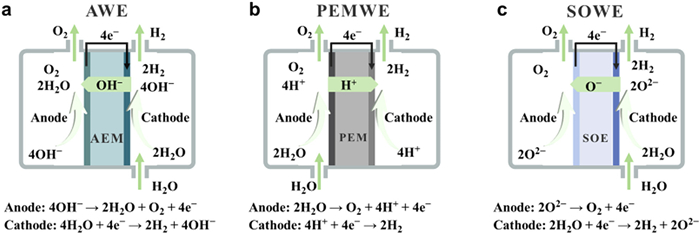
Figure 1.
Schematic diagrams of (a) AWE, (b) PEMWE, and (c) SOWE.
Recent advancements in noble-metal electrocatalysts for alkaline hydrogen evolution reaction
Guoliang Gao , Guangzhen Zhao , Guang Zhu , Bowen Sun , Zixu Sun , Shunli Li , Ya-Qian Lan

In recent decades, there has been an excessive exploitation of fossil fuels in order to meet the escalating global energy demand. However, the need for clean and renewable energy is on the rise as a result of escalating environmental pollution and climate change, primarily attributable to the greenhouse effect [1-5]. Hydrogen has garnered recognition as an optimal energy source due to its substantial calorific value, elevated energy density, absence of carbon emissions, and recyclability [6-8]. Hence, numerous scholarly investigations have prioritized the examination of electrocatalytic hydrogen evolution as a promising approach for hydrogen production [9-11].
Based on the equipment structure, electrolyte composition, and operating parameters, the predominant methods for electrolytic water hydrogen production can be categorized into three types: Alkaline water electrolysis (AWE, Fig. 1a), polymer electrolyte membrane water electrolysis (PEMWE, Fig. 1b), and solid oxide water electrolysis (SOWE, Fig. 1c) [12-14]. A comparative analysis of their primary operating parameters, advantages, and disadvantages is presented in Table 1. The PEMWE system employs a proton exchange membrane (PEM) to facilitate the transportation of protons and to separate the anode and cathode electrodes. This technology surpasses the limitations of the AWE system by achieving a higher current density, thereby enabling enhanced efficiency, rapid reaction rates, and the ability to adjust hydrogen production levels [15,16]. Nevertheless, the current state of PEMWE's electrode catalysts heavily relies on substantial quantities of noble metals [17,18], while the stability of membrane materials and manufacturing costs remain significant obstacles that need to be addressed. The SOWE device is capable of converting electrical and thermal energy into chemical energy at elevated temperatures [19,20]. In comparison to conventional water electrolysis conducted at ambient temperatures, SOWE exhibits superior energy conversion efficiency. However, the substantial capital investment required for equipment and the limited operational lifespan pose challenges. It is anticipated that SOWE may emerge as a viable technical pathway for large-scale hydrogen production once advanced nuclear energy becomes abundantly available in the future. However, it is important to note that the current state of SOWE technology is not yet mature and commercialization remains pending. The commercialization of AWE has spanned over a century, establishing it as the most mature and cost-effective technology extensively employed in the industrial sector, serving as the predominant method for hydrogen production through electrolytic water splitting [21-23]. Moving forward, the continuous advancement in material innovation holds the potential for AWE technology to address concerns related to corrosion and durability, thereby enhancing efficiency and reliability. Furthermore, AWE possesses the advantageous attribute of swift deployment and application in tackling the prevailing issue of renewable energy consumption. In recent years, significant advancements have been made in the development of non-noble metal catalysts for AWE [24,25]. However, a discernible disparity persists between their performance and that of catalysts based on noble metals [26-29]. From a catalytic point of view, it is feasible to further improve AWE's efficiency and reduce its cost.
The electrocatalyst, being an integral element of electrochemical water splitting technology, plays a pivotal role in determining the efficiency of the HER in terms of energy conversion. An optimal electrocatalyst for the HER should demonstrate the following key attributes: (1) Remarkable electrocatalytic performance across a broad pH spectrum in electrolyte solutions; (2) Exceptional durability and consistent activity in intricate electrolytes; (3) Cost-effectiveness, straightforward synthesis methodology, and convenient scalability for industrial applications [30,31]. Currently, the electrodes utilized in alkaline electrolyzers predominantly consist of Ni-based materials, which are coated with catalysts to enhance the efficiency of electrolysis. These catalysts primarily comprise Ni-based catalysts such as Raney Ni or noble metal catalysts. Despite the relative maturity of AWE technology, its operational current density remains limited to a range of 0.2–0.6 A/cm2. Noble metal-based materials, including Pt, Ir, and Ru, are widely acknowledged as highly promising electrocatalysts for the HER due to their high exchange current density and low Tafel slope. Some AWE utilizing platinum group metals have demonstrated an operating current density ranging from 0.6 A/cm2 to 0.9 A/cm2. However, the practical application of these electrocatalysts is hindered by their limited water dissociation capacity and the high energy barrier associated with the Volmer step. Consequently, the slow reaction kinetics in alkaline electrolytes diminish the advantages offered by noble metal catalysts to a certain extent. Furthermore, the exorbitant expense associated with noble metal materials further exacerbates the challenges faced by industrial large-scale implementations. In order to ensure the sustainable advancement of alkaline water electrolysis utilizing noble metal-based electrocatalysts, it is imperative to establish methods for achieving controlled synthesis of these catalysts. This serves as the fundamental step in bridging the gap between chemical synthesis and practical application. Consequently, attaining a comprehensive comprehension of classical modification strategies becomes the primary objective in order to accurately regulate the activity centers [32].
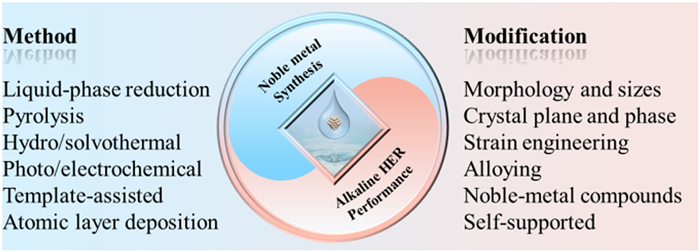
There is a substantial body of commendable literature available on AWE, which offers a comprehensive overview of recent advancements across various domains [33-35]. However, these reviews primarily focus on the emergence of sophisticated non-noble metal electrocatalysts, encompassing transition metal alloys, sulfides, phosphates, and carbides. To our knowledge, the utilization of noble metals in the investigation of alkaline HER has witnessed numerous breakthroughs and experienced rapid development. Nevertheless, a scarcity of comprehensive reviews exists in this particular realm. This paper examines the recent progress in employing noble metals materials as electrocatalysts for the alkaline HER. Initially, we provide a concise overview of the electrocatalytic water splitting mechanism and discuss the merits and drawbacks of noble-metal materials utilized as HER catalysts. Subsequently, our attention is directed towards the prevalent synthesis techniques employed for noble metal-based catalysts and the strategies employed to enhance HER performance (Fig. 2). In conclusion, this study provides a comprehensive overview of the current issues and obstacles encountered in the research domain, specifically focusing on noble-metal HER electrocatalysts. Additionally, we put forth potential avenues for future research in this area.
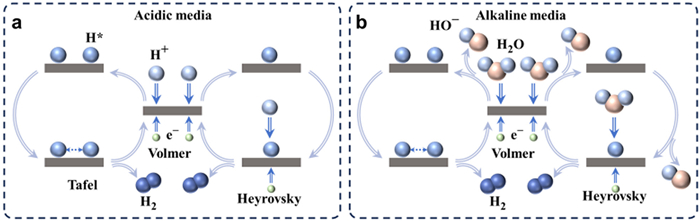
The HER is a straightforward electrocatalytic process that involves the transfer of two electrons. In order to enhance the performance of HER catalysts, it is crucial to have a comprehensive understanding of the reaction mechanism. As a semi-reaction of water splitting, HER consists of two reaction mechanisms (Figs. 3a and b) and encompasses three possible reaction steps (Eqs. 1–5). Taking the alkaline medium as an example, the initial step of HER involves the Volmer reaction between the active site M and H2O, resulting in the formation of adsorbed H (H*, Eq. 2). The second step of the HER involves the electrochemical desorption of H* species, which were formed in the preceding step, with H+ ions present in the solution. This desorption process, primarily facilitated by the dissociation of H2O molecules, ultimately leads to the production of H2 gas. This step is commonly referred to as the Heyrovsky step (Eq. 4). Additionally, another reaction takes place in which two adjacent H* species undergo chemical desorption, resulting in the formation of H2 gas. This reaction process is known as the Tafel step (Eq. 5). The specific pathway, either Volmer–Heyrovsky or Volmer–Tafel step, through which the reactants generate hydrogen is determined by the composition of the catalyst and the reaction medium (Figs. 3a and b) [36]. For instance, Pt-based catalysts typically engage in the Tafel reaction to produce H2, whereas transition metal-based catalysts typically undergo the Heyrovsky reaction to generate H2. This discrepancy arises from the fact that the hydrogen adsorption free energy (ΔGH*) of Pt metal approaches zero, thereby facilitating the formation of a greater H* coverage on the catalyst surface and increasing the likelihood of recombination between adjacent H* species. Conversely, transition metals exhibit a significantly elevated ΔGH*, impeding the adsorption of H+ at the active site [37,38]. In contrast to acidic environments (Fig. 3a), it is evident that in alkaline conditions (Fig. 3b), the generation of H+ necessary for the Volmer step and Heyrovsky step is facilitated by the breaking of the HO-H bond, thereby introducing an additional energy barrier. Consequently, the occurrence of alkaline HER is typically more challenging compared to acidic HER. In order to fulfill the requirements for alkaline HER catalysts, three conditions must be satisfied: (1) A moderate Gibbs free energy (GH*) is necessary to facilitate early H+ adsorption and subsequent H2 desorption; (2) An exceptional ability to dissociate H2O is required to provide H+; (3) The adsorption of OH− should not exhibit excessive strength to prevent the active site from being poisoned. During electrocatalytic testing, the Tafel slope value is commonly employed to ascertain the HER mechanism employed by the catalyst. Specifically, Tafel slope values of 120, 40, and 30 mV/dec correspond to the Volmer, Heyrovsky, and Tafel steps as the rate-determining step, respectively [39,40].
Volmer discharge reaction
|
|
(1) |
|
|
(2) |
Heyrovsky electrochemical desorption reaction
|
|
(3) |
|
|
(4) |
(3) Tafel chemical desorption reaction
|
|
(5) |
Numerous studies have employed noble metals as heterogeneous catalysts due to their exceptional catalytic activity and selectivity in various chemical reactions, which hold significant scientific and industrial implications [41,42]. Nevertheless, the elevated overpotential observed in the HER at the cathode and the oxygen evolution reaction (OER) at the anode restricts the energy conversion efficiency of electrolytic water reactions [43]. The utilization of an electrocatalyst can alleviate the energy barrier of the reaction and enhance catalytic efficiency.
Based on Pt content, electrocatalysts commonly used can be classified into non-Pt catalysts, Pt catalysts, and low-Pt catalysts [44,45]. Among these, Pt is regarded as a superior catalyst for HER, while the oxide of Ir/Ru is considered a superior catalyst for OER. Nevertheless, noble metal-based catalysts face two significant limitations that restrict their extensive industrial applicability: Firstly, noble-metal materials are typically scarce and expensive [46], and secondly, pure noble metal catalysts exhibit sluggish water dissociation rates in alkaline electrolytes, leading to unsatisfactory overall performance [47]. To develop noble metal-based catalysts with diminished noble metal content while preserving their catalytic performance in diverse electrolyte environments, it is imperative to possess a comprehensive comprehension of reaction mechanisms and structure–activity relationships [48,49].
The process of liquid-phase chemical reduction entails the direct reduction of metal salt solutions through the utilization of reducing agents, typically conducted under mild temperature and pressure conditions. Metal precursors, namely chlorides, sulfates, and nitrates, are soluble salts, while reducing agents encompass borohydride, citrate, and alcohol [50,51]. This approach has proven effective in the synthesis of electrocatalysts containing metals such as Pd, Pt, Ru, Rh, Ag, and other metallic elements [52-56]. The primary benefits of employing this methodology include its cost-effectiveness and minimal equipment prerequisites. The liquid-phase reduction procedure can be readily controlled, enabling optimization of the catalyst's crystal surface and size through manipulation of reaction time, temperature, reducing agent, and additional parameters [57].
To prevent agglomeration during preparation and catalysis, as well as to achieve a system with well-dispersed NPs, it is recommended to immobilize small NPs onto carriers [36]. The most straightforward method to regulate the size of metal particles is to synthesize NPs and incorporate them onto the substrate [58]. Numerous investigations have provided evidence that the in-situ synthesis of metal NPs on the surface of the substrate material yields resilient catalysts [51]. Calvo et al. [59] introduced a technique for the in-situ synthesis of supported noble-metal catalysts through the reduction of the noble-metal precursor using NaBH4 in the presence of both the substrate and the noble-metal precursor (Figs. 4a–d). The utilization of this in-situ synthesis approach results in the production of smaller NPs with a more limited size distribution (Figs. 4b and d) in comparison to the conventional ex-situ synthesis method (Figs. 4e and f), which involves the reduction of the noble-metal precursor with NaBH4 followed by fixation on the substrate. The catalyst that was synthesized in situ demonstrated enhanced stability in water at a temperature of 80 ℃, suggesting a more robust interaction between the support and the metal phase.
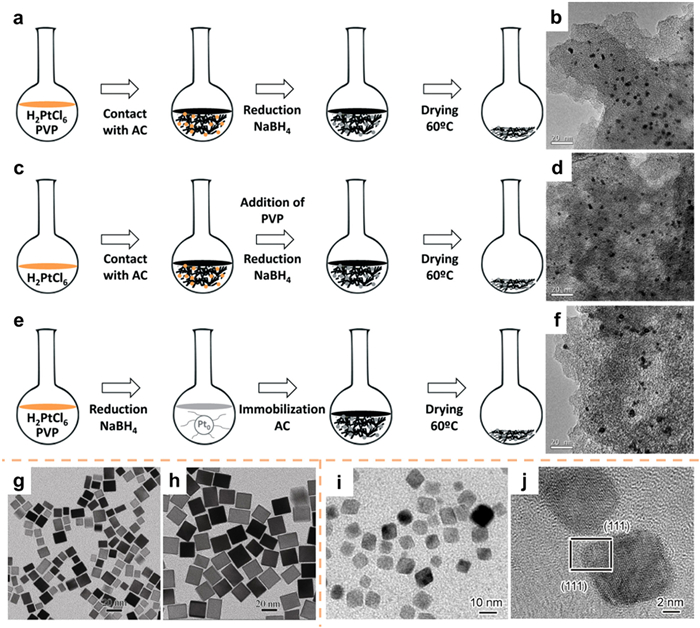
In recent years, scholars have provided evidence illustrating the significant impact of catalyst size and exposed crystal surface on the efficacy and selectivity of certain catalytic reactions [60,61]. Xia's group [62] synthesized Pd cubes featuring exposed (100) crystal planes through the utilization of ascorbic acid as a reducing agent. Extensive research has been conducted to investigate the fundamental mechanism of KBr, a specific capping agent. The growth of nanocrystals typically transpires in two distinct stages: nucleation and growth. In instances where the KBr content is elevated, the reduction rate diminishes, resulting in a reduced formation of seeds during the nucleation stage. Consequently, the noble-metal precursor continues to expand on the existing seeds during the subsequent growth stage. The dimensions of the nanocrystals exhibit a positive correlation with the diminishing size of the seed, while keeping the precursor quantity constant (Figs. 4g and h). In a similar liquid-phase reduction process, Xiong et al. [63] employed citric acid and ascorbic acid as reductants to fabricate Pd octahedrons with exposed (111) crystal planes (Figs. 4i and j).
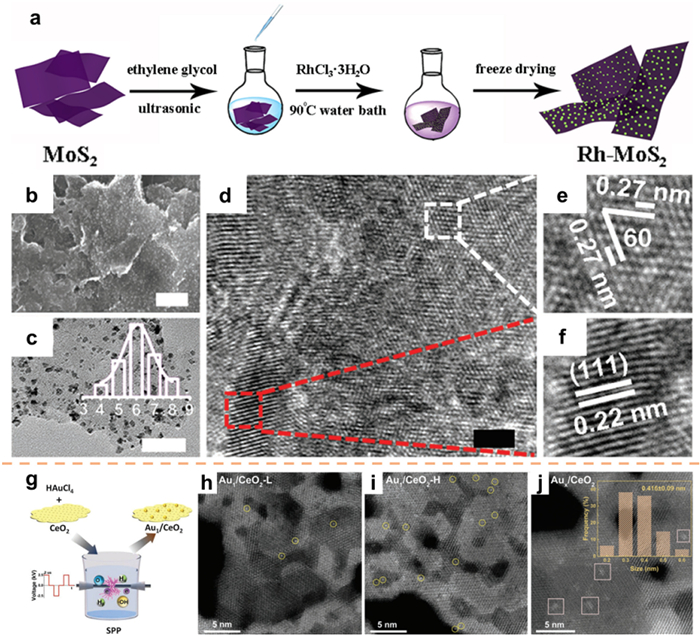
To improve the catalytic performance, it is an efficient strategy to combine catalysts with different advantages through in-situ growth. Shao and co-workers [64] implemented a liquid-phase chemical reduction technique to anchor Rh NPs in situ on a MoS2 substrate (Rh-MoS2). The Rh precursor was introduced into the MoS2 suspension and subsequently reduced to Rh NPs using ethylene glycol at a temperature of 90 ℃ (Fig. 5a). Ultimately, the Rh-MoS2 composite was subjected to freeze-drying. Fig. 5b displays a scanning electron microscopy (SEM) image of the Rh-MoS2, which consist of white Rh NPs and MoS2 nanosheets exhibiting a folded morphology. A significant quantity of Rh NPs was evenly deposited onto the surface of ultrathin MoS2 nanosheets, which possessed an average diameter of 6 nm (Fig. 5c). The crystal structure of the Rh-MoS2 composite was subsequently examined using HRTEM, as depicted in Fig. 5d. The measured plane spacing was determined to be 0.27 nm, corresponding to the (1010) plane (Fig. 5e). Additionally, Fig. 5f showcases another heterogeneous structure with a lattice spacing of 0.22 nm, corresponding to the (111) plane of Rh. In another study, Zhang et al. [65] developed a solution plasma process (SPP) that facilitated the generation of oxygen vacancies in CeO2 nanosheets and the capture of approximately 99% of single atom Au within a time frame of 2 min (Fig. 5g). The presence of a substantial number of electrons and free radicals in the plasma discharge region allows SPP to simultaneously reduce HAuCl4 to monatomic Au and introduce oxygen vacancies in CeO2 as anchoring sites. By adjusting the quantity of Au precursor, samples with Au mass fractions of 0.19, 0.37, and 0.75 wt% were obtained and designated as Au1/CeO2-L, Au1/CeO2-H, and Aux/CeO2, respectively (Figs. 5h–j).
The pyrolysis technique is predominantly employed for the purpose of selecting appropriate precursors for high-temperature pyrolysis under specific temperature conditions and within atmospheres composed of N2, Ar, H2, or NH3 [66-68]. These precursors primarily encompass a combination of carbon skeletons [24], metal complexes [69], metal-organic frameworks (MOFs) [70,71], and polymers [72,73]. Chen and co-workers [74] conducted the preparation of Ru-doped MOFs by employing a Ru3+ absorption technique on a MOF material characterized by a substantial specific surface area. Subsequently, the researchers synthesized RuCo@NC through the annealing process of the absorbed material under an N2 atmosphere (Fig. 6a). The examination of TEM and HRTEM images (Figs. 6b and c) revealed the presence of a composite structure with nitrogen doping, wherein a graphene-like coating enveloped the alloy core. The utilization of the "one-step pyrolysis" approach enables the concurrent carbonization of a carbon precursor and the reduction of a noble metal through a solitary heat treatment, thereby facilitating a simplified process and contributing to the mitigation of production expenses associated with these catalysts.
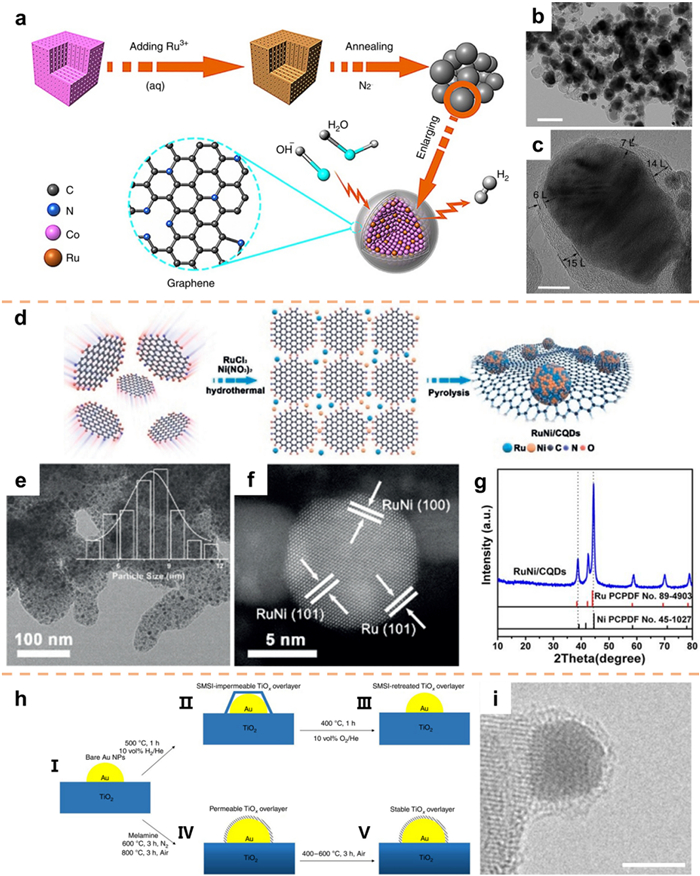
In the preparation of ultrafine catalysts, it is common practice to employ a conductive substrate as a means to secure a metal precursor. This serves to achieve a uniform dispersion and prevent aggregation during the preparation process [75,76]. Carbon quantum dots (CQDs) have proven to be more efficient than other carbon materials such as MOFs and graphene. This is ascribed to the substantial presence of functional groups, which facilitate coordination with metal ions and result in the formation of relatively stable CQDs-metal ion coordination composites [77,78]. Metal NPs are confined within CQDs, resulting in the formation of highly refined nanocrystals with a stable structure that effectively inhibits NPs aggregation during the reaction. Accordingly, Lu et al. [79] developed a comprehensive approach to fabricate carbonized polymer dots and nanocrystalline hybrid materials using CQDs as fundamental building units (Fig. 6d). Through pyrolysis, the disordered RuNi transformed into an ordered RuNi alloy, while neighboring CQDs formed graphene layers enriched with nitrogen. The TEM image in Fig. 6e demonstrates the uniform dispersion of RuNi NPs on the surface of CQDs. The successful preparation of RuNi alloy is demonstrated through the utilization of HAADF-STEM images (Fig. 6f) and X-Ray Diffraction (XRD) analysis (Fig. 6g).
Supported Au catalysts have gained significant popularity in various industrial applications because of their excellent catalytic properties. Nevertheless, the low Tammann temperature of Au renders it susceptible to sintering during high-temperature reactions, thereby compromising the stability of the catalytic process. In an attempt to address this issue, Wang et al. [80] developed anti-sintered Au catalysts by employing melamine in a high-temperature oxidation atmosphere to coat Au NPs with a permeable TiOx thin layer (Fig. 6h). Remarkably, this coating process was observed to occur in an oxidizing atmosphere (Fig. 6i). The crucial factor contributing to this phenomenon lies in the application of melamine modification, followed by treatment in a high temperature N2 atmosphere, and subsequent roasting in an air atmosphere at 800 ℃. Of particular significance is the fact that the TiOx coating remained intact even when exposed to further oxidation atmospheres ranging from 400 ℃ to 600 ℃ (Fig. 6h).
Hydrothermal/solvothermal synthesis is a significant technique employed in the synthesis of nanomaterials owing to its straightforward, expedient, and expeditious operation [81,82]. The primary procedure entails the formulation of a solution utilizing the raw material, subsequent transfer to a hydrothermal reaction kettle, and heating to a specific temperature, while ensuring the synthetic system remains within a predetermined pressure range. Within this non-equilibrium synthetic system, liquid-phase reactions frequently yield materials exhibiting exceptional and distinctive properties [83,84]. For example, Yin and co-workers [85] employed a straightforward hydrothermal technique to fabricate a nanosheet-structured IrNi-FeNi3 hybrid catalyst (Fig. 7a). The TEM and HRTEM images (Figs. 7b and c) revealed lattice spacings of 0.222, 0.203, and 0.204 nm, corresponding to the (111) plane of Ir, Ni, and FeNi3, respectively. The amalgamation of IrNi alloy and FeNi3 alloy generates the IrNi-FeNi3 hybrid, thereby enhancing the activity and stability of the catalysts.
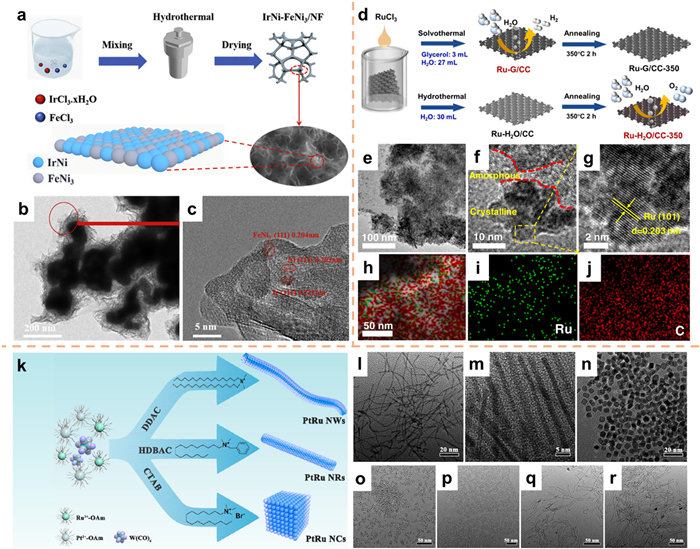
On conductive substrates such as nickel foam (NF) and carbon cloth (CC), in-situ synthesis of self-supporting catalysts can improve the working stability of catalysts. Chen et al. [86] employed glycerol as precursors, and combined it with a solution of RuCl3·xH2O to cultivate Ru nanoparticle electrodes with an amorphous/crystalline mixed structure in-situ on CC through a solvothermal method, referred to as Ru-G/CC. Additionally, Ru-H2O/CC-350, which contained a few O vacancies, was grown in-situ on CC using RuCl3·xH2O solution as a precursor through a hydrothermal method, followed by low-temperature annealing (Fig. 7d). The TEM results revealed that Ru-G/CC displayed a combination of crystalline and amorphous Ru. The scanning transmission electron microscopy-energy dispersive X-ray (STEM-EDX) analysis provided elemental mapping, confirming the uniform distribution of Ru and C on Ru-G/CC (Figs. 7e–j).
Despite the demonstrated exceptional methanol oxidation activity of PtRu alloys in both experimental and theoretical calculations, the successful synthesis of precisely defined face-closed PtRu nanocrystals remains a formidable task. In their study, Dong's group [87] employed the solvothermal method to produce ultra-thin PtRu nanocrystals with diverse morphologies by manipulating surfactant and reaction durations (Figs. 7k–r).
The photochemical deposition method is frequently employed for the integration of co-catalysts into photocatalysts. This technique entails the utilization of photogenerated electrons to reduce precursor ions, leading to the deposition of NPs onto semiconductor substrates [88,89]. This approach is extensively utilized for the preparation of small-scale noble-metal electrocatalysts [90,91]. To reduce the load of noble metal cocatalysts, many atomic-scale Pt materials have been developed. For example, Ling et al. [92] utilized wet chemistry and photodeposition techniques to synthesize Pt atomic clusters that were incorporated within TiO2 nanocrystals/graphene nanosheets (Pt−T/G) (Fig. 8a). The SEM images of Pt−T/G-150 provided evidence of the graphene sheet architecture being well-preserved. Furthermore, the HAADF-STEM image indicated the presence of Pt atoms dispersed along the periphery of anatase crystals (Figs. 8b and c).
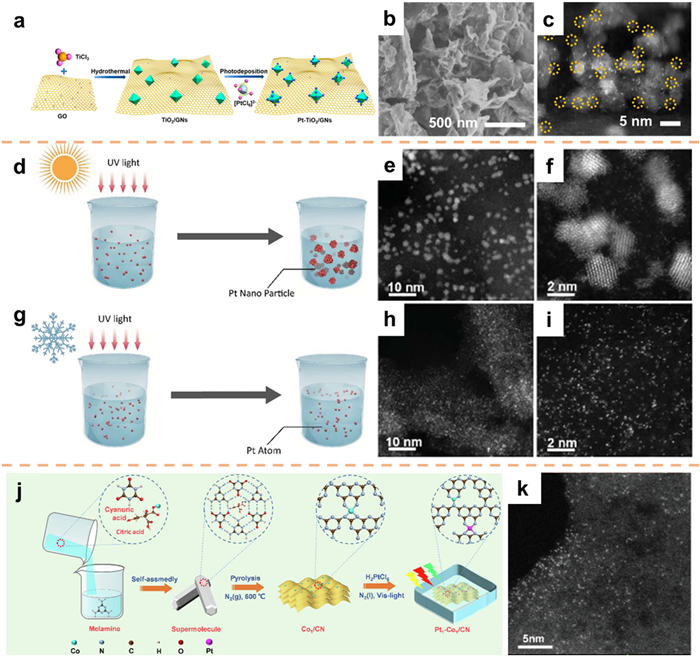
Similarly, Wu's group [93] utilized a photochemical methodology to produce dispersed Pt single atoms at a significantly low temperature of −60 ℃. Conventionally, Pt atoms are formed via photochemical reduction of Pt ion precursors, resulting in the formation of Pt nanocrystals through nucleation and growth processes (Fig. 8d–f). In contrast, Wu et al. deviated from the standard room temperature photochemical reduction process by subjecting an H2PtCl6 antifreeze solution to UV light irradiation at −60 ℃, thereby conducting a photochemical reaction under non-conventional conditions (Figs. 8g–i). The nucleation process is subject to significant influence from the reaction temperature, with a decrease in temperature presenting heightened obstacles to nucleation. In the specific context of this ultralow-temperature reaction, nucleation is effectively inhibited, resulting in the creation of atomically dispersed Pt.
In their study, Sun et al. [94] utilized a supramolecular self-assembly approach to synthesize carbon nitride (CN) material and subsequently incorporated a single Co atom to effectively fabricate the Co1/CN catalyst. Additionally, they employed a freezing-visible-light-deposition technique (Fig. 8j) to prepare the Pt1-Co1/CN bimetallic single-atom catalyst. The HAADF-STEM image analysis confirmed the presence of Pt and Co metal species on the CN surface, thus validating their atomic distribution (Fig. 8k).
Electrodeposition is an electrochemical technique employed to reduce metal ions found in electrolytes, leading to the production of corresponding metals and alloys [95-97]. This process presents numerous benefits, such as its rapidity and cost-effectiveness, the capacity to manipulate the pathway with ease, enabling the creation of uniformly dispersed electrocatalysts, and the elimination of the necessity for supplementary terminating or reducing agents, thereby facilitating the satisfaction of industrial requirements [98]. Furthermore, the manipulation of the catalyst's composition and structure can be achieved through the regulation of electrodeposition parameters, including the electrolyte composition, current density, conductive substrate composition, deposition time, and temperature [99,100].
At high potentials, Pt atoms can dissolve from the Pt anode and then transfer to the cathode, inspired by this, Luo and co-workers [101] proposed a novel potential-cycling technique for the synthesis of a catalyst consisting of Pt single-atom catalysts (SACs) dispersed on CoP-based nanotube (NT) arrays supported by NF, referred to as PtSA-NT-NF (Fig. 9a). This method involved the utilization of a three-electrode pool containing phosphate buffers (PBS), in which the Pt foil acts as the anode and the CoP-based nanotube acts as the cathode. The morphology of the CoP nanotube array was examined using SEM (Fig. 9b), and further analysis was conducted using atomic-resolution (AR) HAADF imaging combined with aberration-corrected STEM, revealing a significant presence of well-dispersed single Pt atoms on the CoP NTs (Fig. 9c).
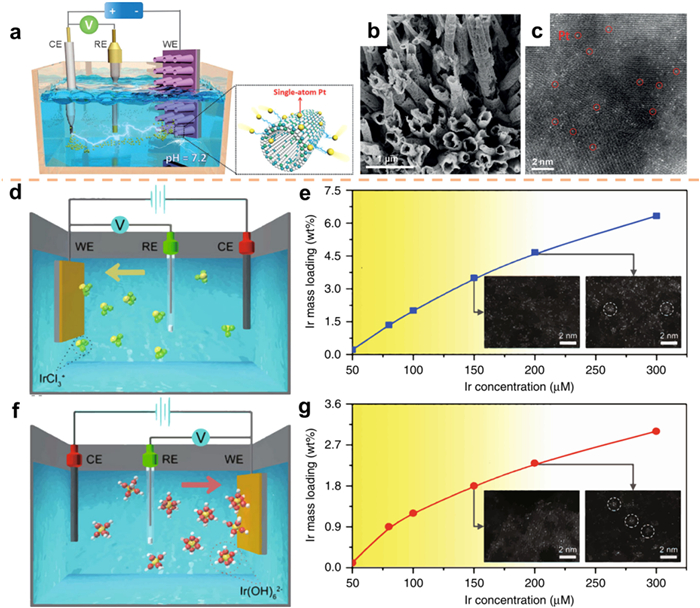
Zeng et al. [102] introduced a comprehensive electrochemical deposition technique to produce single-atomic electrocatalysts. In the process of cathode deposition, the application of an electric field enables the migration of Ir ions towards the cathode, leading to their deposition onto the support (Figs. 9d and e). Conversely, during anode deposition, the electric field drives the Ir anions towards the anode (Figs. 9f and g). To gain a comprehensive understanding of the deposition process of SACs, Zeng et al. conducted a series of experiments wherein they manipulated the concentration of Ir precursors. The cathodic deposition was carried out in a 1 mol/L KOH electrolyte over a span of 10 scanning cycles, with the concentration of Ir ranging from 50 µmol/L to 300 µmol/L. The findings presented in Fig. 9e demonstrate a clear positive relationship between the concentration of Ir and the resulting mass loading of Ir. Even when the Ir concentration was as low as 150 µmol/L, the formation of Ir single atoms was achieved, with a mass loading of 3.5%. However, as the concentration was further increased to 200 µmol/L, the presence of Ir clusters became apparent, resulting in a higher mass concentration of 4.7%. Therefore, it can be deduced that the upper limit for achieving SACs falls within the range of 3.5%–4.7% mass loading. Various observations were made during the deposition of anodes, including the examination of variations in Ir concentration, scanning cycle, and scanning rate (Fig. 9g). This methodology demonstrates its compatibility with diverse metals and catalyst supports, thereby enabling the deposition of more than 30 distinct SACs concurrently on both the cathode and anode through the manipulation of the metal precursor and support.
From the above examples, it can be seen that photoelectrochemical deposition is an efficient method for synthesizing atomic-size catalysts. On this basis, the researchers devised a freeze-assisted photoreduction to achieve nanospatial confinement and even prepared neighboring Pt-Ru monomers catalysts [103]. However, we also need to realize that the expansion of industrial production of this method is a big challenge. At present, the synthesized single atom catalyst of milligram yield level is more suitable for “model catalyst”. Since the active sites of SACs are clear and uniform, researchers use this feature to deeply understand the reaction mechanism at the molecular level [104,105].
The template method involves the utilization of a material with a well-defined three-dimensional structure and a shape that can be easily controlled, serving as a template. By employing physical, chemical, or biological techniques, atoms or ions of the desired material are deposited onto the surfaces or within the cavities of the template. Following this, the template is removed, leading to the acquisition of nanostructured materials that exhibit the desired characteristics [106-109]. It is noteworthy that materials fabricated through this methodology commonly demonstrate structural characteristics similar to those of the template [110-112].
Two-dimensional (2D) graphene nanosheets (GNSs) are ideal support due to their large surface area and excellent electrical conductivity. However, 2D GNSs are prone to accumulation and aggregation during long-term catalysis, which reduces the number of exposed active sites [113,114]. 3D graphene hollow nanospheres (GHSs) have high mechanical robustness, which can inhibit the occurrence of the above situation to a certain extent. Inspired by this, Li et al. [115] utilized a template-assisted approach for the synthesis of a new hybrid Ru nanoparticle anchored to 3D GHSs. The procedure involved the initial adsorption of graphene onto the SiO2 surface via electrostatic interaction, followed by the introduction of a solution containing RuCl3. Subsequent prolonged agitation facilitated the complete adsorption of Ru3+ ions onto the graphene surface, leading to the generation of a stable interface between Ru and graphene upon calcination. Following that, the SiO2 template underwent etching, resulting in the creation of Ru/GHSs possessing a hollow structure (Fig. 10a). The TEM analysis (Fig. 10b) further substantiated the unique hollow structure characteristics of Ru/GHSs. HRTEM measurements (Fig. 10c) unveiled lattice fringe spacing of 0.21 nm and 0.34 nm, corresponding to the (101) crystal plane of hexagonal Ru and the (002) crystal plane of graphitic carbon, respectively. The presence of Ru NPs was indicated by the black region. The even dispersion of Ru NPs contributes to improved accessibility of active sites and increased efficiency of metal utilization. GHSs exhibit the ability to not only hinder the aggregation or dissolution of Ru NPs but also ensure the stability of the hybrid system during electrolysis.
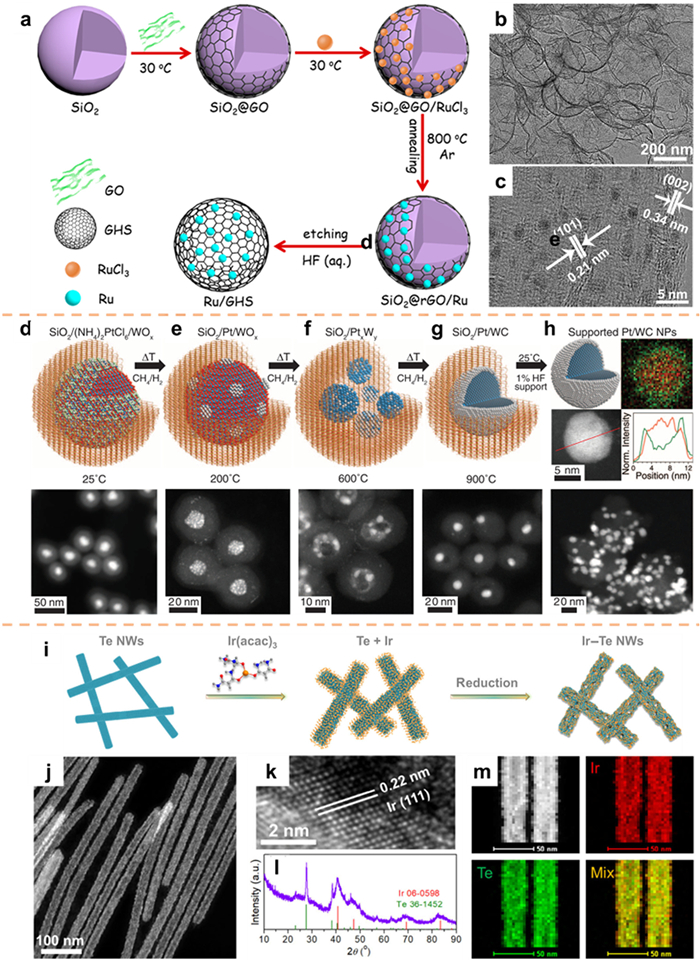
The deposition of noble metals onto transition-metal nuclei has been found to enhance catalyst activity and reduce costs. However, it is frequently observed that these structures undergo alloying or deactivation during preparation or reaction processes [116,117]. In a recent study by Hunt et al. [118], core-shell catalysts were prepared using the template method, which involved the utilization of single atomic layers of metallic carbide surfaces loaded with noble metals. The procedure entailed the encapsulation of a noble-metal salt and an excess of metal oxide within a SiO2 template, which was subsequently subjected to a gradual carbonization process. As a result, the SiO2 template was removed, resulting in the formation of a singular layer of a core-shell catalyst consisting of noble metal, which was supported on the surface of the transition-metal carbide. The detailed process for the preparation of the materials is illustrated in Figs. 10d–h, which includes STEM images depicting each stage. The element distribution maps and linear scanning outcomes (Fig. 10h) offer compelling evidence for the successful synthesis of Pt/WC core-shell structure catalysts. The achievement of self-assembled transition-metal carburization NPs and an atomically thin noble-metal monolayer coating was realized through the carburization process of a mixture comprising noble-metal salts and transition-metal oxides within a silica template. This innovative methodology presents a promising pathway for alleviating the challenges associated with noble-metal catalysts, enhancing their effectiveness, and prolonging their longevity.
The incorporation of low dimensional materials, particularly 1D nanowires, has been observed to augment the reactivity of noble metal nanomaterials while concurrently diminishing the necessary quantity of noble metal [119,120]. Huang and co-workers [121] utilized a hydrothermal technique to fabricate superior Te nanowires (Te NWs) as a framework, subsequently introducing Ir(acac)3 as a precursor. The reduction of Ir(acac)3 by a reducing agent led to the creation of porous Ir-Te NWs (Fig. 10i). The confirmation of the successful synthesis of high-quality 1D Ir-Te nanowires was achieved through morphological and structural characterization. The analysis provided evidence of a physical blending between Ir and Te (Figs. 10j–l). Furthermore, the elemental mapping conducted using energy-dispersive spectroscopy (EDS) showcased a homogeneous dispersion of Ir and Te elements, without any indications of element clustering or separation (Fig. 10m).
The precise construction of catalysts at the atomic level is of great importance in understanding the catalytic mechanism and developing advanced catalysts [122,123]. Atomic layer deposition (ALD), a gas-phase synthesis method similar to chemical vapor deposition (CVD) [124], is utilized for the fabrication of thin film catalysts [125-127]. This technique operates by injecting precursors as vapors and facilitating a self-limiting reaction at the molecular level on the substrate surface, allowing for accurate deposition of the desired material. By manipulating the quantity, sequence, and characteristics of deposition cycles, it becomes feasible to intricately regulate the atomic-level configuration of the active sites of the catalyst. This methodology presents an innovative approach for the meticulous and manageable synthesis of catalysts through a bottom-up paradigm [128,129].
Due to the confinement effect, confined catalysts usually show unique catalytic properties and have received more and more attention [130,131]. Zhang et al. [132] utilized ALD technology to design and synthesize Pt-TiO2 catalysts with varied interface structures. The researchers were able to achieve precise manipulation of the size, position, and wall thickness of Pt metal particles. Through modifications in the deposition sequence and kinetics, a diverse range of localized and supported catalysts were produced. In the specific case of the localized Pt@TiO2 catalyst, ultrafine Pt particles were exclusively confined to the inner surface of TiO2 nanotubes, with no presence of particles observed on the smooth outer surface. Moreover, the absence of notable particle aggregation was observed (Figs. 11a–c). Conversely, in the instance of the Pt/TiO2 catalyst with support, under identical roasting conditions, the dispersion of Pt particles along the external surface of TiO2 nanotubes was observed, accompanied by a substantial presence of larger Pt particles (>3 nm) displaying a diverse range of sizes (Figs. 11d–f).
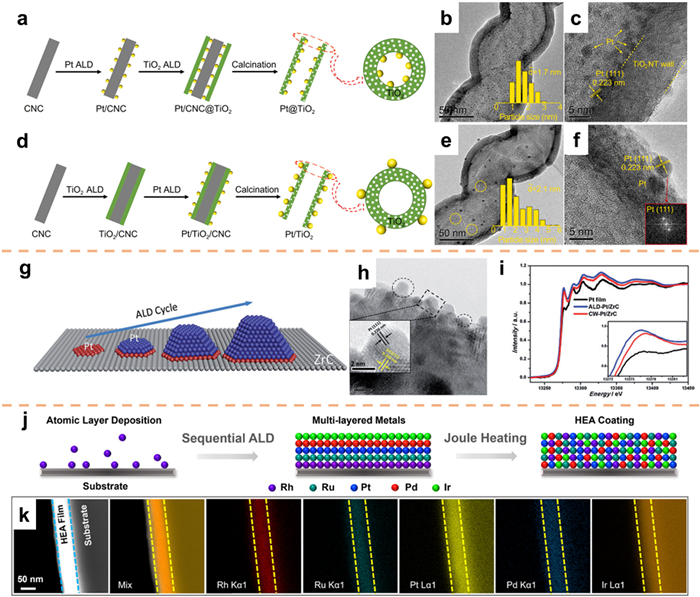
Sun and co-workers [133] employed ALD to produce electrocatalysts with improved catalytic activity and durability. This was achieved by stabilizing Pt catalysts and enhancing interactions through precise manipulation of the metal-support interface at the atomic level. To deposit Pt, ZrC, a carrier material known for its exceptional resistance to corrosion, was utilized. As illustrated in Fig. 11g, the first step involved depositing the Pt precursor onto the surface of ZrC, leading to a strong chemical bonding between the Pt precursor and ZrC. As a consequence of the increased temperature, the initial layer of Pt atoms became incorporated into the ZrC substrate. Through successive cycling, Pt clusters gradually formed on the embedded Pt layer, ultimately leading to the development of Pt NPs on the surface of ZrC (Fig. 11h). Analysis of the XANES revealed a substantial modification in the electronic structure of Pt in the ALD/ZrC catalyst, attributed to its strong interaction with the ZrC support (Fig. 11i). This study affords significant insights for the potential advancement of electrocatalysts with improved stability by enhancing the interactions between catalyst particles and the supporting material.
The synthesis strategies for high entropy alloy (HEA) thin films present substantial challenges in terms of facile preparation, precise thickness regulation, and cost-effectiveness [134,135]. These obstacles are particularly prominent and warrant attention in the realm of noble metal HEA films, as they encounter difficulties in controlling thickness and incurring high expenses (requiring the utilization of a high purity noble metal target) when employing conventional sputtering techniques. Tok et al. [136] produced HEA films containing noble metals using ALD combined with electrical Joule heating (Fig. 11j). The ALD films demonstrated a thickness of approximately 50 nm, indicating precise control over ALD growth at the nanometer scale. The even distribution of metal components in the thickness direction serves as evidence for the effective diffusion of all elements during electrical Joule heating annealing, thereby facilitating the development of a uniform solid solution (Fig. 11k).
In both laboratory investigations and practical applications in industrial settings, the evaluation of the quality of catalysts for the HER is contingent upon two crucial criteria: activity and stability [137,138]. The enhancement of HER activity in catalysts can be attained through the augmentation of active sites and the improvement of the intrinsic activity of the catalyst [139-141]. To achieve this objective, factors such as the adsorption of H+ ions, the strength of the bond between the catalyst and intermediate products, and the desorption of H2 molecules must be duly considered [142,143]. Improving the stability of catalysts for the HER involves various important strategies, including inhibiting catalyst agglomeration during the catalytic process [144-146]. One effective method entails the in-situ growth of noble metals, thereby enhancing the bond strength between noble metals and the substrate [147]. Furthermore, enhancing the corrosion resistance of the catalyst offers an alternative approach to prevent its dissolution and deactivation during the reaction process [148,149].
Metallenes, which are metals with atomically thin layers, have been found to exhibit unique properties [150,151]. In particular, hydride metallenes, which have interstitial hydrogen atoms and a high active surface area, have shown promising potential in hydrogen-related catalysis [152,153]. Cui et al. [154] utilized a seed-mediated growth method to design a highly stable PdHx metallene. The results, as depicted in Fig. 12a, indicate that Ru on PdHx@Ru metallene is primarily located in the surface region. Furthermore, Figs. 12b and c provide evidence of an extended lattice spacing on the surface of Ru, suggesting the presence of tensile strain. The electrochemical test results demonstrate that PdHx@Ru metallene displays remarkable HER catalytic activity in alkaline environments, as evidenced by an overpotential of merely 30 mV at 10 mA/cm2, surpassing both commercial Pt/C and the majority of previously reported Ru-based electrocatalysts (Fig. 12d). Controlled experiments and DFT calculations reveal that the presence of a tensile strained Ru surface layer effectively lowers the energy barrier for H2O dissociation and offers a moderate hydrogen adsorption energy (Fig. 12e).
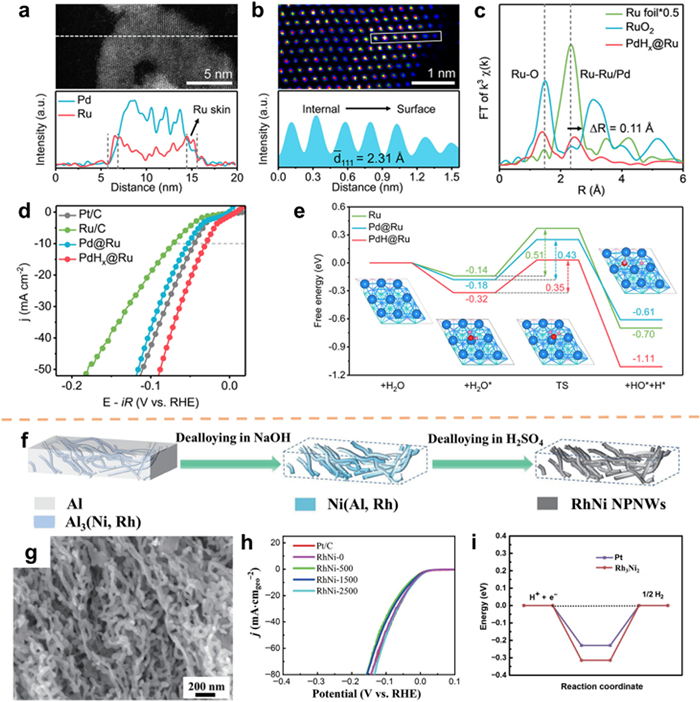
Catalysts incorporating nanostructures possess the capability to amplify catalytic activity by increasing the electrochemical surface area (ECSA) and facilitating mass transfer [155-157]. Among these nanostructures, 1D NWs have attracted significant attention due to their effective utilization in catalysts [157,158]. However, the synthesis of one-dimensional nanostructures typically necessitates the use of surfactants as agents for directing morphology, which presents a challenge in their removal. Moreover, the catalytic activity is adversely affected by the presence of surfactants, as they impede the functionality of active sites [158,159]. In contrast, dealloying is a surfactant-free method that selectively dissolves metal constituents from solid solutions or intermetallic compounds, and has been utilized in the fabrication of nanoporous metals and alloys [160]. The manipulation of precursor phase composition and the dealloying process enables precise control over the desired composition and microstructure of the resulting nanoporous alloy. In accordance with this, Zhang's group [161] introduced a self-templating methodology to fabricate one-dimensional RhNi nanoporous alloy nanowires. This technique integrates metallurgical eutectic solidification, microalloying, and chemical dealloying (Fig. 12f). According to SEM images, the samples exhibited irregular nanowire morphology, with a diameter of approximately 31 nm (Fig. 12g). RhNi catalysts exhibited excellent HER electrocatalytic activity in alkaline media (η10 = 43.1 mV; Fig. 12h). The Gibbs free energy of adsorbed hydrogen atom (ΔGH*) is a crucial parameter that determines the intrinsic activity of the catalyst. As shown in Fig. 12i, ΔGH* corresponding to Rh3Ni2(111) is −0.315 eV (close to −0.229 eV of Pt(111)), indicating that Rh3Ni2 exhibits the same catalytic activity as Pt, consistent with its electrocatalytic performance in HER.
In recent years, the progress in electrocatalyst development has been credited for the improvement of electrocatalytic reactions. A viable approach to enhance electrochemical efficiency and cost reduction entails optimizing the size of electrocatalysts [162,163]. From an engineering perspective, the utilization of nano-sized electrocatalysts can increase the surface area of electrodes and improve performance. Additionally, altering the particle size of a catalyst can impact its intrinsic activity [164-166].
The manipulation of ligands can alter the decomposition temperature of the relevant Ru-based precursor, thus exerting control over the processes of nucleation, growth, and anchoring of metal sites during pyrolysis, consequently impacting the size and dispersion of Ru. By investigating various forms of Ru-based precursors, Liu et al. [167] effectively engineered and synthesized a diverse range of Ru-based NPs possessing distinctive morphological characteristics, including size and dispersion, which were securely affixed to the outer surface of hollow porous carbon shells (Fig. 13a). The STEM analysis conducted on u-Ru-1/C demonstrated that the underlying surface was bolstered by extensively dispersed ultrafine Ru NPs, with an approximate diameter of 1.5 nm (Figs. 13b and c). The TEM images and size distributions of the a-Ru-2/C samples are presented in Figs. 13d and e. The dispersion of particles in the a-Ru-2/C catalyst appeared to be disordered, displaying a wide range of sizes. Upon utilization in the electrocatalytic HER in 1.0 mol/L KOH, the u-Ru-1/C catalyst exhibited a higher electrochemical double layer capacitance (Cdl) value of 23.9 mF/cm2 compared to the a-Ru-2/C catalyst. Additionally, the u-Ru-1/C catalyst demonstrated a small overpotential of 31 mV at 10 mA/cm2 (Figs. 13f and g).
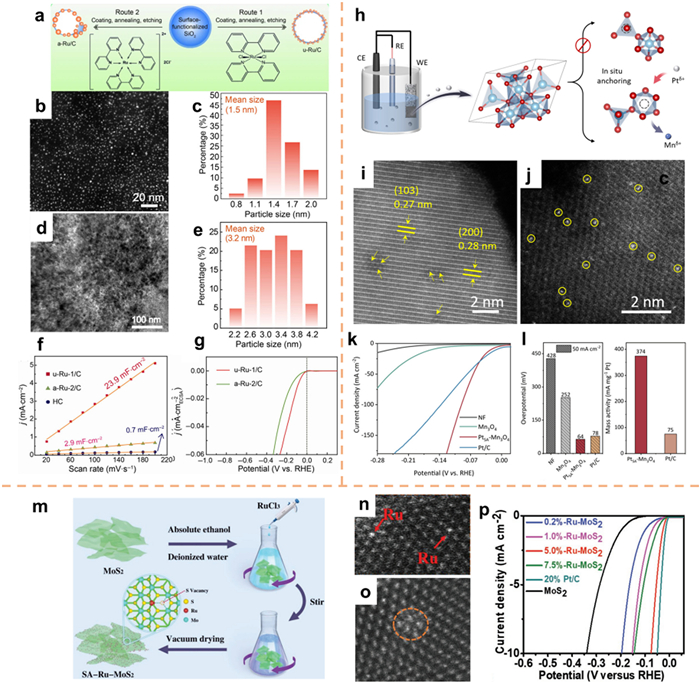
SACs have demonstrated the ability to enhance performance and optimize the utilization of metal centers in comparison to aggregated clusters or NPs [168-170]. Liu and co-workers [171] employed a potential cycling method to fabricate porous Mn3O4 nanosheets with Mn3+ vacancies (VMn-Mn3O4). This process led to the partial dissolution of the Mn3+ site, creating a vacancy suitable for anchoring Pt single atoms (PtSA-Mn3O4, Fig. 13h). The HAADF-STEM image depicted in Fig. 13i revealed the presence of a small Mn vacancy within Mn3O4, while the bright spot observed in Fig. 13j corresponded to atomically dispersed Pt species. The overpotential of PtSA-Mn3O4 is 24 mV at 10 mA/cm2, and the mass activity of PtSA-Mn3O4 (374 mA/mgPt) is 5 times that of Pt/C at 50 mV, which considerably increases the utilization rate of noble metals and reduces costs (Figs. 13k and l). The application of DFT calculations revealed that the electron transfer from the Pt atom to the O atom caused a redistribution of charge, resulting in an increase in the band occupancy of the catalyst near the Fermi level. Consequently, this facilitated the adsorption of H* and subsequent desorption of H2.
The extensive utilization of 2D layered MoS2 nanosheets in the field of HER is primarily attributed to the active sites located at the edges, while the reactivity of in-plane atoms is minimal [172-174]. Therefore, the incorporation of Ru single atoms, anchored by S on the MoS2 surface, is expected to activate the in-plane S atoms, converting them into active sites and optimizing the utilization of Ru. In a study conducted by Cao's group [54], a single-atom Ru catalyst supported on MoS2 (SA-Ru-MoS2) was effectively synthesized at ambient temperature using a direct impregnation technique (Fig. 13m). The presence of single-atom Ru was verified through HAADF-STEM analysis, which also unveiled the occurrence of defects and lattice distortions arising from the incorporation of Ru atoms (Figs. 13n and o). This phenomenon could plausibly be ascribed to the formation of sulfur vacancies during the Ru doping process. The HER performance of MoS2 materials is significantly enhanced by the collective impact of S-vacancy, Ru single atom, and the phase transition of MoS2. It is worth mentioning that in an alkaline environment, the overpotential of SA-Ru-MoS2 at 10 mA/cm2 is only 76 mV (Fig. 13p).
The catalytic reaction primarily occurs at the surface/interface of the catalyst, where the catalytic performance is influenced by the geometric structure, electronic structure, and coordination environment of the catalyst surface atoms [175-177]. Numerous investigations have demonstrated that differences in the surface structures of different crystal planes result in variations in the adsorption energy of reactants and products on the catalyst [178-180]. Therefore, it is crucial to manipulate the active crystal planes and design the crystal structure of the electrocatalyst in order to attain efficient HER [181,182].
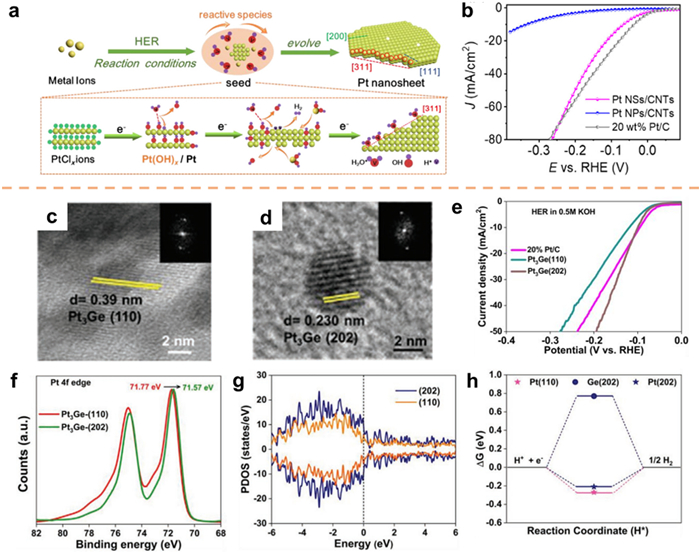
In HER process, Wang et al. [183] conducted a study wherein they employed in-situ electrochemical reduction to prepare Pt nanosheets enclosed with (311), (200), and (111) facets on carbon nanotubes (Pt NSs/CNTs, Fig. 14a). As a comparison sample, Pt NPs (Pt NPs/CNTs) were prepared using a chemical reduction method. The electrochemical test results indicated that the alkaline HER activity of Pt NSs/CNTs was similar to that of 20% Pt/C, with a 12.7-fold increase in mass activity (Fig. 14b). The dissociation of water is facilitated by Pt(311) and Pt(200) according to DFT calculations. Additionally, the H* intermediate exhibits a favorable binding affinity and subsequently releases H2 on Pt(111). In a study conducted by Peter's group [184], Pt3Ge alloys with various exposed facets were synthesized using the solvothermal method. The alkaline HER test revealed that Pt3Ge-(202) exhibited superior performance compared to Pt3Ge-(110) (Figs. 14c–e). The X-ray photoelectron spectroscopy (XPS) results depicted in Fig. 14f demonstrated a significant electron transfer from Ge to Pt in Pt3Ge-(202), thereby contributing to the enhancement of HER kinetics. To gain a deeper comprehension of the electronic characteristics of the aforementioned facets, the partial density of states (PDOS) for both samples was computed. It was observed that the states within the (202) plane exhibited greater magnitudes compared to those within the (110) plane. Additionally, it was found that the samples possessing a higher electron count demonstrated superior HER properties (Fig. 14g). Further examination, employing DFT, revealed that the (202) facets exhibited a greater propensity for H2 desorption in comparison to the (110) facets on Pt (Fig. 14h).
The efficacy of phase adjustment as a technique for optimizing the energy barrier in reactions and enhancing catalytic efficiency has been well-established [185,186]. Moreover, phase engineering exhibits the potential to concurrently optimize desired reactions and suppress competitive reactions [187]. Lai et al. [188] achieved the synthesis of PtFe nanoalloys in three distinct crystal phases, namely face-centered cubic (PtFe-fcc), simple cubic (PtFe-scc), and a mixed phase of fcc and scc (PtFe-mix), through the process of hydrogen-assisted calcination. The uniform distribution of Fe and Pt elements on the nanofoam was confirmed through the utilization of HAADF-STEM and EDS analysis (Fig. 15a). In Fig. 15b, the observation of two lattice spacings (1.93 and 2.74 Å) can be attributed to the (200) planes of fcc-phase PtFe and the (110) planes of scc-phase PtFe, respectively, indicating the distinctive mixing phases of PtFe-mix. The PtFe-mix catalyst exhibited exceptional catalytic efficiency in the HER when tested in a 1.0 mol/L KOH solution, achieving a potential of 28 mV at 10 mA/cm2, surpassing the performance of the commercially available Pt/C catalyst (34 mV, Fig. 15c). This investigation presents a valuable methodology for the advancement of PtFe-based catalysts, resulting in improved intrinsic activity.
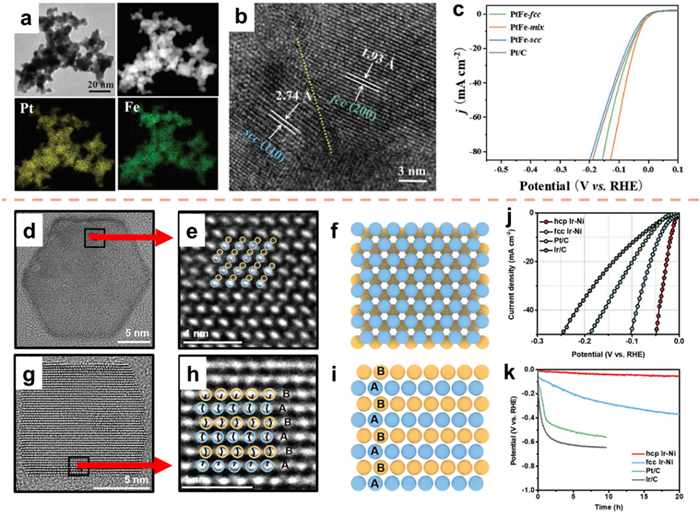
Catalytic reactions demonstrate a significant reliance on the surface, rendering them processes that are sensitive to surface characteristics. The improvement of catalytic performance, which is contingent on the phase, can be accomplished by incorporating homogeneous metastable metal catalysts into the design procedure [189,190]. Shao's group [191] conducted experiments to synthesize hexagonal close-packed (hcp) Ir-Ni and confirmed that the atomic packing arrangement along the [100] direction followed an ABABAB pattern, aligning with the unique hcp crystal phase (Figs. 15d–i). Furthermore, the electrocatalytic performance of hcp Ir-Ni nanoplates was depicted in Fig. 15j, revealing that hcp Ir-Ni required a mere 17 mV at 10 mA/cm2. Subsequent to a 10 h chronopotentiometry test, it was observed that the activity of hcp Ir-Ni remained relatively stable (Fig. 15k). This suggests that hcp Ir-Ni exhibits favorable long-term durability in the alkaline environment.
In addition to composition, morphology, structure, crystal plane, size and dimension, phase has become an important structural parameter of materials, which determines the properties and functions of nanomaterials. Research groups represented by Zhang's team have made a series of advances in the field of phase engineering nanomaterials (PEN), especially phase-based heterostructures, synthesis of unconventional phases, and phase transitions of nanomaterials [192]. With the development of advanced characterization technology and artificial intelligence, there will be greater breakthroughs in the understanding, screening and other aspects of PEN.
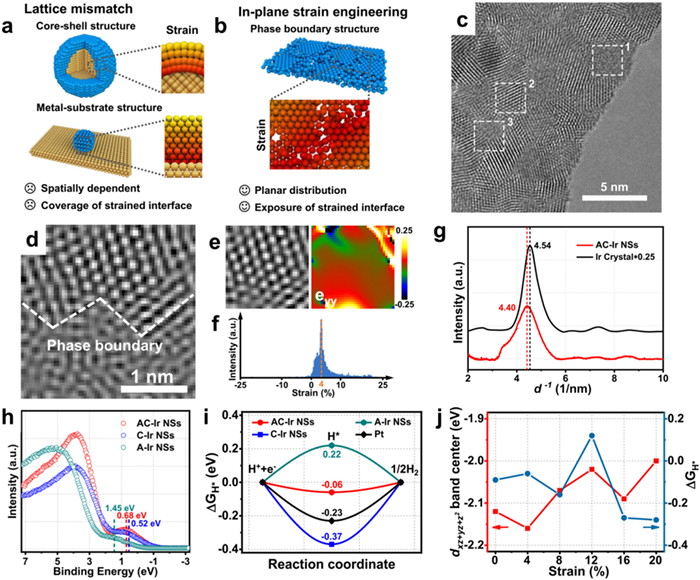
The electronic configuration of the catalyst can be modified by surface strain, resulting in a shift in the d-band center. This shift subsequently affects the adsorption energy and activation energy barrier of the reaction intermediates involved in the catalytic process [193,194]. Surface strain often arises from lattice mismatch, particularly in core-shell nanostructures or metal-substrate interfaces. Since a strained interface is typically confined within core-shell nanostructures or metal-substrate structures, the resulting active interface remains protected from catalytic reactions (Fig. 16a). The achievement of full exposure of the active region of catalysis in ultrathin nanosheets can be accomplished through the utilization of in-plane strain engineering (Fig. 16b). In accordance with this, Wang and co-workers [195] effectively synthesized ultrathin Ir nanosheets composed of amorphous and crystalline phases (AC-Ir NSs). To further investigate the atomic structure of the Ir nanosheets, HRTEM was utilized, which revealed the presence of both crystalline and amorphous structures (Fig. 16c). As shown in Fig. 16d, the amorphous-crystalline phase boundary can be observed. The strain values (eyy) decrease gradually with the distance away from the phase boundary, and the tensile strain in the crystal domain is mainly concentrated around 4% (Figs. 16e–g). As can be seen from Fig. 16h, the maximum value of metal d-band in AC-Ir NSs shows a positive displacement relative to crystalline Ir NSs (C-Ir NSs), and a negative displacement relative to amorphous Ir NSs (A-Ir NSs), indicating that the plane strain in AC-Ir NSs can modate the d-band state at the Ir site. Figs. 16i and j show the optimum value of ΔGH* at a tensile strain of about 4%, indicating that the tensile strain plays an important role in enhancing the intrinsic HER activity.
The Sabatier principle limits the catalytic activity of single element catalyst. Alloying is a highly effective strategy for modifying the catalytic properties, as well as the electronic and geometric states, of active metals by combining them with various elements [196-199]. The geometric effect refers to changes in the catalytic or adsorption behavior resulting from modifications in the geometric environment of the active metal atoms following alloying [200,201]. The electronic effect, conversely, pertains to alterations in the catalytic or adsorption behavior that arise from modifications in the electronic state of the active metal during the alloy structure formation process [202,203].
For example, according to the Brewer-Engel valence bond theory, alloys that are formed from two or more transition metals (one with empty or half-filled d orbitals and the other with paired but not easily bonded d electrons) exhibit good electrocatalytic synergies [204]. Fig. 17a illustrates the synthesis of the alloy catalyst through a one-step hydrothermal process utilizing an ethylene glycol as the reducing agent. The FeIr/NF catalyst demonstrates exceptional durability when subjected to alkaline conditions at elevated current densities, necessitating a mere 6.2 mV of overpotential at 10 mA/cm2 (Figs. 17b and c), which is significantly better than pure noble metal catalysts (Pt/C, Ir/NF) [205].
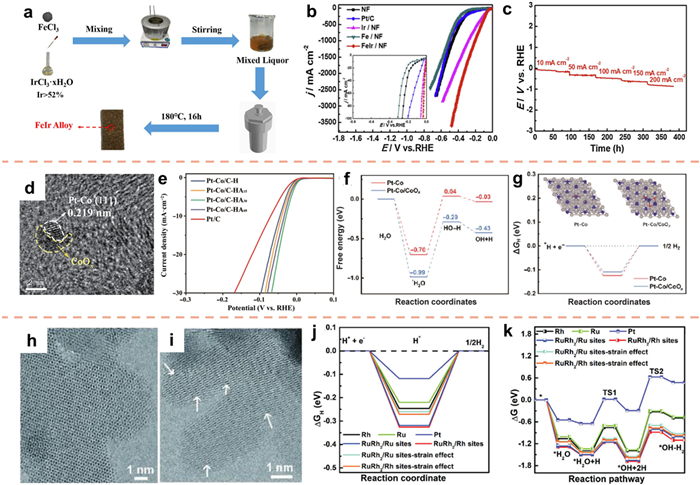
The dissociation of water is acknowledged as a limiting factor in the rate of the alkaline HER, resulting in a notable decrease in the catalytic efficacy of Pt-based catalysts [206-208]. The integration of noble metals with other metals offers a promising approach to achieve improved catalytic activity and efficiency. Extensive investigation has been carried out on alloy catalysts incorporating transition metals (e.g., iron, cobalt, nickel) due to their cost-effectiveness and enduring stability [209,210]. Moreover, the augmentation of water dissociation sites on transition metals amplifies the capacity to rupture H-OH bonds, aligning with the notable hydrogen-adsorption efficacy demonstrated by Pt-group metals [211]. As a result, Cheng and co-authors [212] devised a method to establish an interface between Pt-Co alloy and amorphous cobalt oxide (CoOx) via an in-situ oxidation treatment (Fig. 17d). The amorphous CoOx located at the Pt-Co/CoOx interface functions as active sites for water dissociation, thereby facilitating the Volmer step, and also enables efficient electron transfer due to the presence of Pt-Co. Experimental results indicate that at 10 mA/cm2, the overpotential was measured to be only 28 mV (Fig. 17e). Further analysis using DFT revealed the presence of electron aggregation on cobalt atoms at the Pt-Co/CoOx interface, as well as the formation of a novel hydrolyzed ionization active site. The energy barrier for H2O adsorption, H2O dissociation, and H2 adsorption/desorption at the Pt-Co/CoOx interface was observed to decrease in comparison to Pt-Co, indicating an improvement in the reaction kinetics of the fundamental HER (Figs. 17f and g).
Numerous studies have demonstrated that altering the symmetry of bimetallic catalysts through interface engineering can induce changes in the adsorption state of reaction intermediates [213,214]. Building upon this knowledge, Mu et al. [215] employed a one-pot method to synthesize a RuRh2 bimetallene nanoring with a significant presence of structural defects. The resulting strain effect, alloying effect, and quantum size effect, arising from the nanoring's thinning, can effectively modulate the electronic structure of the catalyst, thereby optimizing the ΔGH and water dissociation (Figs. 17h–k). Consequently, the RuRh2 bimetallene exhibits remarkable alkaline HER activity.
In the case of alloy catalysts featuring a core-shell structure, the interface between the core and shell elements often undergoes the formation of tensile and/or compressive strain as a result of lattice disparities. This strain induces alterations in the surface electronic structure, thereby optimizing the adsorption energy of reaction intermediates [216,217]. Consequently, it is evident that alloying and strain engineering can be simultaneously accomplished within the catalyst system, leading to synergistic advantages and an enhancement in its intrinsic activity.
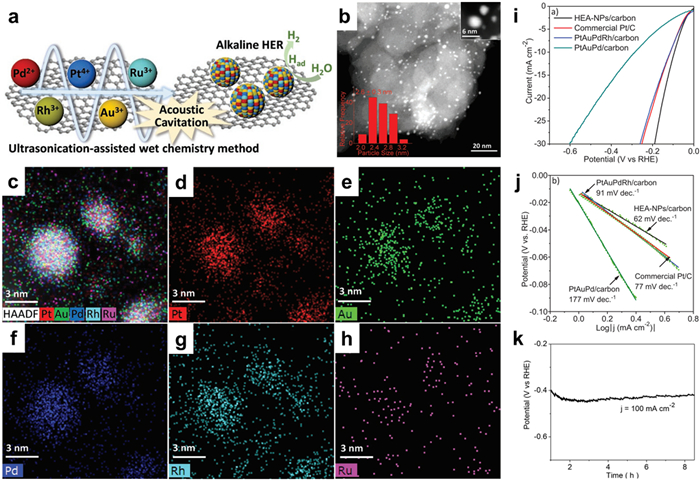
Recently developed noble metal-based high-entropy alloy NPs (NM-HEA NPs) have exhibited excellent catalytic properties, thereby attracting increasing attention from researchers [218,219]. Alloys composed of five or more metallic elements with similar molar quantities may exhibit unique characteristics such as the cocktail effect, high-entropy effect, and lattice distortion effect. The aforementioned characteristics contribute to the improved effectiveness of the catalyst, while simultaneously diminishing the need for noble metal composition [220]. In contrast to binary and ternary alloys, HEAs possess the ability to substantially modify the isoelectronic structure of the d-band center, consequently impacting the adsorption and activation of reactants and intermediates. Dai et al. [221] utilized an ultrasonically-assisted wet chemical technique to synthesize a diverse range of multi-component alloy NPs. These NPs included PtAuPdRhRu HEA NPs, quaternary PtAuPdRh alloy NPs, and ternary PtAuPd alloy NPs (Fig. 18a). Furthermore, the TEM images presented in Fig. 18b illustrate the dispersion of the high-entropy alloy NPs on a carbon substrate (XC-72) following calcination, exhibiting an average particle size of 2.6 ± 0.3 nm. The EDS element map illustrates the uniform dispersion of Pt, Au, Pd, Rh, and Ru throughout the nanospheres, thereby validating the successful establishment of the alloy structure (Figs. 18c–h). As a result of the notable synergistic effect, the electrocatalytic activity of the HER was observed to be considerably greater when using HEA NPs supported on the carbon catalyst, in comparison to the quaternary alloy, ternary alloy, and Pt/C. In the case of HEA-NPs/carbon-700 ℃, a current density of 30 mA/cm2 was attained at a potential of −0.19 V, accompanied by a Tafel slope value of 62 mV/dec. Additionally, the durability of this catalyst was assessed under 100 mA/cm2, and no substantial deterioration was detected during an 8-h timeframe (Figs. 18i–k).
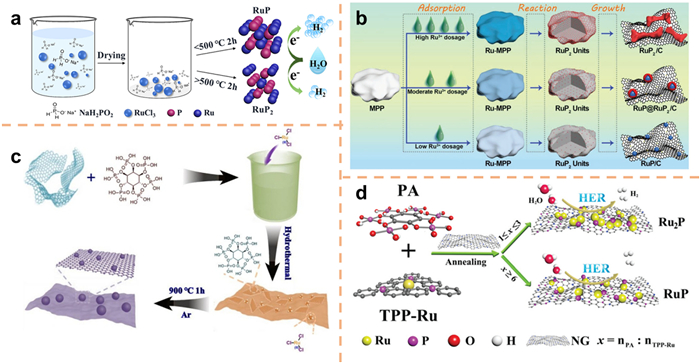
The combination of noble metals and nonmetallic anions can reduce the amount of noble metals on the one hand, and expand the types of catalysts on the other hand to develop high-efficiency electrocatalysts [222]. For example, noble-metal phosphide materials, comprising of Pt-group elements (e.g., Pt, Pd, Rh, Ru, and Ir) and phosphorus (P), constitute a novel class of electrocatalysts for the HER with a broad range of applications [223-227]. The current study elucidates the high catalytic activity of noble metal phosphides by primarily considering two factors. Firstly, the presence of phosphorus effectively and expeditiously modulates the surface electronic structure and hydrogen adsorption free energy of noble metals, thereby significantly enhancing the catalytic activity of the HER [228]. In contrast, the incorporation of P atoms into the noble metal structure facilitates the transfer of electrons from the metal site to the P site, resulting in the electronegativity of the latter. During the electrochemical HER, the negatively charged P species effectively sequesters the positively charged protons present in the solution [229]. Among these materials, ruthenium phosphate (RuPx) stands out due to its exceptional catalytic activity and relatively lower cost compared to other noble metals. RuPx exists in four distinct phases, namely Ru2P, RuP, RuP2, and RuP4 [230], with the first three phases primarily employed as electrocatalysts. The diverse physical, chemical, and electronic properties arise from the variations in structure and composition [231,232]. In their study, Fan et al. [233] utilized RuCl3 as the Ru precursor and NaH2PO2·H2O as the P source, ultimately obtaining the desired catalyst through hydrogen annealing reduction. The annealing temperature manipulation allows for control over the proportion of Ru in RuPx. The manipulation of the annealing temperature allows for control over the proportion of Ru in RuPx. It has been observed that annealing temperatures below 500 ℃ tend to lead to the formation of RuP, whereas temperatures above 500 ℃ promote the formation of RuP2 (Fig. 19a). In a separate study, Huang et al. [234] utilized melamine polyphosphate as the source of phosphorus and employed the pyrolysis method to obtain three distinct variations of RuPx with varying morphologies by adjusting the dosage of the Ru precursor. At higher concentrations of Ru3+, the initial phase involves the formation of RuP2, which triggers the aggregation and enlargement of RuP2, ultimately resulting in the formation of a significant coral-shaped structure. In contrast, at lower concentrations of Ru3+, the initial generation of RuP2 gradually converts into RuP due to the influence of nanosize effects, resulting in a composite material primarily composed of the RuP phase. At intermediate concentrations of Ru3+, the transition and growth of RuP2 units exhibit a favorable equilibrium, resulting in the formation of a core-shell structure comprising RuP@RuP2 (Fig. 19b). Hu et al. [235] employed a two-step hydrothermal pyrolytic strategy to synthesize three types of Ru phosphides (Ru2P, RuP, and RuP2) NPs on graphene nanosheets. By controlling the quantity of phytic acid utilized as a phosphorus source (Fig. 19c), they achieved comparable topography, size, load density, and electrochemical surface area. Similarly, Qin et al. [236] utilized the pyrolysis method and varied the amount of pyritic acid to obtain two distinct metal compounds, Ru2P and RuP, using different Ru precursors and P sources (Fig. 19d).
Ru is the most cost-effective element within the Pt-group, with a price that amounts to only 4% of Pt. Ruthenium phosphide material exhibits exceptional catalytic properties for the HER and stands as the most extensively investigated noble metal phosphide electrocatalyst. Consequently, it holds significant potential for large-scale applications in water electrolysis. Currently, the prevalent approach for synthesizing metal phosphide involves a gas-solid reaction, wherein an excess of phosphoric acid or phosphate serves as the P source. This source undergoes high-temperature decomposition to generate a P-containing gas, which subsequently reacts with metal precursors to yield the desired phosphide compound [223,237]. However, the conventional source of P and the high temperature phosphating process are subject to various limitations, including high risk, time and energy consumption, and potential harm to the environment. Consequently, the advancement of novel P sources and corresponding preparation methods assumes a crucial role in facilitating the widespread implementation of phosphide in the domain of HER [238,239]. In the case of metal phosphide catalysts, the stoichiometric ratio of metal to phosphorus (M/P) assumes particular significance. The presence of negatively charged P ions enhances the likelihood of capturing positively charged protons during the HER, thereby promoting the acceleration of the Volmer step [240,241]. The metallic phase, characterized by M-M bonds, possesses a higher concentration of free electrons and displays exceptional electrical conductivity [242,243]. Consequently, in order to obtain metal phosphide with desirable properties, it is imperative to carefully regulate the M/P ratio during the preparation process and, if necessary, employ suitable modification technique.
Noble-metal electrocatalysts present significant advantages in the water splitting process; nevertheless, their high costs and complex catalytic environments impede their widespread adoption. Additionally, conventional methods for fabricating powder catalysts require the use of adhesives to cover electrode surfaces, resulting in insufficient mechanical resilience, particularly when subjected to high current densities [244]. The existence of an interface gap between the electrode and the active material has a detrimental effect on electron transmission, and the adhesive can also obstruct specific catalytic active sites [245,246]. Hence, it is of utmost importance to advance the development of electrocatalysts with reduced noble-metal content, heightened activity, and enhanced stability. The technique of in-situ deposition of the desired material onto conductive substrates, such as NF and CC, can significantly enhance the interface between the catalysts and the electrode substrate, impede the aggregation of catalysts with diminutive particle sizes, and facilitate the diffusion of electrolyte and the liberation of gaseous products that follow [247,248].
For supported noble metal-based catalysts, it is important to find a suitable supports material, which can increase the dispersion of noble metal materials, and optimize the catalytic activity center of the supported noble metal. Peng et al. [249] introduced a novel spontaneous redox approach to immobilize ultrafine noble-metal NPs onto Ni foam-supported Ni-MOF nanosheets (referred to as M@Ni-MOF, where M denotes Ru, Ir, Pd; refer to Fig. 20a). The TEM analysis demonstrates that the Ru NPs exhibit a uniform distribution on the surface of Ni-MOF, with a size range of 2–4 nm. This can be attributed to the confinement effect of the nanosheet structure and the immobilization of O atoms (Fig. 20b). Figs. 20c and d show that Ni-MOF forms Ni-O-Ru bonds with Ru NPs, which helps to accelerate electron transfer at the interface. The electrochemical results confirm that the Ru@Ni-MOF electrocatalyst, when in its optimal state, exhibits exceptional HER activity. In alkaline electrolytes, the catalyst demonstrated a HER activity of 10 mA/cm2, accompanied by an overpotential of 22 mV (Fig. 20e). The DFT calculation results show that the hydrogen adsorption energy can be reduced by interfacial bond induced charge transfer (Fig. 20f). Furthermore, this approach proves to be suitable for the synthesis of other noble metals with reduced dimensions, such as Ir@Ni-MOF and Pd@Ni-MOF.
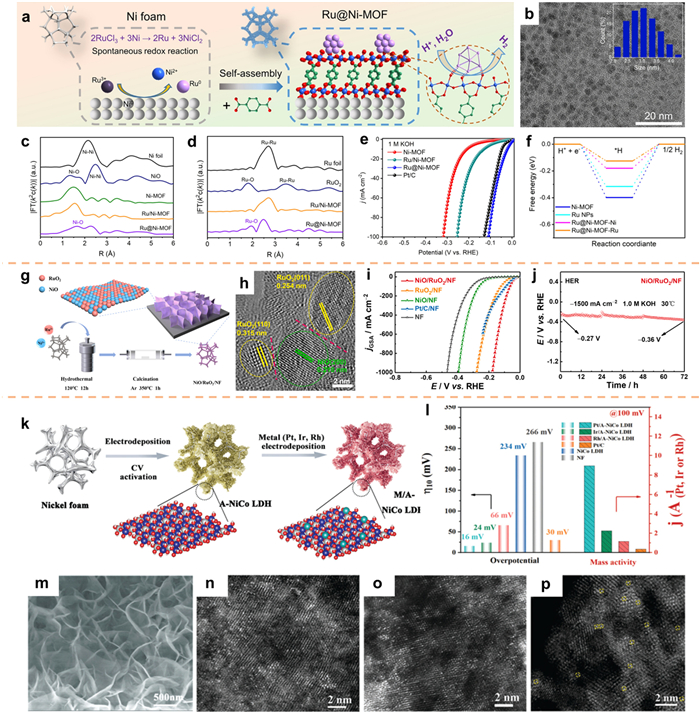
Among Ru-based catalysts, RuO2 has been widely studied as an OER catalyst because of its ideal binding energy with oxygen intermediates. Because the reaction mechanisms of OER and HER are different, it is difficult for a single active site to efficiently catalyze both reactions. By constructing the heterojunction, the advantages of the different components on the adsorption and desorption of the reaction intermediates can be synthesized. Hereby, Yin et al. [250] utilized a hydrothermal technique followed by calcination to fabricate a NiO/RuO2 nanosheet catalyst on Ni foam (NiO/RuO2/NF) (Fig. 20g). The HRTEM analysis exhibited the existence of lattice fringes corresponding to RuO2 and NiO. Moreover, the identification of an interface between distinct lattice fringes of the two materials substantiates the formation of heterojunctions (Fig. 20h). The attainment of significant HER activity in NiO/RuO2/NF, along with the enhancement of its stability under high current density, is achieved through the synergistic implementation of the heterojunction construction strategy and nanoarray integration (Figs. 20i and j).
In order to optimize the utilization efficiency of noble metals, it is crucial to focus on the correlation between material structure and catalytic activity. The rational design of a cost-effective electrocatalyst for the HER holds paramount importance in the context of sustainable development in alkaline water electrolysis. Deng et al. [251] employed a two-step electrochemical deposition technique to immobilize three noble metals onto a 2D NiCo layered double hydroxide (NiCo LDH) support (Fig. 20k), and test the electrocatalytic performance (Fig. 20l). The implementation of this methodology led to the development of a singular-atom HER electrocatalyst denoted as M/A-NiCo LDH, where M denotes Pt, Ir, and Rh (Figs. 20m–p). Among these variants, the Pt/A-NiCo LDH electrocatalyst demonstrated the most significant alkaline HER catalytic capability, exhibiting a minimal overpotential of 16 mV at a current density of 10 mA/cm2, and a mass activity approximately 24.8 times higher than that of commercially available Pt/C (Fig. 20l).
After conducting a comprehensive analysis of numerous self-supporting catalysts, it becomes evident that the catalyst synthesized through the hydrothermal method (Section 3.4) exhibits a greater propensity for acquiring a nanoarray structure. Similarly, the process of electrodeposition proves to be more efficient in obtaining the precursor coating on the substrate (Section 3.5) [252]. The utilization of a 3D self-supported nanoarray electrode presents a significant opportunity to enhance the material's specific surface area, promote the exposure of active sites, and facilitate efficient diffusion of electrolyte and release of gas. In order to harness the potential of the nanoarray, the researchers employed a two-step electrochemical deposition technique, wherein a nanoarray electrode served as a substrate for the preparation of a noble metal catalyst supported on its surface [101,251].
The catalytic performance of the aforementioned examples in Section 4 is presented in Table 2. Collectively, these examples showcase the viability and efficacy of employing these modification strategies to enhance the performance and minimize the expenses associated with noble metal catalysts.
 DownLoad:
CSV
DownLoad:
CSV

|
This paper offers a thorough analysis of the synthesis techniques and modification approaches utilized in the fabrication of noble-metal catalysts. Despite the considerable body of research documenting the preparation and application of noble metal-based catalysts, their widespread production remains elusive. Additionally, this paper explores the challenges and potential opportunities linked to the advancement and industrial integration of noble metal-based electrocatalysts for the HER (Fig. 21). The primary emphasis in the investigation of noble metal alkaline HER catalysts revolves around three key areas: (1) The design and synthesis of catalysts, with a particular focus on achieving industrial-scale production of nanostructured catalysts synthesized in laboratory settings, thereby expanding their production from milligram to potentially kilogram quantities [253]; (2) The structural modulation of the active center; (3) The analysis of the relationship between catalyst structure and activity [254].

The adsorption and activation of reactant molecules, as well as the desorption of products, are dependent on the electronic structure and geometric morphology of the catalyst surface/interface. These factors subsequently influence the catalytic activity and selectivity. Therefore, the precise and controllable synthesis of catalysts is crucial in comprehending the "structure-activity" relationship and the microscopic mechanism of catalytic reactions. However, the application of conventional wet chemical reduction techniques for managing the reduction process is impeded by the substantial variability in inherent reduction potentials among different metal salts. This constraint leads to the complex and heterogeneous composition and structure of the resultant catalyst, as well as a restricted scope for controlling catalyst components. Furthermore, the incorporation of surfactants during wet chemical synthesis tends to diminish catalyst activity. In recent scholarly investigations, scholars have utilized the ALD technique referred to as "selective deposition" to effectively accomplish the meticulous and regulated assembly of transition-metal monometallic to bimetallic NPs, employing a “bottom-up” methodology [255-257].
The optimization of noble metal catalyst is mainly to realize the tradeoff of activity, stability and cost. Although researchers have explored a variety of excellent catalysts, however, the design and selection of catalysts suitable for large-scale industrial production is still very challenging. For example, in the synthesis of catalysts, especially the nucleation process, the synthesis conditions and ligands all have a great influence on the final catalyst. At present, there is no universal method or formula for the synthesis and modification of catalysts. According to the existing relatively mature experimental scheme, the use of artificial intelligence (AI) to simulate the synthesis path may greatly improve the efficiency and accuracy of catalyst screening, and can in turn guide the synthesis of specific catalysts [258-260].
The application of water electrolysis for the generation of environmentally friendly hydrogen has become increasingly popular as a solution to address the worldwide energy challenge. However, the large-scale deployment of water splitting requires achieving high levels of current density surpassing 200 mA/cm2. As a result, it is crucial to design and obtain electrocatalysts that demonstrate exceptional performance under these demanding conditions in order to effectively utilize electrolytic water in hydrogen production technology.
Currently, the field of industrial electrolysis water technology encounters two main limitations. Firstly, commercially utilized catalysts primarily rely on noble metals like Pt, Ir, Ru, which are distinguished by their exorbitant expense and restricted accessibility. Moreover, the energy consumption and catalyst load increase in direct proportion to the rise in current density, leading to higher costs associated with hydrogen production. Moreover, the catalyst exhibits instability when subjected to high current densities, particularly at the ampere level, resulting in a stronger adhesion between the catalyst and the bubble compared to the catalyst and the substrate. As a consequence, the catalyst becomes detached from the substrate. In order to address this issue, the utilization of catalysts on conductive substrates, such as Ni foam, via in-situ growth, has been discovered to alleviate the detachment of active substances and improve stability. Consequently, this approach leads to enhanced performance under high current densities.
Noble-metal SACs have attracted considerable interest in the field of catalysis research due to their remarkable atom utilization and unique performance characteristics. However, despite the potential of electrochemical techniques, such as ALD, CVD, and other synthesis strategies, to manipulate and potentially arrange atoms in order to achieve noble-metal single-atomic configurations, they have not adequately addressed the challenges related to the high loading capacity and superior stability of SACs. Moreover, the substantial expenses associated with production, complex procedures, and restricted outputs (typically below a few hundred milligrams) present practical limitations for the industrial-scale production of SACs. Consequently, it is crucial for research efforts to prioritize the incorporation of supplementary defect sites on carriers as a viable approach to securely immobilize individual atoms. Additionally, the development of spatial confinement structures should be pursued to hinder the aggregation of these solitary atoms, while simultaneously augmenting the thermal and chemical stability of the carrier. Ultimately, the implementation of mass production techniques is imperative to enable the efficient execution of extensive manufacturing processes.
Model catalysts have a unique structural composition and employ more advanced characterization techniques in comparison to nanocatalysts. The examination of model catalysts, which showcase well-defined structures, enables the discernment of the structure-activity relationship of catalysts at the atomic or molecular level. This relationship serves as a fundamental principle for the development of practical catalysts. For example, the ability to exert precise control allows for the targeted exposure of noble-metal NPs ' single-crystal surfaces, thus circumventing the limitations imposed by the heterogeneous nature of catalyst materials commonly used in commercial applications. The application of this catalyst as a representative model facilitates the examination of the selectivity of crystal surfaces in relation to catalytic products. It is important to acknowledge that certain conclusions drawn from model catalysts cannot be directly applied to real-world systems due to the disparity in size between model catalysts and nanocatalysts. Therefore, the crucial significance lies in the discovery of a model catalyst that bears a striking resemblance to real-world systems, as it enables a comprehensive understanding of the fundamental principles that govern the intricate interplay between catalytic activity and structure.
On the one hand, SACs exhibit a high specific mass catalytic activity, enabling the efficient utilization of noble metal atoms and minimizing material costs. However, despite the relatively straightforward controlled synthesis of SACs, their industrial-scale preparation remains a formidable challenge, thereby limiting their practical application. In contrast, the active center of SACs exhibits a consistent and typically low coordination state, resulting in limited interaction with neighboring atoms or molecules. Notably, a higher degree of unsaturation in the coordination of the active center atom enhances the likelihood of contact with the substrate molecule. Consequently, SACs possess inherent benefits in enhancing catalyst activity and product selectivity, rendering them as exemplary model catalysts with broader applicability.
The catalytic efficacy of noble metal-based catalysts in alkaline electrolytes is markedly diminished, by a magnitude of two to three orders, in comparison to their performance in acidic electrolytes. This disparity can be ascribed to the impact of diverse factors, including the free energy of hydrogen adsorption, dissociation of water adsorption, and desorption capacity of hydroxyl groups on the adsorbent, on the overall kinetics of the catalysts. The reaction mechanism of electrolytic water encompasses a multitude of intricate intermediates that exhibit limited abundance and brief lifespans.
Numerous research findings indicate that the electrode-electrolyte interface is intricately linked to nearly all issues encountered in practical electrochemical systems [261-263]. This interface serves as the site for charge transfer, species and energy conversion and storage, ion transport, and various other processes. The physical and chemical attributes of this interface assume a pivotal role in the entirety of the electrochemical process and the overall performance of the electrochemical device. Moreover, the concurrent occurrence of these factors complicates the examination and understanding of the reaction mechanism in alkaline media, as it entails the simultaneous reconstruction of the catalyst surface. The electrochemical interface encompasses intricate species and structures, exhibiting dynamic alterations and non-uniform distribution of electrochemical activity [264-266]. It is imperative to analyze the nanoscale perspective, molecular level, and in-situ dynamics in order to comprehensively understand the electrochemical process of the interface structure. Subsequently, it becomes possible to elucidate the dynamic alterations occurring in the interface structure, electrochemical activity state, and intermediate products throughout the electrochemical process.
Traditional ex-situ techniques for characterization are limited to capturing the stable initial and final states, thus hindering the observation of dynamic changes occurring during the catalytic process. Consequently, it is imperative to conduct in-situ and operando characterization in order to obtain a comprehensive understanding of the genuine electrochemical process. Additionally, the utilization of vacuum interconnection technology in testing and characterization systems enables the achievement of (quasi) in-situ testing, thereby preserving the original surface structure of the model catalyst and effectively preventing oxidation and atmospheric contamination [267,268].
The global constraint on freshwater resources has been significantly worsened by the adoption of water electrolysis for hydrogen production. However, the ample availability of seawater presents a potential remedy for this problem. It is widely acknowledged that sustainable energy sources such as solar and wind energy are predominantly found in arid inland, desert, or coastal regions that suffer from limited access to fresh water resources. Consequently, the large-scale production of green hydrogen, a key component of sustainable energy, remains unattainable. Currently, the utilization of desalination technology to enable indirect seawater electrolysis faces challenges in meeting the purity standards demanded by electrolyzers, particularly those employing PEM technology. Moreover, this approach significantly escalates the overall cost of the process. Hence, the utilization of sustainable energy to facilitate direct seawater or saline-alkali water splitting is deemed more advantageous for the advancement of green hydrogen energy. Furthermore, the utilization of hydrogen derived from seawater as a primary resource can engender the production of potable water via fuel cell apparatus, thereby offering a viable solution to address the scarcity of local freshwater resources [269].
In terms of desalination needs, electrolytic hydrogen production from seawater can be categorized into two types: Direct electrolytic hydrogen production from seawater and electrolytic hydrogen production following seawater desalination. The distillation and membrane treatment methods, which are frequently utilized for desalination, are accompanied by significant expenses. While the process flow of the direct hydrogen production route is uncomplicated, the viability of seawater electrolysis in an industrial setting is limited due to various constraints. Firstly, the direct electrolytic hydrogen production route is hindered by the oxidation of chloride ions present in seawater, resulting in the generation of chlorine gas and hypochlorite. This chemical reaction poses a considerable risk of corrosion within the electrolytic cell. Secondly, the accumulation of insoluble substances on the surfaces of the ion-exchange membrane and catalyst leads to the deactivation of the catalyst. Lastly, the electrolytic efficiency is significantly constrained by the limited concentrations of hydrogen ion (H+) and hydroxide ion (OH−) in seawater [270-272].
The aforementioned analysis reveals that the primary bottleneck issue in seawater electrolysis lies in the destabilizing effect of the complex ionic environment on the catalyst. Given the current challenges associated with seawater electrolysis, it is recommended to prioritize research efforts towards the advancement of catalysts and the design of desalination systems for seawater electrolysis cells. In a recent study, Qiao et al. [273] developed a nickel-iron layered double hydroxide (NiFe-LDH) and demonstrated that the presence of Cl− on the Fe site effectively hindered Fe leaching, resulting in an increased number of Ni sites. This phenomenon ultimately enhanced the long-term stability and activity of the catalyst.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We gratefully acknowledge the financial support by the National Natural Science Foundation of China (No. 52102241), Doctor of Suzhou University Scientific Research Foundation (Nos. 2022BSK019, 2020BS015), the Primary Research and Development Program of Anhui Province (No. 201904a05020087), the Natural Science Research Project in Universities of Anhui Province in China (Nos. 2022AH051386, KJ2021A1114), and the Foundation (No. GZKF202211) of State Key Laboratory of Biobased Material and Green Papermaking Qilu University of Technology.
M.I. Jamesh, X.M. Sun, J. Power Sources 400 (2018) 31–68. doi: 10.1016/j.jpowsour.2018.07.125
S. Chu, A. Majumdar, Nature 488 (2012) 294–303. doi: 10.1038/nature11475
T.R. Cook, D.K. Dogutan, S.Y. Reece, et al., Chem. Rev. 110 (2010) 6474–6502. doi: 10.1021/cr100246c
X.Q. Fan, Y. Zhou, X.K. Jin, et al., Carbon Energy 3 (2021) 449–472. doi: 10.1002/cey2.113
X. Zhao, L.J. Du, B. You, et al., Catal. Sci. Technol. 10 (2020) 2711–2720. doi: 10.1039/D0CY00453G
S. Wang, A.L. Lu, C.J. Zhong, Nano Converg. 8 (2021) 4. doi: 10.1186/s40580-021-00254-x
Y. Li, X.F. Wei, L.S. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 19550–19571. doi: 10.1002/anie.202009854
R.B. Zhang, Z.A. Tu, S. Meng, et al., Rare Met. 42 (2023) 176–188. doi: 10.1007/s12598-022-02136-5
Q. Qian, W. Wang, G. Wang, et al., Small 18 (2022) e2200242. doi: 10.1002/smll.202200242
S. Sultan, J.N. Tiwari, A.N. Singh, et al., Adv. Energy Mater. 9 (2019) 1900624. doi: 10.1002/aenm.201900624
Z.X. Sun, L.J. Sun, S.W. Koh, et al., Carbon Energy 4 (2022) 1262–1273. doi: 10.1002/cey2.213
M. Chatenet, B.G. Pollet, D.R. Dekel, et al., Chem. Soc. Rev. 51 (2022) 4583–4762. doi: 10.1039/D0CS01079K
H.W. Huang, H. Jung, S.F. Li, et al., Nano Energy 92 (2022) 106763. doi: 10.1016/j.nanoen.2021.106763
G.L. Gao, Z.X. Sun, X.L. Chen, et al., Appl. Catal. B: Environ. 343 (2024) 123584. doi: 10.1016/j.apcatb.2023.123584
S.S. Kumar, H. Lim, Sustain. Energy Fuels 7 (2023) 3560–3583. doi: 10.1039/D3SE00336A
R.T. Liu, Z.L. Xu, F.M. Li, et al., Chem. Soc. Rev. 52 (2023) 5652–5683. doi: 10.1039/D2CS00681B
L.A. Chong, G.P. Gao, J.G. Wen, et al., Science 380 (2023) 609–616. doi: 10.1126/science.ade1499
Z.C. Chen, L. Guo, L. Pan, et al., Adv. Energy Mater. 12 (2022) 2103670. doi: 10.1002/aenm.202103670
S. Biswas, G. Kaur, G. Paul, et al., Int. J. Hydrog. Energy 48 (2023) 12541–12570. doi: 10.1016/j.ijhydene.2022.11.307
H.Y. Li, K. Kamlungsua, J. Shin, et al., J. Clean. Prod. 424 (2023) 138747. doi: 10.1016/j.jclepro.2023.138747
H.G. Xu, X.Y. Zhang, Y.L. Ding, et al., Small Struct. 4 (2023) 2200404. doi: 10.1002/sstr.202200404
H.A. Sun, X.M. Xu, H. Kim, et al., Energy Environ. Mater. 6 (2023) e12441. doi: 10.1002/eem2.12441
B. You, Y.D. Zhang, Y. Jiao, et al., Angew. Chem. Int. Ed. 58 (2019) 11796–11800. doi: 10.1002/anie.201906683
J. Wu, Z. Wang, T. Guan, et al., Carbon Energy 5 (2023) e268. doi: 10.1002/cey2.268
Z.X. Sun, X.H. Wang, H. Zhao, et al., Carbon Energy 2 (2020) 122–130. doi: 10.1002/cey2.16
C.H. An, W. Kang, Q.B. Deng, et al., Rare Met. 41 (2022) 378–384. doi: 10.1007/s12598-021-01791-4
W. Song, M.X. Li, C. Wang, et al., Carbon Energy 3 (2021) 101–128. doi: 10.1002/cey2.85
C. Li, D.H. Zhao, H.L. Long, et al., Rare Met. 40 (2021) 2657–2689. doi: 10.1007/s12598-020-01694-w
M.C. Liu, Z.P. Lei, Q.P. Ke, et al., Carbon Energy 4 (2022) 480–490. doi: 10.1002/cey2.162
J. Yang, W.H. Li, S. Tan, et al., Angew. Chem. Int. Ed. 60 (2021) 19085. doi: 10.1002/anie.202107123
J.S. Qin, D.Y. Du, W. Guan, et al., J. Am. Chem. Soc. 137 (2015) 7169–7177. doi: 10.1021/jacs.5b02688
G.L. Gao, G. Zhu, X.L. Chen, et al., ACS Nano 17 (2023) 20804–20824. doi: 10.1021/acsnano.3c05810
Z.Y. Yu, Y. Duan, X.Y. Feng, et al., Adv. Mater. 33 (2021) 35.
P.S. Liao, J.W. Kang, Y.C. Zhong, et al., Sci. China Chem. 66 (2023) 1924–1939. doi: 10.1007/s11426-022-1545-0
L. Wan, Z. Xu, Q. Xu, et al., Energy Environ. Sci. 16 (2023) 1384–1430. doi: 10.1039/D3EE00142C
Y.H. Liu, Q.L. Wang, J.C. Zhang, et al., Adv. Energy Mater. 12 (2022) 2200928. doi: 10.1002/aenm.202200928
Z.S. Li, B.L. Li, M. Yu, et al., Int. J. Hydrog. Energy 47 (2022) 26956–26977. doi: 10.1016/j.ijhydene.2022.06.049
J.J. Lu, S. Yin, P.K. Shen, Electrochem. Energy Rev. 2 (2019) 105–127. doi: 10.1007/s41918-018-0025-9
Y.L. Zhu, H.A. Tahini, Z.W. Hu, et al., Nat. Commun. 10 (2019) 149. doi: 10.1038/s41467-018-08117-6
J. McAllister, N.A.G. Bandeira, J.C. McGlynn, et al., Nat. Commun. 10 (2019) 370. doi: 10.1038/s41467-018-08208-4
H. Chen, B. Zhang, X. Liang, et al., Chin. J. Catal. 43 (2022) 611–635. doi: 10.1016/S1872-2067(21)63899-8
Y.F. Shi, Z.H. Lyu, M. Zhao, et al., Chem. Rev. 121 (2021) 649–735. doi: 10.1021/acs.chemrev.0c00454
Y. Gong, Z. Xu, H. Pan, et al., J. Mater. Chem. A 6 (2018) 12506–12514. doi: 10.1039/C8TA03163K
W. Moschkowitsch, O. Lori, L. Elbaz, ACS Catal. 12 (2022) 1082–1089. doi: 10.1021/acscatal.1c04948
Z.S. Li, B.L. Li, Y.F. Hu, et al., Mater. Adv. 3 (2022) 779–809. doi: 10.1039/D1MA00858G
Y.M. He, L.R. Liu, C. Zhu, et al., Nat. Catal. 5 (2022) 212–221. doi: 10.1038/s41929-022-00753-y
J.D. Chen, M.K. Qin, S.X. Ma, et al., Appl. Catal. B: Environ. 299 (2021) 120640. doi: 10.1016/j.apcatb.2021.120640
R.L. Zhong, S. Sakaki, J. Am. Chem. Soc. 142 (2020) 16732–16747. doi: 10.1021/jacs.0c07239
Z. Wang, C.P. Wang, S.J. Mao, et al., Nat. Commun. 13 (2022) 3561. doi: 10.1038/s41467-022-31313-4
J.L. Huang, C.C. Du, J.H. Nie, et al., Electrochim. Acta 326 (2019) 134982. doi: 10.1016/j.electacta.2019.134982
C.H. Wang, F. Hu, H.C. Yang, et al., Nano Res. 10 (2017) 238–246. doi: 10.1007/s12274-016-1281-9
J.Y. Gao, C.X. Ren, X.C. Huo, et al., ACS ES&T Eng. 1 (2021) 562–570.
H. Eom, I.H. Hwang, D.Y. Lee, et al., Ind. Eng. Chem. Res. 59 (2020) 15489–15496. doi: 10.1021/acs.iecr.0c02059
J.M. Zhang, X.P. Xu, L. Yang, et al., Small Methods 3 (2019) 1900653. doi: 10.1002/smtd.201900653
S.F. Yang, X.B. Yang, X.Q. Cui, et al., SusMat 2 (2022) 689–698. doi: 10.1002/sus2.85
M. Haneda, A. Towata, Catal. Today 242 (2015) 351–356. doi: 10.1016/j.cattod.2014.05.044
Z.Q. Yuan, Z.Y. Chen, J.X. Mao, et al., Chin. J. Chem. Eng. 39 (2021) 135–143. doi: 10.1016/j.cjche.2020.09.029
H.H. Wei, K. Huang, D. Wang, et al., Nat. Commun. 8 (2017) 1490. doi: 10.1038/s41467-017-01521-4
J. Lemus, J. Bedia, L. Calvo, et al., Catal. Sci. Technol. 6 (2016) 5196–5206. doi: 10.1039/C6CY00403B
C.X. Zhang, C. Jiang, Q. Tang, et al., J. Energy Chem. 78 (2023) 438–446. doi: 10.1016/j.jechem.2022.12.023
B. Lim, Y.J. Xiong, Y.N. Xia, Angew. Chem. Int. Ed. 46 (2007) 9279–9282. doi: 10.1002/anie.200703755
M.S. Jin, H.Y. Liu, H. Zhang, et al., Nano Res. 4 (2011) 83–91. doi: 10.1007/s12274-010-0051-3
R. Long, D. Wu, Y.P. Li, et al., Nano Res. 8 (2015) 2115–2123. doi: 10.1007/s12274-015-0722-1
Y.F. Cheng, S.K. Lu, F. Liao, et al., Adv. Funct. Mater. 27 (2017) 1700359. doi: 10.1002/adfm.201700359
Y.M. Xing, C.H. Wang, D.S. Li, et al., Adv. Funct. Mater. 32 (2022) 2207694. doi: 10.1002/adfm.202207694
L. Cao, Z.H. Li, K.M. Su, et al., J. Energy Chem. 54 (2021) 595–603. doi: 10.1016/j.jechem.2020.06.053
J.J. Gao, Y.X. Hu, Y. Wang, et al., Small 17 (2021) 2104684. doi: 10.1002/smll.202104684
Y. Yang, Y.M. Qian, Z.P. Luo, et al., Nat. Commun. 13 (2022) 7225. doi: 10.1038/s41467-022-34976-1
J.N. Chen, H.J. Zhang, J. Yu, et al., Carbon Energy 4 (2022) 77–87. doi: 10.1002/cey2.156
H.X. Chen, H. Xu, Z.R. Song, et al., Rare Met. 42 (2023) 155–164. doi: 10.1007/s12598-022-02121-y
G.L. Gao, X.L. Chen, L. Han, et al., Coordin. Chem. Rev. 503 (2024) 215639. doi: 10.1016/j.ccr.2023.215639
P. Yi, L. Bingzhang, C. Shaowei, Adv. Mater. 30 (2018) 1801995. doi: 10.1002/adma.201801995
Y.Z. Wang, Y.M. Ding, C.H. Zhang, et al., Rare Met. 40 (2021) 2785–2792. doi: 10.1007/s12598-021-01765-6
J.W. Su, Y. Yang, G.L. Xia, et al., Nat. Commun. 8 (2017) 14969. doi: 10.1038/ncomms14969
Z.L. Jiang, S.J. Song, X.B. Zheng, et al., J. Am. Chem. Soc. 144 (2022) 19619–19626. doi: 10.1021/jacs.2c09613
D. Wu, X.W. Wang, X.Z. Fu, et al., Appl. Catal. B: Environ. 284 (2021) 119723. doi: 10.1016/j.apcatb.2020.119723
D.Q. Song, H.Z. Guo, K. Huang, et al., Mater. Today 54 (2022) 42–51. doi: 10.1016/j.mattod.2022.02.011
Y. Liu, Y.P. Yang, Z.K. Peng, et al., Nano Energy 65 (2019) 104023. doi: 10.1016/j.nanoen.2019.104023
Y. Liu, X. Li, Q.H. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 1718–1726. doi: 10.1002/anie.201913910
S.F. Liu, W. Xu, Y.M. Niu, et al., Nat. Commun. 10 (2019) 5790. doi: 10.1038/s41467-019-13755-5
X.H. Jiang, Y.N. Duan, Y. Tian, et al., Rare Met. 41 (2022) 406–414. doi: 10.1007/s12598-021-01810-4
P.P. Yang, X.L. Yuan, H.C. Hu, et al., Adv. Funct. Mater. 28 (2018) 1704774. doi: 10.1002/adfm.201704774
Z.H. Yu, H.Q. Yan, C.A. Wang, et al., Front. Chem. Sci. Eng. 17 (2023) 437–448. doi: 10.1007/s11705-022-2228-1
J.Y. Wang, J.R. Feng, Y.Y. Li, et al., Adv. Sci. 10 (2023) 2206952. doi: 10.1002/advs.202206952
Y.M. Wang, G.F. Qian, Q.L. Xu, et al., Appl. Catal. B: Environ. 286 (2021) 119881. doi: 10.1016/j.apcatb.2021.119881
M.Z. You, X. Du, X.H. Hou, et al., Appl. Catal. B: Environ. 317 (2022) 121729. doi: 10.1016/j.apcatb.2022.121729
L. Huang, X.P. Zhang, Q.Q. Wang, et al., J. Am. Chem. Soc. 140 (2018) 1142–1147. doi: 10.1021/jacs.7b12353
H. Zhao, Q.Y. Mao, L. Jian, et al., Chin. J. Catal. 43 (2022) 1774–1804. doi: 10.1016/S1872-2067(22)64105-6
Y.R. Li, P.J. Ren, D.S. Zhang, et al., ACS Catal. 11 (2021) 4338–4348. doi: 10.1021/acscatal.1c00102
T.F. Li, J.J. Liu, Y. Song, et al., ACS Catal. 8 (2018) 8450–8458. doi: 10.1021/acscatal.8b02288
J.P. Ji, Y.P. Zhang, L.B. Tang, et al., Nano Energy 63 (2019) 103849. doi: 10.1016/j.nanoen.2019.06.045
J.P. Ji, Z.H. Li, C.C. Hu, et al., ACS Appl. Mater. Interfaces 12 (2020) 40204–40212. doi: 10.1021/acsami.0c06931
H.H. Wei, H.B. Wu, K. Huang, et al., Chem. Sci. 10 (2019) 2830–2836. doi: 10.1039/C8SC04986F
M. Yang, J. Mei, Y.J. Ren, et al., J. Energy Chem. 81 (2023) 502–509. doi: 10.1016/j.jechem.2023.03.020
Z. Cai, J.D. Wang, Z.H. Lu, et al., Angew. Chem. Int. Ed. 61 (2022) e202116560. doi: 10.1002/anie.202116560
J. Mo, Y. Ko, Y.S. Yun, et al., Energy Environ. Sci. 15 (2022) 3815–3829. doi: 10.1039/D2EE01510Bamint.2021.07.196|
H. Jiang, Y.J. Sun, B. You, Acc. Chem. Res. 56 (2023) 1421–1432. doi: 10.1021/acs.accounts.3c00059
M.B. Kale, R.A. Borse, A.G.A. Mohamed, et al., Adv. Funct. Mater. 31 (2021) 2101313. doi: 10.1002/adfm.202101313
Y.L. Wang, L.Y. Zhao, Y. Zhao, et al., Adv. Mater. 30 (2018) 1805686. doi: 10.1002/adma.201805686
K. Ye, G.X. Wang, X.H. Bao, Chin. J. Struct. Chem. 39 (2020) 206–213.
L.H. Zhang, L.L. Han, H.X. Liu, et al., Angew. Chem. Int. Ed. 56 (2017) 13694–13698. doi: 10.1002/anie.201706921
Z.R. Zhang, C. Feng, C.X. Liu, et al., Nat. Commun. 11 (2020) 1215. doi: 10.1038/s41467-020-14917-6
P. Zhou, X.G. Hou, Y.G. Chao, et al., Chem. Sci. 10 (2019) 5898–5905. doi: 10.1039/C9SC00658C
X. Lu, S. Gao, H. Lin, et al., Natl. Sci. Rev. 9 (2022) nwac022. doi: 10.1093/nsr/nwac022
J. Geiger, A. Sabadell-Rendón, N. Daelman, et al., npj Comput. Mater. 8 (2022) 171. doi: 10.1038/s41524-022-00852-1
T.F. Yang, Y. Chen, Y. Liu, et al., Chin. Chem. Lett. 33 (2022) 2171–2177. doi: 10.1016/j.cclet.2021.09.014
M.M. Yan, Z.X. Wei, Z.C. Gong, et al., Nat. Commun. 14 (2023) 368. doi: 10.1038/s41467-023-36078-y
Y.Z. Chen, M. Zhou, Y.F. Huang, et al., Rare Met. 41 (2022) 3170–3179. doi: 10.1007/s12598-022-02039-5
Q. Zhou, R. Sun, Y. Ren, et al., Carbon Energy 5 (2023) e273. doi: 10.1002/cey2.273
M. Huang, B.W. Deng, X.L. Zhao, et al., ACS Nano 16 (2022) 2110–2119. doi: 10.1021/acsnano.1c07746
X.Y. Zhang, S. Zhang, Y. Yang, et al., Adv. Mater. 32 (2020) 1906905. doi: 10.1002/adma.201906905
X.Z. Tian, J.R. Yao, S.Y. Guo, et al., J. Mater. Chem. C 10 (2022) 2575–2580. doi: 10.1039/D1TC04307B
Z.K. Yuan, J. Li, M.J. Yang, et al., J. Am. Chem. Soc. 141 (2019) 4972–4979. doi: 10.1021/jacs.9b00154
J.L. Suter, R.C. Sinclair, P.V. Coveney, Adv. Mater. 32 (2020) 2003213. doi: 10.1002/adma.202003213
J.S. Li, M.J. Huang, L.X. Kong, et al., Inorg. Chem. 59 (2020) 930–936. doi: 10.1021/acs.inorgchem.9b03201
S. Ono, T. Kikegawa, Y. Ohishi, Solid State Commun. 133 (2005) 55–59. doi: 10.1016/j.ssc.2004.09.048
D.L. Wang, H.L.L. Xin, R. Hovden, et al., Nat. Mater. 12 (2013) 81–87. doi: 10.1038/nmat3458
S.T. Hunt, M. Milina, A.C. Alba-Rubio, et al., Science 352 (2016) 974–978. doi: 10.1126/science.aad8471
B.Y. Xia, H.B. Wu, Y. Yan, et al., J. Am. Chem. Soc. 135 (2013) 9480–9485. doi: 10.1021/ja402955t
L.Z. Bu, J.B. Ding, S.J. Guo, et al., Adv. Mater. 27 (2015) 7204–7212. doi: 10.1002/adma.201502725
L.G. Li, P.T. Wang, Z.F. Cheng, et al., Nano Res. 15 (2022) 1087–1093. doi: 10.1007/s12274-021-3603-9
X.N. Wang, L.M. Zhao, X.J. Li, et al., Nat. Commun. 13 (2022) 1596. doi: 10.1038/s41467-022-29276-7
Q.Q. Guan, C.W. Zhu, Y. Lin, et al., Nat. Catal. 4 (2021) 840–849. doi: 10.1038/s41929-021-00679-x
S.T. Wan, H.T. Li, Z.H. Ma, et al., Rare Met. 41 (2022) 1735–1742. doi: 10.1007/s12598-021-01954-3
L.H. Lin, Z. Chen, W.X. Chen, Nano Res. 14 (2021) 4398–4416. doi: 10.1007/s12274-021-3412-9
H. Li, R. Zhao, J.H. Zhu, et al., Chem. Mater. 32 (2020) 8885–8894. doi: 10.1021/acs.chemmater.0c02505
N. Poonkottil, H. Rijckaert, K. Rajendran, et al., Adv. Mater. Interfaces 10 (2023) 2201934. doi: 10.1002/admi.202201934
H.B. Zhang, C.L. Marshall, Chin. J. Catal. 40 (2019) 1311–1323. doi: 10.1016/S1872-2067(19)63321-8
L.Y. Wen, M. Zhou, C.L. Wang, et al., Adv. Energy Mater. 6 (2016) 1600468. doi: 10.1002/aenm.201600468
G.Y. Zhang, L.G. Ma, Y.Y. Dong, et al., J. Colloid Interface Sci. 647 (2023) 188–200.
J. Zhang, M. Li, X. Tan, et al., Appl. Catal. B: Environ. 339 (2023) 123166. doi: 10.1016/j.apcatb.2023.123166
J.K. Zhang, W.L. Yu, D. Feng, et al., Appl. Catal. B: Environ. 312 (2022) 121405. doi: 10.1016/j.apcatb.2022.121405
N.C. Cheng, M.N. Banis, J. Liu, et al., Energy Environ. Sci. 8 (2015) 1450–1455. doi: 10.1039/C4EE04086D
S.Q. Wang, B.L. Xu, W.Y. Huo, et al., Appl. Catal. B: Environ. 313 (2022) 121472. doi: 10.1016/j.apcatb.2022.121472
Y. Xu, G. Li, Y. Xia, Appl. Surf. Sci. 523 (2020) 146529. doi: 10.1016/j.apsusc.2020.146529
L. Jing, Y.M. Zou, R. Goei, et al., Langmuir 39 (2023) 3142–3150. doi: 10.1021/acs.langmuir.2c03367
J.X. Han, C.H. Gong, C. He, et al., J. Mater. Chem. A 10 (2022) 16403–16408. doi: 10.1039/D2TA05241E
X.K. Wu, Z.C. Wang, D. Zhang, et al., Nat. Commun. 12 (2021) 4018. doi: 10.1038/s41467-021-24322-2
H. Chen, X. Liang, Y.P. Liu, et al., Adv. Mater. 32 (2020) 2002435. doi: 10.1002/adma.202002435-2
F. Li, G.F. Han, H.J. Noh, et al., Energy Environ. Sci. 11 (2018) 2263–2269. doi: 10.1039/C8EE01169A
J. Xu, Z.L. Zhao, W. Wei, et al., Chem. Eur. J. 27 (2021) 15992–15999. doi: 10.1002/chem.202102803
X.M. Wang, G.F. Long, B. Liu, et al., Angew. Chem. Int. Ed. 62 (2023) e202301562. doi: 10.1002/anie.202301562
Y. Zhu, K. Fan, C.S. Hsu, et al., Adv. Mater. 35 (2023) 2301133. doi: 10.1002/adma.202301133
W.C. Zhan, Q. He, X.F. Liu, et al., J. Am. Chem. Soc. 138 (2016) 16130–16139. doi: 10.1021/jacs.6b10472
L. Gong, J.W. Zhu, F.J. Xia, et al., ACS Catal. 13 (2023) 4012–4020. doi: 10.1021/acscatal.2c06340
H. Jin, B. Ruqia, Y. Park, et al., Adv. Energy Mater. 11 (2021) 2003188. doi: 10.1002/aenm.202003188
T.Q. Yu, Q.L. Xu, G.F. Qian, et al., ACS Sustain. Chem. Eng. 8 (2020) 17520–17526. doi: 10.1021/acssuschemeng.0c06782
W. Zhang, J.F. Chang, Y. Yang, SusMat 3 (2023) 2–20. doi: 10.1002/sus2.108
X.K. Wang, X.K. Zhou, C. Li, et al., Adv. Mater. 34 (2022) 2204021. doi: 10.1002/adma.202204021
M.C. Luo, Z.L. Zhao, Y.L. Zhang, et al., Nature 574 (2019) 81–85. doi: 10.1038/s41586-019-1603-7
Q. Dang, H.P. Lin, Z.L. Fan, et al., Nat. Commun. 12 (2021) 6007. doi: 10.1038/s41467-021-26336-2
J.C. Fan, J.D. Wu, X.Q. Cui, et al., J. Am. Chem. Soc. 142 (2020) 3645–3651. doi: 10.1021/jacs.0c00218
Z.C. Wang, H. Zhao, J. Liu, et al., Chem. Eng. J. 450 (2022) 137951. doi: 10.1016/j.cej.2022.137951
J.C. Fan, Z.P. Feng, Y.J. Mu, et al., J. Am. Chem. Soc. 145 (2023) 5710–5717. doi: 10.1021/jacs.2c11692
J.W. Yin, J. Wang, Y.B. Ma, et al., ACS Mater. Lett. 3 (2021) 121–133. doi: 10.1021/acsmaterialslett.0c00473
C.Y. Ye, Y. Yang, X.X. Wu, et al., Prog. Chem. 34 (2022) 1896–1910.
Z. Zeng, R. Xu, H. Zhao, et al., Mater. Today Nano 3 (2018) 54–68. doi: 10.1016/j.mtnano.2018.11.005
H.T. Niu, C.F. Xia, L. Huang, et al., Chin. J. Catal. 43 (2022) 1459–1472. doi: 10.1016/S1872-2067(21)63862-7
E.P. Lee, Z.M. Peng, W. Chen, et al., ACS Nano 2 (2008) 2167–2173. doi: 10.1021/nn800458p
J. Erlebacher, M.J. Aziz, A. Karma, et al., Nature 410 (2001) 450–453. doi: 10.1038/35068529
Z.H. Zhai, Y. Wang, C.H. Si, et al., Nano Res. 16 (2023) 2026–2034. doi: 10.1007/s12274-022-4861-x
M. Schumann, M.R. Nielsen, T.E.L. Smitshuysen, et al., ACS Catal. 11 (2021) 5189–5201. doi: 10.1021/acscatal.0c05002
L.C. Bai, X. Wang, Q. Chen, et al., Angew. Chem. Int. Ed. 55 (2016) 15656–15661. doi: 10.1002/anie.201609663
T.Z. Wu, M.Y. Han, Z.J. Xu, ACS Nano 16 (2022) 8531–8539. doi: 10.1021/acsnano.2c04603
D.D. Wang, Z.W. Chen, K.Z. Gu, et al., J. Am. Chem. Soc. 145 (2023) 6899–6904. doi: 10.1021/jacs.3c00276
L. Zhang, H.M. Chen, Z.D. Wei, ChemCatChem 13 (2021) 2541–2558. doi: 10.1002/cctc.202001260
Z.W. Che, X.Y. Lu, B.F. Cai, et al., Nano Res. 15 (2022) 1269–1275. doi: 10.1007/s12274-021-3645-z
X. Liang, N.H. Fu, S.C. Yao, et al., J. Am. Chem. Soc. 144 (2022) 18155–18174. doi: 10.1021/jacs.1c12642
J.F. Zhang, H.J. Zhang, Y.M. Wu, et al., J. Mater. Chem. A 10 (2022) 5743–5757. doi: 10.1039/D1TA07910G
X. Zhao, D.P. He, B.Y. Xia, et al., Adv. Mater. 35 (2023) 2210703. doi: 10.1002/adma.202210703
J.X. Wei, K. Xiao, Y.X. Chen, et al., Energy Environ. Sci. 15 (2022) 4592–4600. doi: 10.1039/D2EE02151J
T.F. Jaramillo, K.P. Jorgensen, J. Bonde, et al., Science 317 (2007) 100–102. doi: 10.1126/science.1141483
L. Li, Z.D. Qin, L. Ries, et al., ACS Nano 13 (2019) 6824–6834. doi: 10.1021/acsnano.9b01583
M. Shao, P.C. Wang, Y.M. Wang, et al., Green Energy Environ. 6 (2021) 858–865.
X. Liu, P. Wang, X. Liang, et al., Mater. Today Energy 18 (2020) 100524. doi: 10.1016/j.mtener.2020.100524
F.F. Chang, J.C. Wei, Y.P. Liu, et al., Appl. Surf. Sci. 611 (2023) 155773. doi: 10.1016/j.apsusc.2022.155773
Y.H. Wang, J. Wei, P. Radjenovic, et al., Anal. Chem. 91 (2019) 1675–1685. doi: 10.1021/acs.analchem.8b05499
T.D. Daff, N.H. de Leeuw, J. Mater. Chem. 22 (2012) 23210–23220. doi: 10.1039/c2jm34646j
X. Zhao, X.L. Zhou, S.Y. Yang, et al., Nat. Commun. 13 (2022) 7869. doi: 10.1038/s41467-022-35633-3
J.Y. Duan, T.Y. Liu, Y.H. Zhao, et al., Nat. Commun. 13 (2022) 2039. doi: 10.1038/s41467-022-29699-2
L.L. Zhao, H.F. Yuan, D.H. Sun, et al., Nano Energy 77 (2020) 105056. doi: 10.1016/j.nanoen.2020.105056
J. Wu, R. Zhao, H. Xiang, et al., Appl. Catal. B: Environ. 292 (2021) 120200. doi: 10.1016/j.apcatb.2021.120200
X.B. Bao, Y.T. Gong, X.Z. Zheng, et al., J. Energy Chem. 51 (2020) 272–279. doi: 10.1016/j.jechem.2020.03.064
S. Mondal, S. Sarkar, D. Bagchi, et al., Adv. Mater. 34 (2022) 2202294. doi: 10.1002/adma.202202294
J.H. Zhuang, X.L. Liu, Y.J. Ji, et al., J. Mater. Chem. A 8 (2020) 22143–22154. doi: 10.1039/D0TA07985E
W. Tong, B.L. Huang, P.T. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 2649–2653. doi: 10.1002/anie.201913122
B.B. Fan, H.Z. Wang, H. Zhang, et al., Adv. Funct. Mater. 32 (2022) 2110783. doi: 10.1002/adfm.202110783
Y. Shi, D. Zhang, H. Huang, et al., Small 18 (2022) 2106947. doi: 10.1002/smll.202106947
J.T. Zhang, X.Z. Liu, Y.J. Ji, et al., Nat. Commun. 14 (2023) 1761. doi: 10.1038/s41467-023-37406-y
H.Y. Jin, T. Song, U. Paik, et al., Acc. Mater. Res. 2 (2021) 559–573. doi: 10.1021/accountsmr.1c00115
S.Z. Geng, Y.J. Ji, J.Q. Su, et al., Adv. Sci. 10 (2023) 2206063. doi: 10.1002/advs.202206063
Y. Chen, Z.C. Lai, X. Zhang, et al., Natl. Rev. Chem. 4 (2020) 243–256. doi: 10.1038/s41570-020-0173-4
Z.H. Xia, S.J. Guo, Chem. Soc. Rev. 48 (2019) 3265–3278. doi: 10.1039/C8CS00846A
Z.H. Hu, Z.T. Wu, C. Han, et al., Chem. Soc. Rev. 47 (2018) 3100–3128. doi: 10.1039/C8CS00024G
G. Wu, X. Han, J.Y. Cai, et al., Nat. Commun. 13 (2022) 4200. doi: 10.1038/s41467-022-31971-4
Y. Nakaya, S. Furukawa, Chem. Rev. 123 (2023) 5859–5947. doi: 10.1021/acs.chemrev.2c00356
Y. Wang, B. Yu, M. He, et al., Nano Res. 15 (2022) 4820–4826. doi: 10.1007/s12274-021-4059-7
Z.H. Pu, T.T. Liu, G.X. Zhang, et al., Adv. Energy Mater. 12 (2022) 2200293. doi: 10.1002/aenm.202200293
T.Y. Jeon, S.H. Yu, S.J. Yoo, et al., Carbon Energy 3 (2021) 375–383. doi: 10.1002/cey2.82
Y.X. Liao, J. Li, S.M. Zhang, et al., Chin. J. Catal. 42 (2021) 1108–1116. doi: 10.1016/S1872-2067(20)63735-4
D.Y. Liu, Y. Zhang, H. Liu, et al., Carbon Energy 5 (2023) e324. doi: 10.1002/cey2.324
R. Zhou, X. Fan, X.X. Ke, et al., Nano Lett. 21 (2021) 4092–4098. doi: 10.1021/acs.nanolett.1c01113
N. Han, M.Z. Sun, Y. Zhou, et al., Adv. Mater. 33 (2021) 2005821. doi: 10.1002/adma.202005821
M.M. Jaki, Int. J. Hydrog. Energy 11 (1986) 519–532. doi: 10.1016/0360-3199(86)90019-4
F. Shen, Y.M. Wang, G.F. Qian, et al., Appl. Catal. B: Environ. 278 (2020) 119327. doi: 10.1016/j.apcatb.2020.119327
J.D. Wu, J.C. Fan, X. Zhao, et al., Angew. Chem. Int. Ed. 61 (2022) e202207512. doi: 10.1002/anie.202207512
J.D. Chen, C.H. Chen, M.K. Qin, et al., Nat. Commun. 13 (2022) 5382. doi: 10.1038/s41467-022-33007-3
J. Jin, J. Yin, H.B. Liu, et al., Angew. Chem. Int. Ed. 60 (2021) 14117–14123. doi: 10.1002/anie.202104055
Y. Wang, C. Wang, L.J. Jin, et al., Colloid Surf. A: Physicochem. Eng. Asp. 604 (2020) 125313. doi: 10.1016/j.colsurfa.2020.125313
W. Wu, Y. Wu, D. Zheng, et al., Electrochim. Acta 320 (2019) 134568. doi: 10.1016/j.electacta.2019.134568
Q. Chen, B. Wei, Y. Wei, et al., Appl. Catal. B: Environ. 301 (2022) 120754. doi: 10.1016/j.apcatb.2021.120754
Y.D. Wang, W. Wu, R.Z. Chen, et al., Nano Res. 15 (2022) 4958–4964. doi: 10.1007/s12274-022-4128-6
P.C. Sercel, A. Shabaev, A.L. Efros, Nano Lett. 17 (2017) 4820–4830. doi: 10.1021/acs.nanolett.7b01662
M.J. Walsh, W.M. Tong, H. Katz-Boon, et al., Acc. Chem. Res. 50 (2017) 2925–2935. doi: 10.1021/acs.accounts.7b00313
X.Q. Mu, J.N. Gu, F.Y. Feng, et al., Adv. Sci. 8 (2021) 2002341. doi: 10.1002/advs.202002341
P. Wang, W. Nong, Y. Li, et al., Appl. Catal. B: Environ. 288 (2021) 119999. doi: 10.1016/j.apcatb.2021.119999
M.G. Li, Z.L. Zhao, Z.H. Xia, et al., Angew. Chem. Int. Ed. 60 (2021) 8243–8250. doi: 10.1002/anie.202016199
D. Zhang, H. Zhao, X.K. Wu, et al., Adv. Funct. Mater. 31 (2021) 2006939. doi: 10.1002/adfm.202006939
Y.C. Qin, F.Q. Wang, X.M. Wang, et al., Rare Met. 40 (2021) 2354–2368. doi: 10.1007/s12598-021-01727-y
D.B. Miracle, O.N. Senkov, Acta Mater. 122 (2017) 448–511. doi: 10.1016/j.actamat.2016.08.081
M.M. Liu, Z.H. Zhang, F. Okejiri, et al., Adv. Mater. Interfaces 6 (2019) 1900015. doi: 10.1002/admi.201900015
R. Hao, Q.L. Feng, X.J. Wang, et al., Rare Met. 41 (2022) 1314–1322. doi: 10.1007/s12598-021-01877-z
S. Liu, E. Zhang, X. Zhang, et al., Sci. Sin. Chim. 50 (2020) 1001–1014. doi: 10.1360/SSC-2020-0058
C.T. Zhang, Q. Liu, J.W. Zhu, et al., Mater. Today Phys. 31 (2023) 100996. doi: 10.1016/j.mtphys.2023.100996
F. Luo, Q. Zhang, X.X. Yu, et al., Angew. Chem. Int. Ed. 57 (2018) 14862–14867. doi: 10.1002/anie.201810102
Y.H. Liu, J. Ding, F.H. Li, et al., Adv. Mater. 35 (2023) 220711.
Z.H. Pu, J.H. Zhao, I.S. Amiinu, et al., Energy Environ. Sci. 12 (2019) 952–957. doi: 10.1039/C9EE00197B
X. Wang, L.C. Bai, J. Lu, et al., Angew. Chem. Int. Ed. 58 (2019) 19060–19066. doi: 10.1002/anie.201911696
K. Guo, D.P. Fan, J.C. Bao, et al., Adv. Funct. Mater. 32 (2022) 2208057. doi: 10.1002/adfm.202208057
Y.H. Wang, R.Q. Li, H.B. Li, et al., Rare Met. 40 (2021) 1040–1047. doi: 10.1007/s12598-020-01665-1
J. Yu, Y.N. Guo, S.X. She, et al., Adv. Mater. 30 (2018) 180004.
J.Y. Yu, G.X. Li, H. Liu, et al., Adv. Funct. Mater. 29 (2019) 1901154. doi: 10.1002/adfm.201901154
Q.B. Chang, J.W. Ma, Y.Z. Zhu, et al., ACS Sustain. Chem. Eng. 6 (2018) 6388–6394. doi: 10.1021/acssuschemeng.8b00187
H.F. Du, Z.Z. Du, T.F. Wang, et al., Adv. Mater. 34 (2022) 2204624. doi: 10.1002/adma.202204624
T.T. Liu, J.M. Wang, C.Y. Zhong, et al., Chem. Eur. J. 25 (2019) 7826–7830. doi: 10.1002/chem.201901215
Y.T. Li, F.Q. Chu, Y.F. Bu, et al., Chem. Commun. 55 (2019) 7828–7831. doi: 10.1039/C9CC03668G
Z.H. Pu, T.T. Liu, W.Y. Zhao, et al., ACS Appl. Mater. Interfaces 12 (2020) 11737–11744. doi: 10.1021/acsami.9b23426
X.L. Wang, G.X. Zhang, W. Yin, et al., Carbon Energy 4 (2022) 246–281. doi: 10.1002/cey2.182
Y. Zhang, D. Zhang, Y.N. Qin, et al., J. Energy Chem. 72 (2022) 108–115. doi: 10.1016/j.jechem.2022.05.001
X.X. Wang, C. Sun, F. He, et al., ACS Appl. Mater. Interfaces 11 (2019) 32460–32468. doi: 10.1021/acsami.9b07975
J.Y. Cai, Y. Song, Y.P. Zang, et al., Sci. Adv. 6 (2020) eaaw8113. doi: 10.1126/sciadv.aaw8113
Y. Wang, G. Yang, W. Yan, Chem. Ind. Eng. Prog. 42 (2023) 3532–3549.
A. Ray, S. Sultana, L. Paramanik, et al., J. Mater. Chem. A 8 (2020) 19196–19245. doi: 10.1039/D0TA05797E
M.T. Jin, X. Zhang, S.Z. Niu, et al., ACS Nano 16 (2022) 11577–11597. doi: 10.1021/acsnano.2c02820
H.M. Sun, Z.H. Yan, F.M. Liu, et al., Adv. Mater. 32 (2020) 1806326. doi: 10.1002/adma.201806326
H.M. Sun, X.B. Xu, Z.H. Yan, et al., J. Mater. Chem. A 6 (2018) 22062–22069. doi: 10.1039/C8TA02999G
J. Li, J. Zhang, J.K. Shen, et al., Mat. Chem. Front. 7 (2023) 567–606. doi: 10.1039/D2QM00931E
S. Chandrasekaran, M. Khandelwal, F. Dayong, et al., Adv. Energy Mater. 12 (2022) 2200409. doi: 10.1002/aenm.202200409
L.M. Deng, F. Hu, M.Y. Ma, et al., Angew. Chem. Int. Ed. 60 (2021) 22276–22282. doi: 10.1002/anie.202110374
T.Q. Yu, Q.L. Xu, L. Luo, et al., Chem. Eng. J. 430 (2022) 133117. doi: 10.1016/j.cej.2021.133117
B.B. Fan, H.Z. Wang, X.P. Han, et al., Chem. Commun. 58 (2022) 8254–8257. doi: 10.1039/D2CC02732A
F.H. Yuan, M.R. Mohammadi, L.L. Ma, et al., Rare Met. 41 (2022) 2624–2632. doi: 10.1007/s12598-022-01967-6
S.H. Liu, S.Z. Geng, L. Li, et al., Nat. Commun. 13 (2022) 1187. doi: 10.1038/s41467-022-28888-3647
T. Yang, X.N. Mao, Y. Zhang, et al., Nat. Commun. 12 (2021) 6022. doi: 10.1038/s41467-021-26316-6
K. Cao, J.M. Cai, R. Chen, Chem. Mater. 32 (2020) 2195–2207. doi: 10.1021/acs.chemmater.9b04647
Z.R. Zhang, C. Feng, D.D. Wang, et al., Nat. Commun. 13 (2022) 2473. doi: 10.1038/s41467-022-30148-3
Y. Zuo, Z. Wang, H. Zhao, et al., ACS Appl. Mater. Interfaces 14 (2022) 14466–14473. doi: 10.1021/acsami.2c00009
K. McCullough, T. Williams, K. Mingle, et al., Phys. Chem. Chem. Phys. 22 (2020) 11174–11196. doi: 10.1039/D0CP00972E
B.R. Goldsmith, J. Esterhuizen, J.X. Liu, et al., AIChE J. 64 (2018) 2311–2323. doi: 10.1002/aic.16198
Q. Zhu, Y. Huang, D. Zhou, et al., Nat. Synth. 3 (2024) 319–328.
X. Hua, H.L. Xia, Y.T. Long, Electrochim. Acta 431 (2022) 141085. doi: 10.1016/j.electacta.2022.141085
J. Wang, J.X. Tian, G.X. Liu, et al., Small Methods 7 (2023) 2300392. doi: 10.1002/smtd.202300392
W.L. Zhou, J.J. Jiang, W.R. Cheng, et al., Chin. J. Struct. Chem. 41 (2022) 2210004–2210015.
C.L. Brosseau, A. Colina, J.V. Perales-Rondon, et al., Nat. Rev. Method Prim. 3 (2023) 80. doi: 10.1038/s43586-023-00272-5
X. Wang, S.C. Huang, T.X. Huang, et al., Chem. Soc. Rev. 46 (2017) 4020–4041. doi: 10.1039/C7CS00206H
X.X. Lv, G.Q. Li, G.T. Zhang, et al., Chin. J. Struct. Chem. 41 (2022) 2210016–2210028.
C.F. Zhu, Z.G. Chen, W.B. Gong, et al., Appl. Surf. Sci. 605 (2022) 154669. doi: 10.1016/j.apsusc.2022.154669
Z.G. Chen, Y.F. Xu, D. Ding, et al., Nat. Commun. 13 (2022) 763. doi: 10.1038/s41467-022-28413-6
H.M. Yu, J. Wan, M. Goodsite, et al., One Earth 6 (2023) 267–277. doi: 10.1016/j.oneear.2023.02.003ty||53||548|2020|||
S. Dresp, F. Dionigi, M. Klingenhof, et al., ACS Energy Lett. 4 (2019) 933–942. doi: 10.1021/acsenergylett.9b00220
H.Y. Wang, C.C. Weng, J.T. Ren, et al., Front. Chem. Sci. Eng. 15 (2021) 1408–1426. doi: 10.1007/s11705-021-2102-6
W.J. He, X.X. Li, C. Tang, et al., ACS Nano 17 (2023) 22227–22239. doi: 10.1021/acsnano.3c08450
H. Liu, W. Shen, H.Y. Jin, et al., Angew. Chem. Int. Ed. 62 (2023) e202311674. doi: 10.1002/anie.202311674

Figure 2 Strategy for the synthesis method and modification of noble-metal electrocatalysts for alkaline HER.

Figure 3 Volmer, Heyrovsky, and Tafel reactions during HER in acidic (a) and alkaline electrolytes (b).

Figure 4 Steps of synthesis of Pt/C catalysts: (a) In situ synthesis Step 1 and (b) corresponding transmission electron microscopy (TEM) image. (c) In situ synthesis Step 2 and (d) corresponding TEM image. (e) Ex-situ synthesis and (f) corresponding TEM image. Reproduced with permission [59]. Copyright 2016, Royal Society of Chemistry. (g) TEM images of Pd cubes obtained with 300 mg of KBr and (h) 600 mg of KBr. Reproduced with permission [62]. Copyright 2010, Springer Nature. (i) TEM and (j) high resolution transmission electron microscopy (HRTEM) image of Pd Octahedrons. Reproduced with permission [63]. Copyright 2015, Springer Nature.

Figure 5 (a) Schematic of the preparation of Rh-MoS2. (b) SEM image and (c) TEM image of Rh-MoS2; Scale bar: 100 and 50 nm, respectively. (d) HRTEM image of Rh-MoS2 indicating MoS2 nanosheets and Rh nanoparticles. Scale bar: 2 nm. (e, f) Enlarged area denoted in (d) HRTEM images corresponding to MoS2 and Rh, respectively. Reproduced with permission [64]. Copyright 2017, Wiley-VCH. (g) Schematic illustration of the preparation process. (h–j) HAADF images and the elemental distributions of Au1/CeO2-L, Au1/CeO2-H, and Aux/CeO2, respectively. Reproduced with permission [65]. Copyright 2022, Wiley-VCH.

Figure 6 (a) Schematic illustration of the preparation method of the RuCo nanoalloys. (b) TEM and (c) HRTEM images of the RuCo@NC hybrid. Reproduced with permission [74]. Copyright 2017, Springer Nature. (d) Schematic illustration of the preparation of the RuNi/CQD. (e) TEM and (f) HAADF-STEM image of RuNi/CQDs. (g) XRD spectra of RuNi/CQDs. Reproduced with permission [79]. Copyright 2020, Wiley-VCH. (h) SMSI and melamine-induced TiOx overlayer structure and behavior. (i) HRTEM image of Au/TiO2@M-N-800. The scale bar corresponds to 5 nm. Reproduced with permission [80]. Copyright 2019, Springer Nature.

Figure 7 (a) Synthesis flow, (b) TEM, (c) HRTEM of IrNi-FeNi3/NF nanosheets. Reproduced with permission [85]. Copyright 2021, Elsevier. (d) Schematic diagram of Ru-G/CC and Ru-H2O/CC-350. (e) TEM and (f) HRTEM images of Ru-G/CC, (g) enlarged view of the marked area in (f), (h–j) EDX elemental mapping images of Ru-G/CC. Reproduced with permission [86]. Copyright 2022, Elsevier. (k) Schematic illustration of the synthetic route. Representative TEM images of (l) PtRu nanowires (PtRu NWs), (m) PtRu nanorods (PtRu NRs), and (n) PtRu nanocubes (PtRu NCs). TEM images of the PtRu NWs collected at (o) 30 min, (p) 1 h, (q) 2 h and (r) 3 h of the reaction times, respectively. Reproduced with permission [87]. Copyright 2017, American Chemical Society.

Figure 8 (a) Schematic showing the preparation of Pt−T/G. SEM image (b) and Atomic-resolution HAADF-STEM image (c) of Pt−T/G-150. Reproduced with permission [92]. Copyright 2020, American Chemical Society. (d, g) Schematic of the synthesis of Pt. (e, f) STEM images of Pt nanoparticles prepared at room temperature. (h, i) STEM images of atomically dispersed Pt prepared at −60 ℃. Reproduced with permission [93]. Copyright 2019, Royal Society of Chemistry. (j) The schematic for the synthesis process of Pt1-Co1/CN photocatalyst. (k) Aberration-corrected HAADF-STEM image of Pt1-Co1/CN sample. Reproduced with permission [94]. Copyright 2023, Elsevier.

Figure 9 (a) Schematic of the synthesis process with Pt as the CE. (b) SEM images of a piece of PtSA-NT-NF. (c) HAADF image of the PtSA-NT-NF. Reproduced with permission [101]. Copyright 2017, Wiley-VCH. Schematic of (d) cathodic and (f) anodic deposition of Ir species. Mass loadings as a function of Ir concentration in the 1 mol/L KOH electrolyte for (e) cathodic and (g) anodic deposition. Reproduced with permission [102]. Copyright 2020, Springer Nature.

Figure 10 (a) Schematic illustration of the Fabrication Procedure of Ru/GHS Catalyst. (b) TEM, (c) HRTEM images of Ru/GHSs. Reproduced with permission [115]. Copyright 2019, American Chemical Society. Schematic illustration and STEM images of (d) silica-encapsulated (NH4)2PtCl6/WOx NPs synthesized at room temperature and subsequently heated to (e) 200 ℃, (f) 600 ℃, and (g) 900 ℃. (h) STEM image, EDX map, and line scan of a resulting core-shell Pt/WC, and a STEM image of Pt/WC. Reproduced with permission [118]. Copyright 2016, American Association for the Advancement of Science. (i) Schematic illustration of the synthesis of 1D Ir–Te NWs. (j) High-magnification STEM images, (k) high-magnification TEM images, (l) XRD pattern, (m) EDS elemental mapping of Ir–Te NWs. Reproduced with permission [121]. Copyright 2021, Springer Nature.

Figure 11 Synthesis of (a) confined Pt@TiO2 and (d) supported Pt/TiO2 catalysts. TEM and HRTEM images of the (b, c) Pt@TiO2 and (e, f) Pt/TiO2 catalysts. Reproduced with permission [132]. Copyright 2022, Elsevier. (g) Schematic illustration of ALD-Pt growth on ZrC support. (h) High-magnification TEM of ALD-Pt/ZrC. (i) Normalized XANES spectra of Pt nanoparticles on the composite catalyst support at the Pt L2 edge. Reproduced with permission [133]. Copyright 2015, Royal Society of Chemistry. (j) Schematic illustration of the fabrication approach. (k) HAADF-STEM image showing the HEA thin film cross section. Reproduced with permission [136]. Copyright 2023, American Chemical Society.

Figure 12 (a) EDX line scan profile of PdHx@Ru. (b) Atomic resolution HAADF-STEM image (top) of PdHx@Ru metallenes and the corresponding measured intensity profiles. (c) FT-EXAFS spectra of Ru K-edge for Ru foil, RuO2, and PdHx@Ru. (d) Alkaline HER polarization curves. (e) Reaction free energy pathways of H2O dissociation for the Ru, Pd@Ru, and PdH@Ru surfaces. Reproduced with permission [154]. Copyright 2023, American Chemical Society. (f) Schematic illustration showing the two-step dealloying process of the Al97.8Ni2Rh0.2 precursor. (g) SEM images of the RhNi-2500. (h) Polarization curves. (i) Gibbs free energy profiles. Reproduced with permission [161]. Copyright 2022, Springer Nature.

Figure 13 (a) Schematic of the controlled synthesis of u-Ru/C and a-Ru/C. (b) STEM and (c) histogram indicating the nanoparticle size distribution of the u-Ru-1/C sample. (d) TEM and (e) histogram indicating the nanoparticle size distribution of the a-Ru-2/C sample. (f) Cdl and (g) normalized HER polarization curves. Reproduced with permission [167]. Copyright 2021, Springer Nature. (h) Schematic diagram of the synthesis process. (i) HAADF-STEM images of Mn3O4. (j) HAADF-STEM image. (k) HER polarization curves. (l) Comparison of overpotentials. Reproduced with permission [171]. Copyright 2022, Royal Society of Chemistry. (m) The synthesis illustration of the SA-Ru-MoS2 electrocatalysts. (n) HRTEM images of SA-Ru-MoS2. (o) High-angle annular dark field-scanning TEM images of SA-Ru-MoS2. (p) LSV curves Reproduced with permission [54]. Copyright 2019, Wiley-VCH.

Figure 14 (a) Schematic diagram of the formation of Pt NSs/CNTs. (b) HER polarization curves. Reproduced with permission [183]. Copyright 2020, Elsevier. HR-TEM images of Pt3Ge-(110) (c) and Pt3Ge-(202) (d) nanoparticles. (e) Comparison of HER polarization curve in 0.5 mol/L KOH electrolyte. (f) XPS of Pt 4f edge for Pt3Ge-(202) and Pt3Ge-(110). (g) PDOS for Pt3Ge-(202) and Pt3Ge-(110). (h) Energy profile diagram for H* adsorption. Reproduced with permission [184]. Copyright 2022, Wiley-VCH.

Figure 15 (a) HAADF-STEM and corresponding EDX maps of PtFe-mix. (b) HRTEM images of PtFe-mix. (c) HER polarization curves. Reproduced with permission [188]. Copyright 2021, Wiley-VCH. (d) The spherical aberration corrected HAADF-STEM image of hcp Ir-Ni from [001] direction. (e) High-resolution HAADF image taken from the selected area in (d). (f) A model of hcp Ir-Ni from [001] direction. (g) The spherical aberration corrected HAADF-STEM image of hcp Ir-Ni from [100] direction. (h) High-resolution HAADF image taken from the selected area in (g). (i) A model of hcp Ir-Ni from [100] direction. (j) HER polarization plots in 1.0 mol/L KOH, (k) chronopotentiometry curves. Reproduced with permission [191]. Copyright 2023, Wiley-VCH.

Figure 16 (a) Strain engineering through lattice mismatch. (b) In-plane strain engineering through constructing amorphous-crystalline phase boundaries. (c) HRTEM image of AC-Ir NSs. (d) HRTEM image of amorphous–crystalline boundary. (e) HRTEM image (left) and strain values (eyy) strain component (right) of crystalline domain. (f) The histogram of strain distribution. (g) Electron diffraction profile of AC-Ir NSs and Ir crystal. (h) The synchrotron-based valence band spectrum of AC-Ir NSs, C-Ir NSs and A-Ir NSs. (i) The calculated ΔGH* on AC-Ir NSs with 4% tensile strain, C-Ir NSs and A-Ir NSs. (j) The dxz+yz+z2 band centers of the active Ir sites and the calculated ΔGH* on AC-Ir NSs with different degrees of lattice expansion. Reproduced with permission [195]. Copyright 2022, Springer Nature.

Figure 17 (a) Schematic diagram. (b) HER polarization curves and (c) corresponding chronopotentiometry curves. Reproduced with permission [205]. Copyright 2020, Elsevier. (d) HRTEM images of Pt-Co/C-HA30 catalysts. (e) HER polarization curves. (f) Reaction energy diagram of water dissociation. (g) Hydrogen absorption Gibbs free energy. Reproduced with permission [212]. Copyright 2022, Springer Nature. (h, i) TEM images of the RuRh2 bimetallene nanoring. (j) Free energy diagrams for H adsorption. (k) Water dissociation potential (associated with strain effect). Reproduced with permission [215]. Copyright 2020, Wiley-VCH.

Figure 18 (a) Schematic illustration of the synthesis of HEA-NPs/carbon, (b) STEM image of HEA-NPs/carbon. HAADF image and EDS elemental maps of (c) mixed, (d) Pt, (e) Au, (f) Pd, (g) Rh, and (h) Ru for HEA-NPs/carbon (selected area in (b) inset). Particle size analyses of HEA-NPs/carbon (inset in (b)). (i) Polarization curves and (j) Tafel plots. (k) Chronopotentiometry curves of HEA-NPs/carbon-700 ℃ samples at 100 mA/cm2. Reproduced with permission [221]. Copyright 2019, Wiley-VCH.

Figure 19 (a) Schematic of the synthesis of RuPx for HER. Reproduced with permission [233]. Copyright 2018, American Chemical Society. (b) Schematic of the generation of RuP2/C, RuP@RuP2/C, and RuP/C [234]. (c) Schematic of the synthesis of Ru phosphides. Reproduced with permission [235]. Copyright 2019, Wiley-VCH. (d) Schematic of the controlled fabrication of RuP nanocrystals on N- and P-codoped graphene. Reproduced with permission [236]. Copyright 2019, Royal Society of Chemistry.

Figure 20 (a) Schematic illustration of the formation of Ru@Ni-MOF. (b) TEM images of Ru@Ni-MOF. (c, d) The corresponding k3-weighted Fourier transformes. (e) LSV polarization curves. (f) Free energy diagram of ΔGH*. Reproduced with permission [249]. Copyright 2021, Wiley-VCH. (g) Synthesis diagram of NiO/RuO2/NF. (h) HRTEM images of NiO/RuO2/NF. (i) HER polarization curves. (j) Chronopotentiometry curve of HER. Reproduced with permission [250]. Copyright 2021, Elsevier. (k) Synthesis illustration. (l) HER comparison overpotentials at a current density of 10 mA/cm2 and mass activity at 100 mV overpotential. (m) SEM image of Pt/A-NiCo LDH. Aberration-corrected HADDF STEM images of (n) Pt/A-NiCo LDH, (o) Ir/A-NiCo LDH and (p) Rh/A-NiCo LDH. Reproduced with permission [251]. Copyright 2022, Royal Society of Chemistry.
Table 2. Summary of the alkaline HER performance of recent reported noble metal-based electrocatalysts.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: