Citation:
Xiangkang Jiang, Zhixing Wang, Hong Dong, Xiang Zhang, Jin Hu, Manman Chu, Yanshuai Hong, Lei Xu, Wenjie Peng, Xiqian Yu, Jiexi Wang. An in-depth understanding of Al doping homogeneity affecting the performance of LiCoO2 at cut-off voltage over 4.6 V[J]. Chinese Chemical Letters,
2024, 35(12): 109553.
doi:
10.1016/j.cclet.2024.109553
An in-depth understanding of Al doping homogeneity affecting the performance of LiCoO2 at cut-off voltage over 4.6 V
English
An in-depth understanding of Al doping homogeneity affecting the performance of LiCoO2 at cut-off voltage over 4.6 V
Engineering Research Center of the Ministry of Education for Advanced Battery Materials, School of Metallurgy and Environment, Central South University, Changsha 410083, China
b.
BASF Shanshan Battery Materials Co., Ltd., Changsha 410205, China
c.
Beijing Advanced Innovation Center for Materials Genome Engineering, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
Received Date:
29 November 2023 Accepted Date:
19 January 2024 Revised Date:
21 December 2023 Available Online:
15 December 2024
Abstract:
The increasing demand for energy density pushes LiCoO2 (LCO) to work at higher voltage (≥4.5 V), which brings a series of problems including detrimental phase transition and structural instability. Various elemental doping has been proven an effective strategy to improve its structure stability. However, the understanding of elemental doping homogeneity effect is not enough, whether in terms of the controllability of doping homogeneity or its complex consequences. In this work, LCO powders with different Al doping homogeneity were synthesized and tested under high voltage (≥4.5 V) in both half and full cell at room and high temperature, respectively. The results show that the Al homogeneously doped LCO showed better cycling stability and rate performance compared to the inhomogeneous LCO sample. Particularly, the discharge capacity of Al homogeneously doped LCO after 500 cycles under 4.5 V in full cells could reach 160.1 mAh/g at 1.0 C with 94.1% capacity retention. Postmortem characterization demonstrates that a better doping homogeneity favors the stability of both the bulk and interface as well as the kinetic conditions. This study provided new insights about LCO performance fading, which sheds new light on the development of high-voltage LCO products
The ever-increasing demand for energy density pushes the development of next generation of Lithium ion batteries (LIBs) with higher energy density and power density to an extremely urgent level [1–3]. Considering those electrochemically active components, cathode materials are the key factor that determine the energy density of LIBs. Specifically, the charging cut-off voltage and reversible capacity of the cathode materials determine its capacity for accommodating lithium ions [4,5]. LiCoO2 is the 1st generation commercialized cathode material and still dominates the field of consumer electronics over 30 years due to its superior volumetric energy density and excellent rate performance [6–8]. Its crystal structure is typical layered α-NaFeO2 type in a R3̅m space group. During the charge/discharge process, lithium ions are extracted from and inserted into the layered CoO2 skeleton, while the crystal structure will go through a series of phase transitions. The higher the charge cut-off voltage, the higher the discharge capacity. In the early times, the discharge specific capacity of LCO is about ~140 mAh/g and ~160 mAh/g under 4.2 V and 4.35 V (versus Li/Li+), respectively, which are still much below its theoretical capacity of 274 mAh/g. However, although the discharge capacity has been significantly improved, the structural stability problem becomes more pronounced. Specifically, when the voltage is above 4.5 V or even 4.6 V, the irreversible phase transition from O3 to H1–3 will occur and cause huge lattice contraction and expansion [9]. Moreover, the ester-based electrolyte stabilization window will be broken down and the electrolyte will be oxidized and decompose, resulting in severe interfacial side reactions between electrolyte and LCO particles. Besides, other factors such as surface reconstruction and lattice oxygen release together with gas evolution and transition metal dissolution will also exacerbate the degradation.
In order to solve the aforementioned issues and achieve long cycling stability of high voltage LCO, diverse modification strategies have been put forward from different levels including bulk element doping [10–12], surface coating [13–16], microstructure design [17,18], interfacial engineering [19–21], and electrolyte optimizing [22–24], etc. Among them, element doping is a widely used and effective modification strategy which can improve the stability down to the natural electronic structure scale. For example, a La-Al co-doping method was reported in 2018 and achieved a high initial specific capacity of 190 mAh/g under 4.5 V (versus Li/Li+) with 96% capacity retention over 50 cycles [25]. In addition, a Ti-Mg-Al trace co-doping approach was investigated in 2019 and realized a higher initial capacity of 202 mAh/g under 4.6 V (versus Li/Li+) and a high capacity retention of 86% after 100 cycles [26].
Although element doping has been widely adopted in the commercial high-voltage LCO products, whether the alien element is doped homogeneously or not has not been systematically investigated. Even whether the positive effects are caused by doping is still unknown. For instance, Wang et al. first proposed a synergistic strategy involving Al & Ti bulk co-doping and Mg gradient surface doping, which contributed to a high capacity retention of 78% after 200 cycles under 4.6 V (versus Li/Li+) with an initial discharge capacity of 224.9 mAh/g [27]. According to the subsequent characterization results, the surface gradient Mg doping relieved the side reactions and stabilized the material surface. In order to investigate the limitations and the intrinsic functions of Mg doping in high voltage LCO material modification, Xu et al. studied the influence of Mg doping concentration, demonstrating that Mg can narrow the band gap and there is a proper doping window of Mg, exceeding which will cause randomly distributed MgO islands and decrease the discharge plateau [28]. However, some doping elements have very low solubility and will segregate even with a very low doping concentration. Titanium has such a low solubility and tends to be enriched at LCO particle surfaces and interfaces, constructing a 3D network and regulating the micro-strain distribution [29,30]. Other low solubility doping elements such as W, Nb, Te and Y are also adopted in synergistic doping strategies for high voltage LCO modification [31,32]. Recent research shows that the size effect of lanthanide has an important influence on the difficulty of doping into the lattice [33].
In this work, two kinds of LCO with different Al doping homogeneity were successfully prepared by controlling the distribution of Al element in LCO precursor. The effect of doping homogeneity on the crystal structure and electrochemical properties over 4.6 V (versus Li/Li+) high voltage was further studied. Compared to the heterogeneously Al doped LCO, the homogeneously Al-doped LCO showed better structural stability and improved lithium-ion diffusion behavior. Therefore, it demonstrated better cycling stability and rate performance, both at room temperature and high temperature, and both in half cells and full cells. Subsequent analysis indicated the function mechanism of homogeneous Al doping, which included stabilizing the bulk phase structure, mitigating the bulk phase transition, stabilizing the surface interface and improving lithium-ion diffusion. This work fills the gap in the study of the element doping homogeneity effect, and reveals the influence of Al doping homogeneity on high voltage LCO materials, which can guide the high voltage LCO material design and pave the way for new generation high voltage LCO product development in the industry.
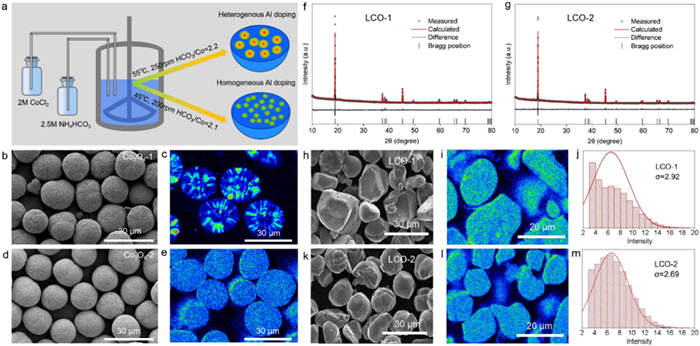
For high-voltage LCO material design, elements doping is necessary to improve the structural stability. It is well known that the most efficient strategy for Al-doping is Al & Co co-precipitation into a CoCO3 matrix in a carbonate precipitation system. In this work, two kinds of Al-doped CoCO3 are prepared through NH4HCO3 precipitation with different preparation condition and then convert to Al-doped Co3O4 precursors (Fig. 1a). Heterogeneous Al doped Co3O4 (Co3O4–1) and homogeneous Al-doped Co3O4 (Co3O4–2) precursors were synthesized through a combination of CoCO3 co-precipitation and following Co3O4 sintering step. Figs. 1b and d compare the morphologies of Co3O4–1 and Co3O4–2 precursors. The Al-segregation can be observed on the surface of Co3O4–1, indicating the Al inhomogeneous distribution during Al & Co co-precipitation. It can be clearly observed from the EPMA images (Figs. 1c and e) that the Al distribution in Co3O4–2 is much more homogeneous than that of Co3O4–1, which can be mainly attributed to the Al-precipitation kinetics is improved by fine-tunning the precipitation parameters. After high-temperature sintering with lithium source, the X-ray diffraction (XRD) patterns of LCO-1 and LCO-2 (Figs. 1f and g) indicate that both have pure hexagonal α-NaFeO2 type phase structure with high crystallinity, and the high peak intensity ratio of (003)/(104) also indicates that both have highly ordered layered structure [34,35]. Al doping has no obvious effect on its crystal structure even for LCO-1, which is confirmed by Rietveld refinements (Table S1 in Supporting information). Figs. 1h and k show that LCO-1 and LCO-2 present similar microscopic morphology, both of which consist of micron-sized irregular spherical particles between 10-30 µm in size. The detailed SEM images are shown in Fig. S1 (Supporting information), and the doping concentration is above 7000 ppm as characterized in Table S2 (Supporting information). Although LCO-1 and LCO-2 are prepared by two different processes, there is no significant difference between them in microscopic morphology. Besides, the Al doping concentration of LCO-1 and LCO-2 is also much close (Table S2 in Supporting information). The homogeneity of Al doping distribution in LCO-1 and LCO-2 is quantificationally compared by electron probe microanalysis (EPMA). As shown in Figs. 1i and l, the cross-sectional surface scan results verify the homogeneity of Al doping in LCO-2 and the heterogeneity in LCO-1. In Figs. 1j and m, the Al doping homogeneity is quantified as the standard deviation (σ) of Al signal intensity, which is statistically determined based EPMA images. The detailed statistical results are shown in Table S3 (Supporting information). The σ value is 2.92 and 2.69 for LCO-1 and LCO-2, respectively, indicating that the Al doping homogeneity of LCO-2 is superior to LCO-1. To summarize, LCO-1 and LCO-2 have few differences in crystal structure, microscopic morphology, and doping concentration except doping homogeneity.
Figure 1
Figure 1.
Characterization of (a-e) Co3O4 precursors and (f-m) corresponding LCOs. (a) The schematics of preparation process of Co3O4 precursors by precipitation. Scanning electron microscope (SEM) images of (b) Co3O4 −1 and (d) Co3O4 −2. EPMA images of Al-distribution in (c) Co3O4 −1 and (e) Co3O4 −2. The XRD patterns of (f) LCO-1 and (g) LCO-2. SEM images of (h) LCO-1 and (k) LCO-2. Cross-sectional EPMA mappings of (i) LCO-1 and (l) LCO-2. Statistics of Al signal intensity of EPMA image of (j) LCO-1 and (m) LCO-2.
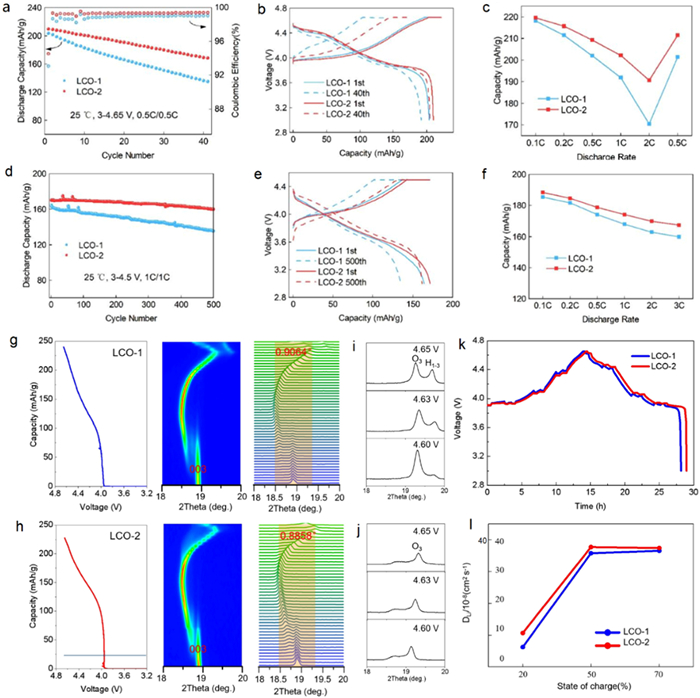
To systematically investigate the influence of Al doping homogeneity on electrochemical performance, both LCO-1 and LCO-2 are assembled into half cells (LCO/Lithium) and full cells (LCO/Graphite) for electrochemical testing at 25 ℃ and 45 ℃. For half cells test results (Fig. 2a), LCO-2 exhibits higher discharge capacity and more stable cycling performance than LCO-1 at 25 ℃ under 4.65 V. Specifically, LCO-1 shows an initial discharge capacity of 203.9 mAh/g for the first activation cycle at 0.1C, whereas LCO-2 releases a higher initial discharge capacity of 209.4 mAh/g. During the following 40 cycles test at 0.5 C (Fig. 2b), LCO-1 exhibits a significant capacity fade from 201.7 mAh/g to 135.1 mAh/g with a capacity retention of 67.0%, while LCO-2 still discharges 168.3 mAh/g after 40 cycles with 80.5% capacity retention. In addition, the rate performance at different current densities of LCO-2 is also superior to that of LCO-1 (Fig. 2c), suggesting the homogeneous Al doping can improve the lithium-ion diffusion kinetics of LCO. Moreover, the full cells test results further prove that LCO-2 outperforms LCO-1 in a long cycling test up to 500 cycles at 1 C under 25 ℃ in 3.0–4.5 V. Concretely, as shown in Figs. 2d and e, the discharge capacity of LCO-1 attenuates from 164.5 mAh/g to 135.6 mAh/g with a retention rate of 82.4%, whereas LCO-2 demonstrates higher discharge capacities of 170.1 mAh/g for the first cycle and 160.1 mAh/g after 500 cycles with 94.1% capacity retention. Besides, the comparison of rate performance between LCO-1 and LCO-2 (Fig. 2f) is consistent with that of half cells, also indicating that LCO-2 has higher discharge capacities and better dynamic conditions. Furthermore, high temperature electrochemical tests carried out in both half cells and full cells (Fig. S3 in Supporting information) also demonstrate that LCO-2 has a higher discharge capacity and better cycling stability. Overall, it is apparent that LCO-2 has better discharge capacity, cycling stability and rate performance than LCO-1. Thus, it can be inferred that the homogeneous Al doping can effectively improve the thermodynamical structural stability and diffusion kinetics of LCO. To better understand the superior electrochemical performance of LCO-2 with homogeneous Al doping, comprehensive characterizations including in-situ XRD, ex-situ XRD, electrochemical impedance spectroscopy (EIS), cross-sectional SEM were applied to investigate the fundamental mechanism. Figs. 2g-j illustrate the in-situ XRD results for the first charging process of LCO-1 and LCO-2, which can directly present the phase transition and structure evolution behavior during the delithiation process. Figs. 2g and h focus on the (003) Bragg diffraction peak shifts in 3.0–4.65 V charging process of LCO-1 and LCO-2, respectively. It is clear that LCO-1 and LCO-2 exhibit similar structure evolution trend, yet the variation amplitude of (003) peak for LCO-1 is 0.9064°, which is larger than that of LCO-2 with 0.8858° The weakened peak shift means that the phase transition and the associated cell volume changes are restrained within LCO-2. Synchronously, the (003) peak splits at the end of the charge for LCO-1, indicating the occurrence of irreversible O3 to H1–3 phase transition and the structural stability will be seriously threatened (Fig. 2i). Whereas, LCO-2 shows no such peak split (Fig. 2j), implying better structural stability. The XRD refinement results (Fig. S4 and Table S4 in Supporting information) show that the changes of both the cell volume and cell parameters of LCO-2 are smaller than those of LCO-1, strongly demonstrating the effective suppression of phase transition and the improvement of structural stability. The galvanostatic intermittent titration technique (GITT) method was applied to analyze the diffusion kinetics of lithium ions under different state of charge (Fig. 2k). The chemical diffusion coefficient of LCO-2 is always higher than that of LCO-1 (Fig. 2l, Fig. S5 and Table S5 in Supporting information), which contributes to better rate performance of LCO-2.
Figure 2
Figure 2.
Electrochemical performance of LCOs at 25 ℃ and corresponding in-situ XRD and GITT testing results. (a, d) Cycling performance, (b, e) 1st and 40th charge-discharge curves, and (c, f) rate capability (a-c) in half cells under 4.65 V and (d-f) in full cells under 4.5 V. In-situ XRD (003) peaks evolution of (g) LCO-1 and (h) LCO-2 in 3.0–4.65 V. Enlarged XRD curves of (i) LCO-1 and (j) LCO-2 at different voltage stages. (k) GITT curve and (l) corresponding Li+ diffusion coefficients of LCOs.
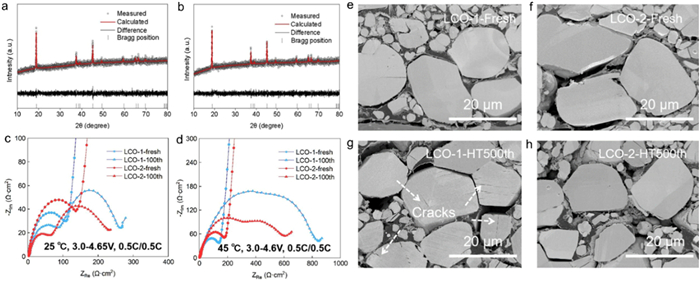
Ex-situ XRD and corresponding refinement are also carried out for structure evolution analysis of LCO after 500 cycles in full cells and the results are shown in Figs. 3a and b. Although LCO-2 outperforms LCO-1 in discharge capacity, cycling stability and rate performance, their XRD patterns after 500 cycles are still both well assigned to the standard PDF card without any extra peaks, indicating that the LCO phase is maintained. Compared with the pristine state before cycling, the peak intensity after cycled is significantly weakened especially for (003) peak, indicating the reduced degree of crystallization. The refinement results (Table S6 in Supporting information) show that the cell lattice parameter a increases while c decreases after cycling for both LCO-1 and LCO-2. As a result, the c/a ratio decreases compared to that before the long cycle. All these refinement parameters exhibit a consistent trend of change, but the c/a ratio for LCO-1 decreases more than that of LCO-2, meaning that the deterioration of layered structure is more serious within LCO-1 than LCO-2, which reconfirms the structural stability effect of homogeneous Al doping [34]. EIS tests of half cells before and after 100 cycles at both 25 ℃ and 45 ℃ were conducted to provide detailed electrode/electrolyte interface kinetic information [36]. Figs. 3c and d show the Nyquist plots of LCO-1 and LCO-2 tested under 4.65 V at 25 ℃ and 4.6 V at 45 ℃, respectively. It can be seen that all Nyquist plots consist of one or two semicircles in the high frequency region and one diagonal line in the low frequency region, corresponding to the electrode/electrolyte interface resistance (Rsei), the charge transfer resistance (Rct) and the Warburg resistance related to the lithium ions diffusion ability in the electrode bulk, respectively [37]. The values of each impedance are summarized in Table S7 (Supporting information). The Rsei values of LCO-1 after 100 cycles are 85.7 Ω tested at 25 ℃ and 398.8 Ω tested at 45 ℃, whereas the values of LCO-2 are 55.5 Ω and 248.3 Ω, respectively. The lower Rsei values indicate that LCO-2 has a more favorable electrode/electrolyte surface for lithium ion transport [38]. In addition, for the group tested at 25 ℃, the Rct value of LCO-1 increases 84.8 Ω to 179.5 Ω after 100 cycles with a growth rate of 112%. In contrast, this resistance of LCO-2 increases from 107.5 Ω to 142.5 Ω with a smaller growth of 33%. As for the samples tested at 45 ℃, the Rct value of LCO-1 increases 154.4 Ω to 457.7 Ω after 100 cycles, while Rct value of LCO-2 increases from 199.5 Ω to 404.2 Ω, of which the growth rate is consistent with the trend of the results tested at 25 ℃. Above all, both the Rsei values and Rct increases after 100 cycles of LCO-2 are smaller than that of LCO-1 both at 25 ℃ and 45 ℃. The comparison strongly demonstrates that LCO-2 has more favorable electrode/electrolyte interface stability and smaller charge transfer impedance than LCO-1, indicating that the homogeneous Al doping is conductive to facilitating the interface reaction kinetics and stabilizing the electrode/electrolyte interface impedance. To further verify the structure stabilizing effect of homogeneous Al doping, the LCO electrodes before and after 500 cycles test in full cells under 4.5 V at 45 ℃ were characterized by the cross-sectional SEM. High temperature and long cycling are set to intensify the failure behavior. As shown in Figs. 3e and f, the fresh particles in both sampels are tightly packed, and the sections of the particles are intact and clean with almost no cracks. However, after 500 cycles under high cut-off voltage and high temperature, the structural collapse and interior cracks are observed within LCO-1 (Fig. 3g). For the crack formation mechanism, it is believed closely related to the irreversible phase transition of O3 to H1–3 occurring over 4.5 V, which will bring about huge and heterogeneous microscopic stress distribution [39,40]. On the contrary, the cross section of LCO-2 after 500 cycles remains intact with only a few visible cracks (Fig. 3h), proving the mechanical stability of LCO-2 is obviously enhanced. Such results are consistent with the front in-situ XRD analysis and reconfirms that the homogeneous Al doping can effectively stabilize the structure and suppress the detrimental phase transition, thus boosting the electrochemical performance.
Figure 3
Figure 3.Ex-situ XRD characterization of (a) LCO-1 and (b) LCO-2 after 500 cycles. Nyquist plots for LCO half cells before and after 100 cycles (c) under 4.65 V cut-off voltage at 25 ℃ and (d) under 4.6 V cut-off voltage at 45 ℃. Cross sectional SEM characterization results of LCOs: (e) LCO-1 at fresh state; (f) LCO-2 at fresh state; (g) LCO-1 after 500 cycles; (h) LCO-2 after 500 cycles.
To sum up, LCO-1 with heterogeneous Al doping and LCO-2 with homogeneous Al doping were successfully synthesized followed by different precursor process methods but same calcination procedures. In-situ and ex-situ XRD results prove that the homogeneous Al doping can effectively suppress the irreversible phase transition and improve the structural stability. EIS analysis indicates that the homogeneous Al doping is conductive to promoting the reaction kinetics and stabilizing the electrode/electrolyte interface. Furthermore, cross-sectional SEM images reconfirm the structural stabilizing effect of the homogeneous Al doping. As a result, LCO-2 outperforms LCO-1 in all electrochemical tests under high voltage in both half cells and full cells. This work fills in a gap in the research of Al doping homogeneity effect on high voltage LCO and illustrate its stabilization mechanism on both bulk phase transition and electrode/electrolyte interface properties, which provide new insights for doping modification process and developing high voltage LCO cathode materials in industry.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 52122407), the Science and Technology Innovation Program of Hunan Province (No. 2022RC3048), and the Key Research and Development Program of Yunnan Province (No. 202103AA080019). We also thank BASF Shanshan Battery Material Co., Ltd. for financial support.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109553.
[1]
B. You, Z. Wang, F. Shen, et al., Small Methods 5 (2021) e2100234. doi: 10.1002/smtd.202100234
Figure 1
Characterization of (a-e) Co3O4 precursors and (f-m) corresponding LCOs. (a) The schematics of preparation process of Co3O4 precursors by precipitation. Scanning electron microscope (SEM) images of (b) Co3O4 −1 and (d) Co3O4 −2. EPMA images of Al-distribution in (c) Co3O4 −1 and (e) Co3O4 −2. The XRD patterns of (f) LCO-1 and (g) LCO-2. SEM images of (h) LCO-1 and (k) LCO-2. Cross-sectional EPMA mappings of (i) LCO-1 and (l) LCO-2. Statistics of Al signal intensity of EPMA image of (j) LCO-1 and (m) LCO-2.
Figure 2
Electrochemical performance of LCOs at 25 ℃ and corresponding in-situ XRD and GITT testing results. (a, d) Cycling performance, (b, e) 1st and 40th charge-discharge curves, and (c, f) rate capability (a-c) in half cells under 4.65 V and (d-f) in full cells under 4.5 V. In-situ XRD (003) peaks evolution of (g) LCO-1 and (h) LCO-2 in 3.0–4.65 V. Enlarged XRD curves of (i) LCO-1 and (j) LCO-2 at different voltage stages. (k) GITT curve and (l) corresponding Li+ diffusion coefficients of LCOs.
Figure 3Ex-situ XRD characterization of (a) LCO-1 and (b) LCO-2 after 500 cycles. Nyquist plots for LCO half cells before and after 100 cycles (c) under 4.65 V cut-off voltage at 25 ℃ and (d) under 4.6 V cut-off voltage at 45 ℃. Cross sectional SEM characterization results of LCOs: (e) LCO-1 at fresh state; (f) LCO-2 at fresh state; (g) LCO-1 after 500 cycles; (h) LCO-2 after 500 cycles.

 DownLoad:
DownLoad:

 下载:
下载:




 下载:
下载:
