into hard and soft template methods. The former commonly uses molecular sieves, SiO2, etc. as templates, while the latter mainly employs surfactants like F127 and P123 as templates. Recently, interpenetrating polymer networks (IPNs) with different thermostabilities, provide a highly promising strategy to obtain multiscale pore distributions within HPCs [35]. Rather than covalent bonding, physical interpenetrating polymer networks are capable of producing highly interconnected porous architectures, since the macro-phase separation between the carbon sources/pore formers may be prevented during annealing [36,37]. For instance, Zhu et al. successfully obtained HPC electrodes, characterized by a significant specific surface area of 2544 m2/g through pyrolyzing the IPN precursor formed by resorcinol/formaldehyde resin and polyacrylamide [38]. The hierarchically micro-/meso‑/macroporous carbon structure was attributed to the congenerous pore-forming effect of KOH and secondary polyacrylamide network, including polyacrylamide decomposition, and a foaming process formed by partial hydrolysis of polyacrylamide in alkaline medium. To date, studies have been conducted on HPCs synthesis based on organic-organic structured IPNs. However, the synthesis of HPCs utilizing organic-inorganic interpenetrating polymer networks remains an intriguing research area.
In this study, we construct organic-inorganic IPNs by combining resorcinol-formaldehyde (RF) with polyferric chloride (PFC) to craft HPCs for ZHCs cathodes application. The establishment of physically cross-linked IPNs is achieved by Fe3+ hydrolysis into the second network bridged by −OH functionalities, with the aid of electrostatic forces. Of note, this combination occurs without the need for any crosslinking agents or additional additives. During annealing, RF acts as a carbon source to endow HPCs with a robust carbon skeleton, while PFC decomposes serving as a porogenic agent to prevent the macroscopic phase separation and achieve the precise pore generation. Additionally, the introduction of KOH enhances the overall pore development. The synergistic effect of PFC and KOH enables HPC materials with interconnected hierarchical pores, a specific surface area of 1550 m2/g, alongside a 15.11% oxygen content level, which implies HPC electrodes with more exposable sites and favorable wettability. When employed as the ZHCs electrode, the HPC4 material shows a remarkable specific capacity of 215.1 mAh/g at 0.5 A/g accompanied by a high Zn2+ ion diffusion coefficient of 11.1 × 10−18 cm2/s. Moreover, the as-constructed Zn//HPC4 device offers an energy output of 117.0 Wh/kg at a power density of 272.1 W/kg, all while keeping excellent cycle stability. Characterization verifies the alternate adsorption and desorption of Zn2+/SO42− ions on the electrode surface as well as the charge transfer between C = O functional groups and Zn2+. This work provides a novel outlook on the strategic design of synthetic HPCs.
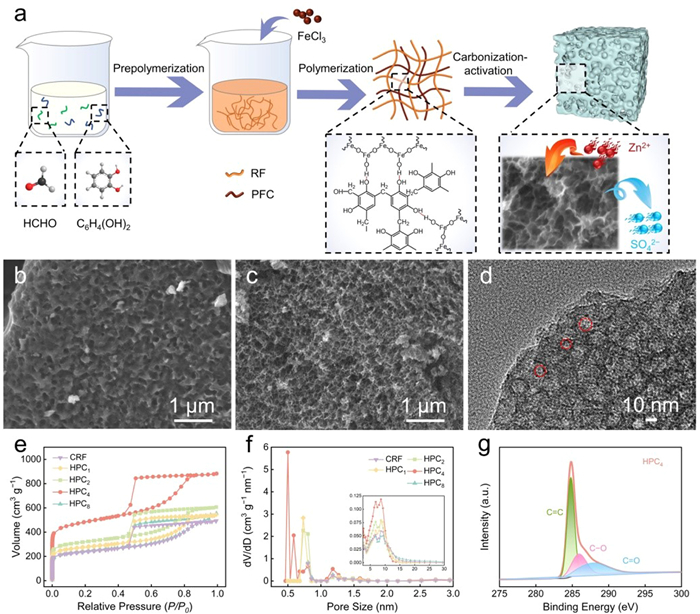
The route for the synthesis of HPCs (denoted as HPCx, where x refers to the mole ratio of RF to PFC) is depicted in Fig. 1a. Briefly, to fabricate the HPCx material, resorcinol and formaldehyde were pre-polymerized under basic condition in the presence of Na2CO3 to form the first network (resorcinol/formaldehyde prepolymer resin). Subsequently, ferric chloride solution was added drop-wise to the prepolymer solution to form PFC, where Fe3+ ions were gradually hydrolyzed under alkaline environment into the second inorganic network bridged by −OH functionalities. Through a swelling process and further hydrothermal polymerization, the inorganic-organic IPNs were formed in which one network was internally entangled and physically intertwined with another. During the thermal treatment, the organic network (resorcinol-formaldehyde resin) in IPNs acted as a carbon skeleton, while the PFC network occupied the internal space and decomposes with the activator KOH at high temperature to synergistically develop pores. Fourier transform infrared spectra confirm that the two networks are physically intertwined with the presence of respective functionalities, indicating the successful fabrication of IPNs. As shown in Fig. S1 (Supporting information), RF and P−IPN4 have analogous absorption peaks at 1610 cm−1, classified as the aromatic C=C stretching, and 1100 cm−1, ascribed to C−OH or symmetric stretching of C − O − C. Meanwhile, P−IPN4 exhibited the characteristic absorptions of Fe−O locating at 554 and 460 cm−1. The solid-state H nuclear magnetic resonance (1H NMR) spectra reveals distinct difference in peak shapes between RF and P−IPN4. The appearance of peak splitting in the 1H NMR spectra of P−IPN4 signifies an alteration in the chemical environment of hydrogen atoms within the entire molecular structure, which is likely attributable to hydrogen bonding interactions occurring between PFC and RF networks (Fig. S2 in Supporting information). Thermogravimetric analysis (TGA) curves provide insights into the thermal behavior (Fig. S3 in Supporting information). As for RF, a noticeable weight loss process occurs around 100 ℃, primarily attributed to water evaporation and hydroxyl pyrolysis on the benzene ring. Subsequently, RF gradually carbonized and underwent weight loss in the temperature range spanning 100–600 ℃, resulting in a 57% residual carbon rate. The TGA curve of P−IPN4 exhibits a similar trend to that of RF, culminating in a final residual carbon rate of 44%. X-ray diffraction (XRD) patterns of the RF and IPNs precursors show the characteristic polymer diffraction peak at approximately 18°, indicating the physically cross-linked structure of RF and PFC (Fig. S4a in Supporting information). Besides, the XRD patterns of the carbonized samples, CRF and HPCx, exhibit two diffraction peaks at 21° and 43° (Fig. S4b in Supporting information). These peaks display broad and weak intensity characteristics, representing (002) and (100) planes, respectively, indicating the presence of amorphous structures [39,40]. In Raman spectra (Fig. S5 in Supporting information), two conspicuous peaks are located at wavenumbers of 1350 and 1590 cm−1, which can be assigned to D band and G band. The D band signifies disordered carbon with lattice defects and the G band is related to E2g symmetric vibration of graphene. The relative intensity ratio of D and G bond, denoted as ID/IG, can be utilized to assess the degree of graphitization [41]. By calculation, the ID/IG values of as-prepared carbon materials are all around 0.8, indicating a high degree of graphitization.
Figure 1
Figure 1.
(a) Typical schematic diagram for the procedures of HPCx. SEM images of (b) CRF, (c) HPC4. (d) TEM image of HPC4. (e) Nitrogen adsorption−desorption isothermal curves. (f) Pore size distributions of CRF and HPCx, and (g) high-resolution C 1s XPS spectra.
Morphology characterizations were assessed via scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Figs. 1b−d). In the absence of the PFC inorganic network within the CRF framework, the etching and foaming effects driven by the activator KOH are constrained to the outer layer of the bulk precursor. This localized interaction results in the creation of undesired sizable pores. In contrast, with the introduction of the secondary interpenetrating network, KOH tends to penetrate the precursor matrix to bestow conspicuous pore-forming attributes. Meanwhile, by facilely modulating the molar ratio of RF to PFC, the pore structure of HPCx can be finely tuned (Fig. S6 in Supporting information). Specifically, an excess of PFC triggers the partial collapse of the pore structure, as observed in HPC1. Reducing the molar ratio of PFC enhances porosity and yields denser pore distribution, culminating in a sponge-like three-dimensional pore structure with neat interconnection and interpenetration. When the molar ratio reaches 8:1 (RF relative to PFC), the efficacy of pore formation within the bulk phase is compromised, leading to the emergence of underdeveloped pores. TEM image of HPC4 also validates the interconnected network structure. These results suggest the effectiveness of two physically interpenetrating polymer networks in preventing macroscopic phase separation, preserving structural integrity during heat treatment, and enabling precise control and flexible manipulation of the pore structure.
Pore structure and surface area are essential factors that contribute to the electrochemical performance of carbon material. Thus, nitrogen adsorption-desorption measurements were performed on CRF and HPCx (Fig. 1e). Notably, the obtained carbon materials all displayed type-IV curves featuring a steep rise in the relative pressure of P/P0 < 0.05 and significant hysteresis loops occurring at 0.4 < P/P0 < 1.0, suggesting the concurrent existence of ample micropores, mesopores, and macropores within the carbon framework. The generation of abundant pores can be credited to the dual synergistic mechanism involving the activator KOH and chemical filler PFC. The etching effect of the former on the bulk precursor induces the release of gases and initiates a comprehensive enhancement in porosity. Concurrently, the latter undergoes decomposition under high heat, giving rise to vacant nanospaces through the in-situ occupancy effect, thereby achieving precise pore formation. Through the adjustment of the PFC content, the specific surface area and total pore volume (Vtotal) can be manipulated (Table S1 in Supporting information). When the PFC content was too high, it caused the collapse of pores, resulting in a lower specific surface area and Vtotal, e.g., HPC1 (892 m2/g, 0.61 cm3/g), HPC2 (1028 m2/g, 0.94 cm3/g), whereas, with a low PFC content, inadequate pore formation also reduced in both the specific surface area and Vtotal, e.g., HPC8 (780 m2/g, 0.86 cm3/g). Accordingly, due to the addition of the optimal percentage of PFC, HPC4 showed the highest nitrogen adsorption with a high specific surface area of 1550 m2/g and a Vtotal of 1.35 cm3/g, indicating a well-developed pore structure. As for CRF, without the addition of the second network PFC, the activator could not penetrate the precursor uniformly during calcination, resulting in a lower specific surface area and low porosity (767 m2/g, 0.76 cm3/g). Also, the pore size distribution curves of CRF and HPCx (Fig. 1f) reveal subtle distinctions. The engagement of the secondary network imparts the HPCx samples with a propensity for heightened occurrences of micropores, in contrast to CRF, which lies within the range of 0.5−0.8 nm, as well as 1.2 nm. Within the mesopore dimension, CRF and HPCx exhibit pore sizes clustered around 6.8−8.6 nm. Thus, according to N2 sorption characterization and SEM results, we can conclude that the intertwined secondary PFC network significantly augments the comprehensive material porosity, providing the means for fine-tuning the nanostructure of the material. Additionally, KOH activation synergistically induces porosity with the pore-forming and foaming effects, consequently expanding the pore structure and facilitating ion diffusion kinetics. X-ray photoelectron spectrometry (XPS), as illustrated in Fig. S7 (Supporting information), was applied to study the elemental composition and chemical state of HPC4. It could be seen that only two peaks, C 284.4 eV and O 532.3 eV, are shown revealing the thorough removal of Fe (Fig. S8 in Supporting information). In the high-resolution XPS spectra of C 1s presented in Fig. 1g, it can be fitted into three characterized peaks corresponding to C=C, C−O, and C=O, respectively, at 284.8, 285.9, and 287.8 eV. Regarding to O species, it can be curve-fitted to C−O (533.4 eV), and C=O (532.1 eV) (Fig. S9 in Supporting information). Notably, the high content of O (15.11%) in HPC4 (Table S2 in Supporting information) can promote surface wettability, contributing to the reduction of interfacial resistance and enhancement of electrochemical properties. Coupled with the highly open, and well-developed channels of HPC4, it can provide abundant adsorption sites allowing rapid ion migration and charge storage.
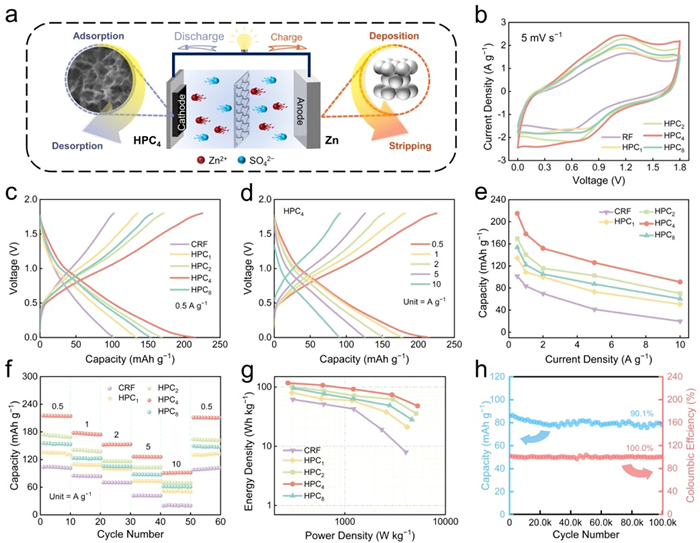
To evaluate the electrochemical performance, HPCx was employed as the cathode for the assemble of Zn-ion hybrid capacitors, with Zn foil as the anode and 2 mol/L aqueous ZnSO4 as electrolyte (Fig. 2a). The cyclic voltammetry (CV) profiles at 5 mV/s (Fig. 2b) presents a quasi-rectangular shape with slight redox peaks, suggesting the coexistence of electric double-layer capacity (EDLC) and extra pseudocapacitive behaviors, which is associated to the presence of O functional sites within the carbon framework [42]. As the voltage approaches 1.8 V, the CV curves rise sharply, implying the presence of polarization on the surface of electrodes and electrolyte. Compared to other samples, it is obvious that the HPC4 has the highest quasi-rectangular area which also means the highest specific capacitance. When using galvanostatic charge/discharge (GCD) measurements to evaluate the capacity performance of ZHCs, the HPC4-based device delivers a discharge capacity of 215.1 mAh/g at 0.5 A/g (Fig. 2c), which significantly surpasses the performance of CRF (101.5 mAh/g), HPC1 (134.3 mAh/g), HPC2 (169.5 mAh/g), and HPC8 (153.7 mAh/g). Moreover, as the current density gradually increases from 0.5 A/g to 10 A/g, the GCD curves maintain symmetrical shapes (Fig. 2d) and exhibit low voltage drops at different current densities (Fig. S10 in Supporting information). The impact of graphite additive on capacitance was assessed, revealing a negligible specific capacitance of 2.5 mAh/g at 0.1 A/g (Fig. S11 in Supporting information). Moreover, HPC4-based device maintains its superior capacitance at various current densities, and even at 10 A/g, it still demonstrates an outstanding capacity retention of 42% (Fig. 2e). The rate performance was depicted in Fig. 2f. Notably, the HPC4-based device demonstrated a remarkable capability for reversible recovery to the initial level when the current density is reverted to 0.5 A/g after successive testing for ten cycles which reflecting the desirable rate performance. The excellent capacitance of HPC4 can be credited to the optimal pore structure, high surface area and pore volume regulated by the PFC content in IPN precursor, which can expose more active sites and promote diffusion kinetics of Zn ions. Besides, the as-constructed Zn//HPC4 device yields a peak energy density of 117.0 Wh/kg at a power density of 272.1 W/kg, alongside the highest power density of 5.29 kW/kg at 48.1 Wh/kg which are surpassing the performance of Zn//CRF and Zn//HPCx devices (Fig. 2g), and very comparable to those of most of the recently reported supercapacitors (Table S3 in Supporting information). Additionally, Zn//HPC4 device exhibits a cycling durability which retains 90.1% of its capacitance after 100,000 cycles at 10 A/g (Fig. 2h). After cycling, the HPC4 cathode shows negligible changes and minimal formation of zinc dendrites, demonstrating the considerable stability of HPC4 (Fig. S12 in Supporting information). Zn//HPC4 device was also employed to power a miniature fan (Fig. S13 in Supporting information), indicating the potential of its application.
Figure 2
Figure 2.
Electrochemical performances of the assembled ZHCs. (a) Diagram of the operation mechanism of CRF/HPCx-based capacitor. (b) CV curves at 5 mV/s. (c) GCD profiles at 0.5 A/g. (d) GCD profiles and (e) the relationship between capacity and current density. (f) Rate capacities at different current densities. (g) Ragone plots. (h) Cycling stability of HPC4-based device at 10 A/g.
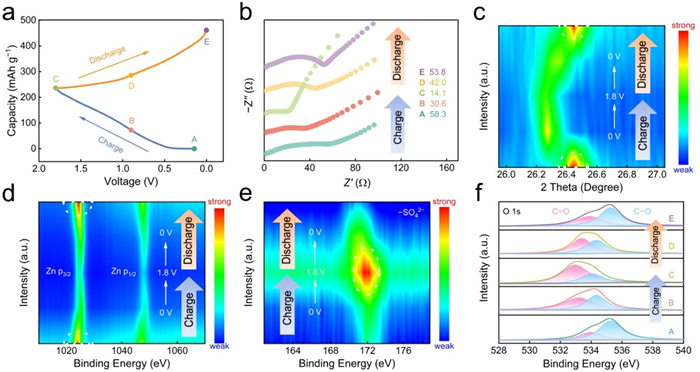
For the purpose of insight into the intrinsic charge storage mechanism, a series of ex-situ spectra characterizations were conducted on the HPC4 cathode. Fig. 3a depicts the initial GCD curve of Zn//HPC4 at 1 A/g, while five representative states (A, B, C, D, and E) are chosen to monitor the HPC4 electrode changes under various charge/discharge conditions [43,44]. The ex-situ electrochemical impedance spectroscopy (EIS) results reveal a reversible variation throughout the electrochemical reaction process (Fig. 3b). The voltage increases alongside the decrease of the charge transfer resistances (Rct) in charging process, while during discharge process, the voltage and Rct have a distinctly different change. The Rct exhibits a range of 14.1–58.3 Ω demonstrating the low ion diffusion resistance and rapid electronic transport kinetics. The ex-situ XRD analyzed the structural changes of the HPC4 cathode (Fig. 3c). The XRD patterns of HPC4 cathode feature a sharp peak at about 26.4°, which corresponds to the by-product Zn4(OH)6SO4·nH2O (ZHS) generated on the cathode surface due to the pH change during (dis)charging [45,46]. The peak intensity gradually weakens in the charging process, followed by strengthening, accompanied by a shift towards lower angle, before returning to the initial angle. The behavior indicates the reversible formation and dissolution of by-products. Significantly, the ex-situ SEM image (Fig. S14 in Supporting information) reveals a limited presence of ZHS, as there was no occurrence of H+ insertion storage. In the ex-situ Raman spectra, a slight shift in both D peak and G peak indicates the EDLC-based storage in the interfacial ion adsorption and desorption (Fig. S15 in Supporting information).
Figure 3
Figure 3.
(a) A GCD curve of Zn//HPC4 device. (b) Nyquist plots. (c) XRD characterization and high-resolution XPS spectra of (d) Zn 2p, (e) S 2p, and (f) O 1s at various discharged/charged courses.
More atomic conversion details are elucidated using ex-situ XPS spectra. The XPS counter map of Zn 2p reveals a reduction in the intensity of the Zn signal in charging process, indicating the removal of Zn2+ ions, and conversely, Zn2+ ions adsorb on the electrode surface during the discharge, with the gradual increment in the peak intensity of the Zn signal (Fig. 3d) [47]. Additionally, a contrasting trend is observed in the peak intensity of S 2p in SO42− when compared to the Zn signal (Fig. 3e). The intensity of S 2p peak initially increases and then decreases in the (dis)charging period (0 V − 1.8 V − 0 V). These distinct trends highlight the alternating processes of cation/anion absorption and release in the HPC4 cathode [48]. The high resolution XPS spectra of O 1s was deconvoluted into two peaks which can be identified as C=O (533.1−533.9 eV) and C−O (534.5−535.3 eV) species (Fig. 3f). Significantly, the intensity of the C=O peak gradually increases in the course of charging and subsequently decreases when discharging, presenting a different trend from the C−O peak. Furthermore, an obvious binding energy shift is accompanied by the entire process, implying the reversible transformation of C=O and C−O groups as electroactive species in the electrochemical system [49]. It is noteworthy that the XPS results of Zn are inverted from those of C=O that the additional pseudocapacitance generation mechanism of Zn//HPC4 device can be reasonably deduced. In discharging process, C=O group within the HPC4 cathode is reduced to C−O−Zn by gaining electrons and adsorbing Zn2+ ions. And the inference can be corroborated by the high-resolution C 1s XPS spectra (Fig. S16 in Supporting information). When the voltage increases from 0 V to 1.8 V, the opposite process to the one described above is followed. Accordingly, in conjunction with the above discussion, the charge storage mechanism of aqueous Zn//HPC4 devices consists mainly of the pseudocapacitance owing to the redox reaction between Zn2+ and C=O/C−O groups, as well as the EDLC contributed by the physical adsorption and desorption of cations and anions on the carbon skeleton, like Zn2+ and SO42−. Expressed briefly as follows:
(1)
(2)
(3)
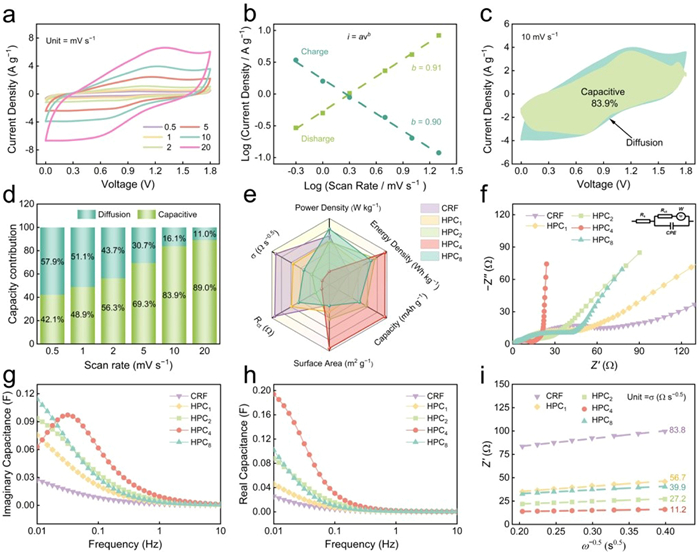
CV profiles were collected under different scan rates to comprehensively assess the electrochemical reaction kinetics. As the scan rate ranged from 0.5 mV/s to 20 mV/s, the CV profiles are gradually distorted while maintaining the quasi-rectangular shape, suggesting the good stability of HPC4 electrodes. Besides, even at 20 mV/s, the redox peaks remain faintly visible, implying the combined contribution of EDLC and pseudocapacity (Fig. 4a). A comprehensive study of the electrochemical reaction kinetics of HPC4 cathode was investigated on the basis of Dunn equation. The mechanism of charge storage can be assessed by investigating the correlation between current (i) and scan rate (v), as outlined by the equation: i = avb, where a value is a constant, and b reflects the specific characteristics of charge storage process [50,51]. A value of 0.5 for b signifies the ionic diffusion-controlled behavior, whereas b = 1 represents the ideal surface-controlled process [52–54]. As illustrated in Fig. 4b, the HPC4-based device exhibits b values of 0.90 and 0.91, indicating the joint diffusion-controlled and surface-controlled process during energy storage. Furthermore, the specific contributions of two different controlled processes can be determined by the power-law equation: i/v1/2 = k1v1/2 + k2 [55,56]. The shaded region depicted in Fig. 4c represents the capacitance contribution at scan rate of 10 mV/s, amounting to 83.9% of the total capacitance. The contribution of capacitance rises from 42.1% to 89.0% as the scan rate is increased from 0.5 mV/s to 20 mV/s, which indicates the gradual dominance of capacitance (Fig. 4d) [57,58]. Meanwhile, the kinetic contributions of zinc-based devices constructed from other samples are illustrated in Fig. S17 (Supporting information). Evidently, the Zn//HPC4 device exhibits superior capacitance contributions compared to the other assembly configurations at varying scan rates. By comparing various parameters (Fig. 4e), the exceptional energy storage performance of HPC4 cathode can be ascribed to the synergistic effect of several factors, including a large specific surface area, a finely tuned pore structure, a robust porous carbon framework and substantial oxygen doping, so that expose abundant adsorption sites, rich oxygen motifs and demonstrate superior electrochemical energy storage performance.
Figure 4
Figure 4.
(a) CV profiles of HPC4-based device at different scan rates. (b) The b values in the anodic and cathodic scanning from 0.5 mV/s to 20 mV/s. (c) Capacitive contribution at 10 mV/s, and (d) capacitive and diffusion-controlled contribution ratios at the scan rates from 0.5 mV/s to 20 mV/s of HPC4-based device. (e) The comparison chart of the six parameters of CRF/HPCx-based devices. (f) Nyquist plots. (g) Real capacitance, and (h) imaginary capacitance versus frequency. (i) The relationship between Z’ and ω−0.5.
Also, the ion/electron transfer kinetics of carbon samples prepared by IPN strategy were further investigated via EIS. The Nyquist plots, as depicted in Fig. 4f, clearly exhibit a semicircle lying in the high-frequency region and a linear line in the low-frequency area, revealing the ion/electron transfer process jointly governed by fast kinetic diffusion and pseudocapacitance. Notably, the equivalent series resistance (Rs), deduced from the intersections on the real axis, displays minimal variation among different samples, ranging from 0.39−2.90 Ω (Table S4 in Supporting information). Regarding the Rct calculated from the fitted semicircle diameter, HPC4 demonstrates a remarkably low value of 10.5 Ω, in contrast to the other assembled ZHCs, which range from 19.0 Ω to 76.0 Ω. This difference can be ascribed to the large pore volume of HPC4 (Vtotal = 1.35 cm3/g, Vmicro = 0.46 cm3/g), facilitating fast charge transfer and ion exchange at the electrode/electrolyte interface. The capacitance model, C(ω) = C′(ω) + jC″(ω), which could be extracted from previous EIS data was utilized to investigate the capacitance variations over a wide frequency range. Within this model, C′(ω) and C″(ω) denote the real and the imaginary part of the capacitance with respect to frequency. The relaxation time constant (τ0) was employed to characterize the temporal duration required for effectively delivering the stored energy and power. It could be mathematically expressed as τ0 = 1/f0, where f0 stands for the knee frequency. As described in Figs. 4g and h, τ0 of HPC4-based device is about 31 s which implies the rapid (dis)charging process. Furthermore, the ion diffusion resistances (σ, Ω s−0.5) were quantified through a linear fit of the correlation between the real part of impedance (Z’) and reciprocal of the square root inverse of angular frequency (ω−0.5). Remarkably, the HPC4-based device displays the lowest σ value of 11.2 Ω s−0.5 (Fig. 4i) [59,60], surpassing the σ values obtained from HPC1, HPC2, HPC8-based device (56.7, 27.2, 39.9 Ω s−0.5) and the σ value recorded from CRF-based device (83.8 Ω s−0.5). To fully validate the rapid Zn2+ ion diffusion kinetics, the Zn2+ ion diffusion coefficient DZn2+ was further calculated (Table S4). The results show that HPC4 cathode exhibits an order of magnitude higher DZn2+ than other cathodes, with a remarkable value of 11.1 × 10−18 cm2/s. The low diffusion resistance and high DZn2+ illustrate that the ca/anions were able to diffuse rapidly in the well-developed pore structure and the extra pseudocapacitive energy storage on the surface of HPC4.
Collectively, the HPC4-based ZHC demonstrates outstanding performance with regard to specific capacitance, energy-power density and fast ion transport, which can be ascribed to several key aspects. Firstly, the inorganic-organic IPN meticulously engineered by PFC and RF is subjected to simultaneous carbonization and activation, resulting in a three-dimensional interconnected porous network. The unique structural configuration affords an impressive ion-accessible specific surface area as high as 1550 m2/g and a pore volume of 1.35 cm3/g. Additionally, 15.11% oxygen doping level not only enhances the material’s surface wettability but also provides abundant active sites for electrochemical energy storage. Lastly, the robust porous carbon framework imparts the exceptional cycling stability to the assembled ZHC device, ensuring a prolonged operational lifespan.
In conclusion, physically intertwined RF and PFC-based precursor is successfully designed through employing the organic-inorganic IPN strategy. Within this IPN structure, PFC network acts as structure-directing agent based on in-situ nanospace occupation and synergizes with the activator KOH to create HPCs with 3D interconnected pores by one-step pyrolysis. The prepared HPCs display a large ion-accessible specific surface area (1550 m2/g) and substantial energy-storage sites (oxygen content of 15.11%), which greatly facilitates fast electrochemical reaction kinetics on the electrode surface. The assembled Zn//HPC4 demonstrates excellent electrochemical performance in ZHC devices, including high discharge capacity (215.1 mAh/g) as well as remarkable energy output (117.0 Wh/kg). The outstanding electrochemical performance can be credited to the joint contribution of the EDLC and pseudocapacitance. Specifically, the alternate adsorption and desorption of anions and cations at the electrode-electrolyte interface, in conjunction with the redox reaction between the electroactive functional group C=O and Zn ions. This study not only provides novel insight in the design of high-performance porous carbon but also demonstrates its immense potential in Zn-based energy storage devices.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work is financially supported by the National Natural Science Foundation of China (Nos. 22272118, 22172111, 21905207, and 22309134), the Science and Technology Commission of Shanghai Municipality (Nos. 22ZR1464100, 20ZR1460300, and 19DZ2271500), China Postdoctoral Science Foundation (No. 2022M712402), Shanghai Rising-Star Program (No. 23YF1449200), Zhejiang Provincial Science and Technology Project (No. 2022C01182), and the Fundamental Research Funds for the Central Universities (Nos. 22120210529 and 2023–3-YB-07).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109559.
[1]
M.S. Javed, T. Najam, I. Hussain, et al., Adv. Energy Mater. 13 (2023) 2202303. doi: 10.1002/aenm.202202303
[2]
H. Jiang, J. Liu, B. Tang, et al., Adv. Funct. Mater. 34 (2024) 2306399. doi: 10.1002/adfm.202306399
S. Zhang, C. Zhao, K. Zhu, et al., Energy Environ. Mater. 6 (2022) e12388.
[55]
Z. Song, L. Miao, L. Ruhlmann, et al., Adv. Funct. Mater. 32 (2022) 2208049. doi: 10.1002/adfm.202208049
[56]
Q. Wang, S. Wang, J. Li, et al., Adv. Electron. Mater. 6 (2020) 2000388. doi: 10.1002/aelm.202000388
[57]
Z. Song, L. Miao, Y. Lv, et al., Angew. Chem. Int. Ed. 62 (2023) e202309446. doi: 10.1002/anie.202309446
[58]
D. Jia, Z. Shen, Y. Lv, et al., Adv. Funct. Mater. 34 (2023) 202308319.
[59]
C. Hu, Y. Qin, Z. Song, et al., J. Colloid Interface Sci. 658 (2024) 856–864. doi: 10.1016/j.jcis.2023.12.144
[60]
Y. Zhang, Z. Song, L. Miao, et al., Angew. Chem. Int. Ed. 63 (2024) e202316835. doi: 10.1002/anie.202316835
Figure 1
(a) Typical schematic diagram for the procedures of HPCx. SEM images of (b) CRF, (c) HPC4. (d) TEM image of HPC4. (e) Nitrogen adsorption−desorption isothermal curves. (f) Pore size distributions of CRF and HPCx, and (g) high-resolution C 1s XPS spectra.
Figure 2
Electrochemical performances of the assembled ZHCs. (a) Diagram of the operation mechanism of CRF/HPCx-based capacitor. (b) CV curves at 5 mV/s. (c) GCD profiles at 0.5 A/g. (d) GCD profiles and (e) the relationship between capacity and current density. (f) Rate capacities at different current densities. (g) Ragone plots. (h) Cycling stability of HPC4-based device at 10 A/g.
Figure 3
(a) A GCD curve of Zn//HPC4 device. (b) Nyquist plots. (c) XRD characterization and high-resolution XPS spectra of (d) Zn 2p, (e) S 2p, and (f) O 1s at various discharged/charged courses.
Figure 4
(a) CV profiles of HPC4-based device at different scan rates. (b) The b values in the anodic and cathodic scanning from 0.5 mV/s to 20 mV/s. (c) Capacitive contribution at 10 mV/s, and (d) capacitive and diffusion-controlled contribution ratios at the scan rates from 0.5 mV/s to 20 mV/s of HPC4-based device. (e) The comparison chart of the six parameters of CRF/HPCx-based devices. (f) Nyquist plots. (g) Real capacitance, and (h) imaginary capacitance versus frequency. (i) The relationship between Z’ and ω−0.5.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
