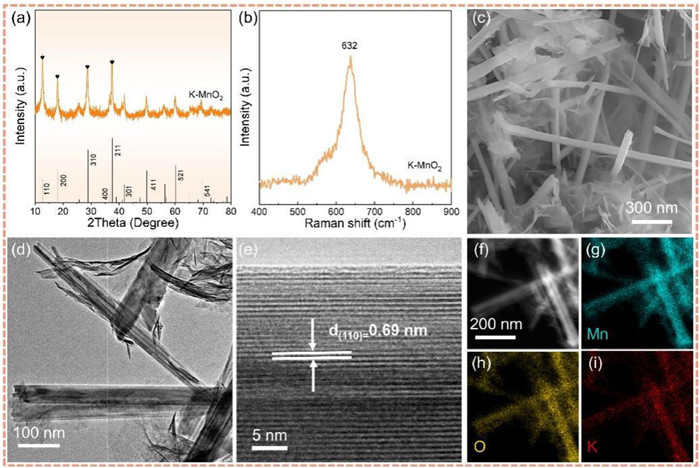
Figure 1.
Morphological and structural characterization of K—MnO2. (a) XRD patterns, (b) Raman spectra, (c) SEM image, (d) TEM image, (e) HRTEM image and (f–i) EDS elemental mapping images.
Potassium ion doped manganese oxide nanoscrolls enhanced the performance of aqueous zinc-ion batteries
Yang Li , Xiaoxu Liu , Tianyi Ji , Man Zhang , Xueru Yan , Mengjie Yao , Dawei Sheng , Shaodong Li , Peipei Ren , Zexiang Shen

The significant growth in smart grids, electric vehicles, and portable electronic devices is driving a growing global demand for electrochemical energy storage [1,2]. In recent years, aqueous zinc-ion batteries (AZIBs) have garnered unprecedented attention due to their low cost, abundant zinc resources, and environmental friendliness [3,4]. Notably, there has been remarkable progress in the development of cathode materials for AZIBs, including Mn- or V-based oxides, Prussian blue analogues and redox-active organic compounds [5]. Among these candidates, MnO2 stands out due to its various crystal structures, cost-effectiveness, non-toxicity, high working voltage range (1.3–1.35 V vs. Zn/Zn2+) and capability to form composites with conductive nanofillers such as graphene, carbon nanotube, and polyaniline [6]. However, the application of Mn-based cathodes is severely hindered by substantial intrinsic issues, especially for Mn2+ dissolution during cycling stability, which is derived from the disproportionation reaction of Mn3+ and structural transformation, leading to severe capacity decay upon cycling. To address these issues, strategies including MnO2 crystal engineering through lattice vacancies or the introduction of foreign ions [7-9], electrolyte engineering through the addition of Mn2+ salts or optimization of Zn2+ solutes have been developed [10]. It is well known that Zn2+ has a smaller ionic radius (0.74 Å) and higher ionic conductivity (1–10 mS/cm) in aqueous zinc ion batteries [11,12], but the host lattice of α-MnO2 exhibits strong electrostatic interactions with Zn2+ leading to slower Zn2+ diffusion and a limited insertion/extraction reversibility. This results in poorer cathode capacity and multiplicity performance. Moreover, the Jahn–Teller effect within the [MnO6] octahedra and the disproportionate dissolution of Mn(Ⅲ) additionally destabilize the host lattice structure, ultimately resulting in the irreversible dissolution of manganese oxides [4,13]. It has been reported that the pre-inserting of metal cations in the tunnel structure is an important way to improve the electrochemical performance of α-MnO2 cathodes [14,15]. By coordinating guest ions with neighboring host atoms, the pre-insertion of metal ions stabilizes the crystal structure of materials and optimizes the charge and electronic bandgap resulting in ultimately improved properties by enhancing the reaction kinetics of the ions in the host structure. Additionally, it is pertinent and suitable for the innovation of new materials [16]. Previous researches have demonstrated the electrochemical characteristics of metal ions, such as Li+ [17], K+ [18,19], Cu2+ [20,21], Al3+ [22], which are pre-inserted within MnO2 electrodes, exhibit distinct electrochemical characteristics. This resulted in an increased gap between the MnO2 layers facilitating rapid ion diffusion, while the column effect strengthened the swollen layer structure. Yuan et al. utilized atomic-level observation techniques and visualization models to assess the influence of the presence of K+ on the tunneling structure of MnO2 and its effect on ion insertion/extraction. Thereby, the interacting mechanism has been enhanced understanding [23-25]. Furthermore, Jiao et al. investigated the relationship among phase stability, structural integrity, and the proportion of pre-inserted ions in K+ doped MnO2 cathodes [26]. Higher concentrations of pre-insertion K+ enhance structural stability, particularly when the K/Mn ratio exceeds 0.26. Consequently, higher K+ concentrations are advantageous for promoting the stability of the cathode. However, it is essential to consider and discuss the relationship between the enhancing structural stability and the electronic state resulting from the presence of K+.
Understanding the charging mechanism of Zn||α-MnO2 batteries is significant for electrode design [27]. Over the past few years, a variety of mechanisms have been proposed based on the different crystal structures and electrolytes of MnO2 [28], including Zn2+insertion/extraction mechanism [29,30], H+/Zn2+ co-insertion/extraction mechanism [31], transformation reaction mechanism [32], and deposition/dissolution mechanism [33]. However, these mechanisms have not been united to insight into the electrochemical reaction of α-MnO2. Achieving reversible intercalation of H+/Zn2+ with high capacity and long cycle life, as well as exploring the electrochemical energy storage mechanism of aqueous Zn-MnO2 cells, remains a significant challenge for aqueous zinc-ion batteries.
Herein, K+ doped manganese dioxide nanoscrolls were prepared by a hydrothermal method, which improved the conventional α-MnO2 synthesis method. The electronic structures were further analyzed by X-ray absorption fine structure (XAFS) and X-ray photoelectron spectroscopy (XPS). The electrochemical reaction mechanism was demonstrated by ex-situ X-ray diffraction (ex-situ XRD) and XPS. Moreover, the comprehension of zinc storage ability was completed by density functional theory (DFT), which revealed the effect of K+ doping on the electronic structure of manganese dioxide. This exploration offers a fresh perception of the incorporation of potassium to improve the zinc storage features in Zn/MnO2 cells, and enhancement of rechargeable aqueous Zn||MnO2 cells.
The crystal structures of synthesized K-MnO2 and α-MnO2 were characterized through X-ray diffraction (Fig. 1a). The distinct peaks observed at 12.7°, 18.1°, 28.8° and 37.5° corresponding to the characteristic planes (110), (200), (310) and (211) of α-MnO2 as documented in the standard reference card (JCPDS No. 44-0141). The XRD pattern of α-MnO2 (Fig. S1 in Supporting information) reveals an unaltered crystal structure post K+ doping. The Raman spectrum (Fig. 1b) depicts a sharp peak at 632 cm−1 for K—MnO2, attributing to the Mn-O stretching vibration within the MnO6 octahedral [20]. SEM images provide a visual revelation of a well-defined wire-like structure, exhibiting a diameter of approximately 100–150 nm and a length of around 5 µm (Fig. 1c). Additionally, discernible nanosheet stacking is observed. TEM images unveil the intricate architecture of K—MnO2, showing curled-up nanosheets arranged in a rolled-up configuration (Fig. 1d). Notably, a homogeneous morphology envelops both the nanoscrolls and the overlaid nanosheets. Comparatively, the MnO2 control sample exhibits nanorods measuring 500 nm to 2 µm in length (Fig. S2 in Supporting information). HR-TEM reveals lattice stripes with a consistent spacing of 0.69 nm, aligning seamlessly with the (110) crystalline surface of α-MnO2 (Fig. 1e). Elemental detection via energy dispersive X-ray spectroscopy (EDS) (Figs. 1f–i) underscores the uniform distribution of Mn, K, and O elements across individual nanoscrolls, affirming the homogeneous doping of K element within the MnO2 nanoscrolls.
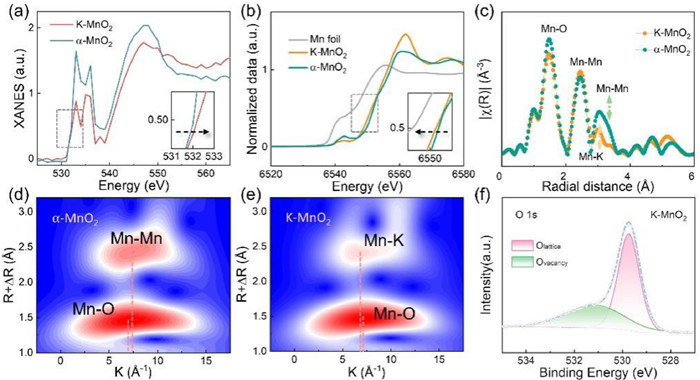
To elucidate the effects of K+ incorporation on the electronic structure of manganese dioxide nanoscrolls, the oxidation states and local coordination environments of Mn atoms are characterized in both K-MnO2 and α-MnO2. Leveraging advanced techniques, X-ray absorption fine structure spectroscopy (XAFS) and XPS, we meticulously standardized the XAFS data to extract the X-ray absorption near-edge structure spectra (XANES), as delineated in Fig. 2a. From the O K-edge XAFS spectra, K—MnO2 exhibited a conspicuous energy shift in energy compared to α-MnO2, signaling a higher valence of O atoms in K—MnO2 and suggesting the presence of oxygen vacancies. As illustrated in Fig. 2b, the absorption edge shift to higher energies correlates with an increase in the average Mn oxidation state. The absorption edge of K—MnO2 nestled between Mn foil and α-MnO2 with a subtle deviation from α-MnO2, signifying the discernible impact of doped K+ on the electronic configuration of Mn atoms [34]. The results delineated indicate that the valence of Mn atoms in K—MnO2 ranges from 0 to +4 [35]. EXAFS spectra of K—MnO2 and α-MnO2 are visually shown after Fourier transforming the XAFS data (Fig. 2c). This process revealed detailed information about the coordination structure (Table S1 in Supporting information) Wavelet transform (WT) analysis on the EXAFS data revealed distinctive peaks corresponding to Mn—O, Mn—Mn, and Mn—K bonds at 1.5 Å, 2.6 Å, and 3.0 Å, respectively. The isochronal plots of K—MnO2 and α-MnO2 (Figs. 2d and e) showed the two bonds of α-MnO2 including Mn—O (7.3 Å−1) and Mn—Mn (7.4 Å−1). While K-MnO2 emphasized the presence of Mn—O (7.3 Å−1) and Mn—K (6.8 Å−1) bonds in K—MnO2 [36]. Furthermore, XPS analyses exhibited the chemical composition and elemental valence states of K—MnO2 (Fig. S3 in Supporting information). Notably, the O 1s XPS spectrum of K—MnO2 (Fig. 2f) unveiled two discernible peaks at 529.8 eV and 530.9 eV, corresponding to lattice oxygen and oxygen defects, respectively. This concurrence with the XAFS analysis confirms the existence of oxygen vacancies. It is suggested that the inclusion of K+ during the synthesis process ostensibly facilitated the genesis of these oxygen vacancies in MnO2 [18].
To illustrate the electrochemical performance between K-MnO2 and α-MnO2 as zinc-ion battery cathodes, ZIBs were assembled that consist of zinc metal anode and aqueous 2 mol/L ZnSO4 + 0.1 mol/L MnSO4 solution as the electrolyte. Fig. 3a shows the electrochemical reaction test model. The electrochemical behavior of Zn||K—MnO2 and Zn||α-MnO2 cathodes was examined using cyclic voltammetry (CV), as illustrated in Fig. 3b. Both Zn||K-MnO2 and Zn||α-MnO2 presented three reversible redox peaks at a scan rate of 0.2 mV/s, indicating that Zn||K—MnO2 and Zn||α-MnO2 possess analogous electrochemical reaction mechanisms. Notably, two reduction peaks imply the energy storage mechanism of Zn2+ and H+ intercalation at 1.372 and 1.268 V, respectively, which corresponds to the reduction of Mn4+ to a lower valence state in the Zn||K-MnO2. Additionally, there is a broad oxidation peak observed at 1.59 V, which relates to the detachment of H+ and Zn2+ from the cathode and oxidation of Mn ions [37]. As shown in Fig. 3b, the K—MnO2 cathode displays higher reduction and oxidation peak current intensities than α-MnO2, demonstrating its fewer side reactions and greater electrochemical capacity [38]. The potential difference (ΔE) of K—MnO2 between reduction and oxidation peaks (0.322 V) is far smaller than α-MnO2 (0.336 V), reflecting that K+ doped cathode is better in electrochemical activation and reaction kinetics. Fig. 3c presents the GCDs of Zn||K—MnO2 and Zn||α-MnO2 under constant-current charge/discharge, within the voltage range 0.8–1.85 V. The GCDs of Zn||K-MnO2 and Zn||α-MnO2 are displayed in Fig. 3c. The initial cycle discharge/charge capacity of K—MnO2 is 250.9/264.1 mAh/g at a current density of 0.2 C, which surpasses that of α-MnO2 (200.3/217.2 mAh/g). This can be attributed to K+ doped that created more defects for Zn ions, resulting in more active sites for zinc ion storage [39]. At the rate of 0.2, 0.5, 1, 2, 3 and 4 C, K—MnO2 cathode delivers mean discharge capacity of 245.9, 285.1, 282.6, 255.1, 232.2, 211.8 mAh/g, respectively, larger than α-MnO2 (Figs. 3d and e). After going back to 0.2 C, the capacity of K—MnO2 reaches 256.8 mAh/g, still larger than the α-MnO2 cathode. The two points collectively demonstrate that the introduction of K+ observably enhances the rate capacity of α-MnO2. Furthermore, the cycle durability possessing K—MnO2 capacity did not show a significant decrease after 100 cycles at 1 C (with a capacity retention rate near 99%) (Fig. 3f). And the cycle performance and capacity retention of K—MnO2 slightly better than other reported MnOx cathodes (Table S2 in Supporting information). Additionally, the capacity's subsequent cycles were enhanced, which owes to the continuous zinc storage reaction and the exposure of more active sites, in line with prior studies [40,41]. Therefore, K—MnO2 demonstrated superior electrochemical performance can be attributed to two aspects: (1) The introduction of K+ into manganese dioxide enhances its structural stability and facilitates the rapid movement of ions, thereby increasing ion transport rates at the electrode interface; (2) The distinctive volume structure of the K—MnO2 nanoscrolls effectively mitigates volume changes during the Zn2+ storage process. All in all, the K—MnO2 nanoscrolls exhibit a higher specific surface area than nanowires, nanorods and nanosheets, and the presence of K+ and oxygen vacancies enhances the electrochemical reaction active sites, resulting in superior electrochemical capacity.

The cyclic voltammetry (CV) curves across scan rates ranging from 0.2 mV/s to 1 mV/s are obtained to illustrate the electrochemical reaction kinetics of K-MnO2 and α-MnO2 (Figs. 4a and d). Notably, these curves exhibit consistent shapes as the scan rate progressively increases. The investigation then delves into the correlation between peak current (i) and scan rate (v) augments the energy storage performance, as per the formulation outlined in the following equations.
|
|
(1) |
|
|
(2) |
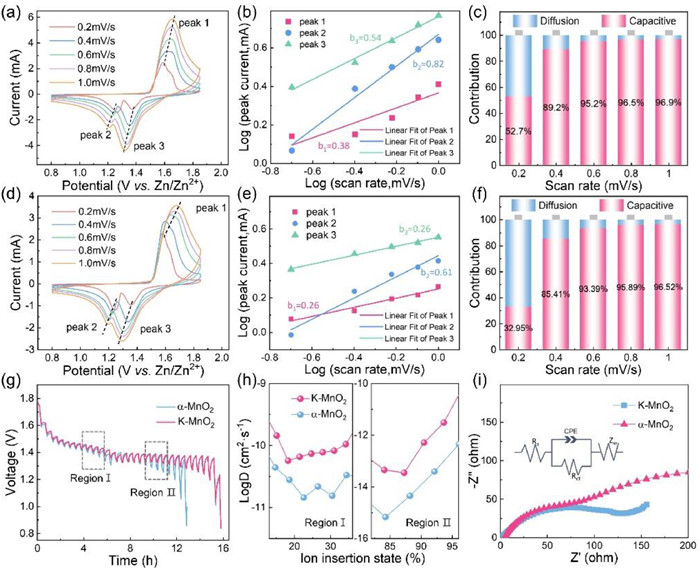
In Eq. 2, there is a linear relationship shown between log(i) and log(v), where a and b are empirical constants. Typically, the value of b approaching 1 suggests the dominant role of the capacitor in the discharge/charging. While the value of b approaching 0.5 indicates a diffusion-controlled discharge/charging process [42]. Figs. 4b and e illustrate the α-MnO2 electrodes corresponding b values of 0.26, 0.61, and 0.26. The linear fitting for log(i) and log(v) (Fig. 4e) reveals b values less than 0.5, for peaks 1 and 3, signifying the involvement of diffusion-limited processes in oxidation/reduction reactions [43]. In contrast, the K-MnO2 displays b values of 0.38, 0.82, and 0.54 at peaks 1, 2 and 3 (Fig. 4b), suggesting that the redox process of K—MnO2 is primarily influenced by a combination of diffusion and pseudocapacitive behavior [44]. To further investigate the Zn2+ storage kinetics of K—MnO2, the cyclic voltammogram (CV) with various scan rates was analyzed. The contributions from intercalation and capacitive reactions in Zn2+ storage can be distinguished through the power-law relationship [45,46].
|
|
(3) |
At various scan rates of 0.2, 0.4, 0.6, 0.8 and 1 mV/s, the respective contributions of the diffusion control component (k2) and pseudocapacitive behavioral control (k1) for the current response at static potential are represented by i(V), where v symbolizes the scan rate, with k1 and k2 denoting defining constants. Specifically, k1 indicates the contribution of pseudocapacitive behavioral control, while k2 represents the contribution of diffusion control, which is proportional to the square root of v. The contributions of K-MnO2 were found to be 52.7%, 89.2%, 95.2%, 96.5%, and 96.9% at scan rates of 0.2, 0.4, 0.6, 0.8, and 1 mV/s respectively (Figs. 4c and f). This reveals that the K-MnO2 cathode exhibited higher pseudocapacitive behavior contributions compared to the α-MnO2 electrode at all scanned rates. However, at high scan rates, the surface capacitance contribution from K-MnO2 dominates the capacity. For instance, at a faster scan rate of 1 mV/s, the K-MnO2 dominates the capacity at high scan rates. The K-MnO2 electrode's pseudocapacitance contribution proportion reaches as high as 96.9%, significantly enhancing the multiplicity performance of K-MnO2. This indicates the successful stimulation of the H+/Zn2+ co-insertion/extraction through K+ doping [37]. The constant current intermittent titration technique (GITT) was utilized to analyze ion diffusion coefficients. Fig. 4g exhibits the relevant GITT curves for K-MnO2 and α-MnO2. The diffusion coefficients during the discharge/charge process were subsequently determined based on Fick's second law:
|
|
(4) |
The GITT profile is similar to the discharge profile with the upper (Region Ⅰ) and lower (Region Ⅱ) plateaus corresponding to H+ and Zn2+ intercalation, respectively, with the diffusion coefficient of K—MnO2 higher than α-MnO2 [47]. Besides, the Rct (120.1 Ω) of K—MnO2 is smaller than those of α-MnO2 Rct (144.1 Ω), reflecting faster transfer kinetics and larger electronic conductivity at the K—MnO2 electrode–electrolyte interface (Fig. 4h) [48].
A systematic study was conducted to elucidate the electrochemical mechanism of K—MnO2 by combining XPS, TEM, and ex-situ XRD. Mn 2p spectra obtained during discharge revealed a decline in Mn (Ⅳ) content and a corresponding increase in Mn(Ⅲ) content, indicating the reduction of Mn4+ to Mn3+ in the discharge process (Figs. 5a–d) [18]. Conversely, there were minimal changes observed in the K 2p1/2 and K 2p3/2 pre- and post-discharge. The O 1s spectra in Fig. 5c illustrated ZnSO4Zn(OH)2·4H2O and Mn—O—H in the fully discharged state, attributed to H+ intercalation, and corroborated by Fig. 5. Furthermore, high-resolution Zn 2p spectra observed post-discharge revealed the synergistic effect of the zinc-containing products generated by Zn2+insertion and the pH alteration following H+ insertion, leading to the formation of ZnSO4Zn(OH)2·4H2O [21,31,49].
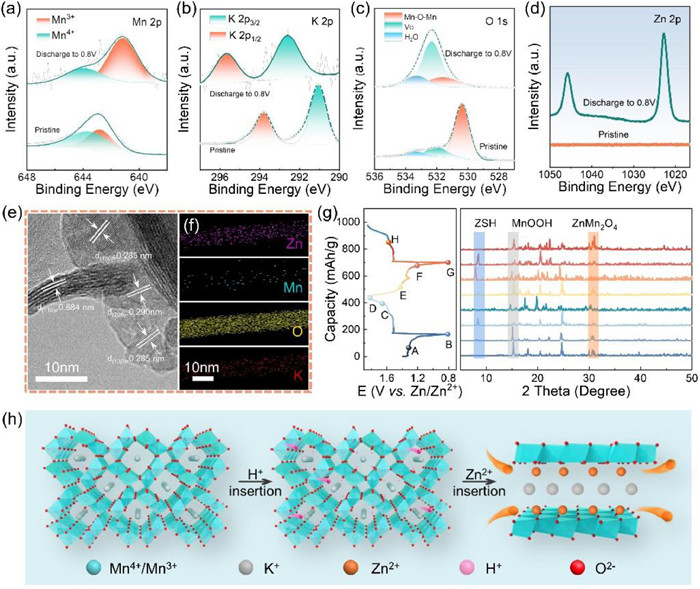
After complete discharge, the material's lattice structure transforms (Fig. 5e and Fig. S4 in Supporting information). there are two types of lattice fringes, which matches well with the d(100) = 0.684 nm plane of tetragonal MnO2 phase, d(130) = 0.285 nm plane of orthorhombic MnOOH phase, and the other interplanar spacing is calculated to be 0.290 nm, which is very close to the d(200) space of ZnMn2O4. The EDS images in Fig. 5f also shows the presence of zinc elements, indicating the successful embedding of Zn2+ in the cathode material with uniform distribution [37,50]. In addition, ex-situ XRD analysis was conducted to examine the material transformations and structural modifications during the initial charging and discharging processes (Fig. 5g). The q2 ~ ln[q3I(q)] curve in Fig. S5 (Supporting information) exhibited a noticeable change from a positive slope to a negative slope in the large q region before and after discharge, indicating a phase transition of the K—MnO2 cathode material. This transition resulted in new substance generation within the material due to the embedding of V+ and Zn2+. When the K—MnO2 cathode is discharged to 1.3 V, new phases of ZnSO4Zn(OH)2·4H2O (JPCDS No. 39-0690), along with MnOOH (JPCDS No. 24-0713) and ZnMn2O4 (JPCDS No. 24-1133) are detected. a discovery supported by TEM observations. The appearance of ZnSO4Zn(OH)2·4H2O is ascribed to a chemical conversion reaction, in which the insertion of H+ triggered the reaction of OH− with Zn2+ and SO42−, while extra OH− react with Zn2+, SO42−, and H2O to produce diffraction peak intensities are enhanced with a further discharge to 0.8 V. These results provide evidence of the electrochemical reaction mechanism resulting from Zn2+ and H+ insertion, leading to the creation of Zn4SO4(OH)6·4H2O and MnOOH. Besides, new diffraction peak signals of ZnMn2O4 (JPCDS No. 24-1133) are both captured. Later, these signals disappear during a followed charge process. The appearance of a new phase of ZnMn2O4 is ascribed to the reaction between Zn2+ and MnOOH, which is regarded as an intercalation reaction. In addition, thus generating K—MnO2 cathode is under a fully charged state to 1.85 V, the phase of Zn4SO4(OH)6·4H2O completely vanishes, and the K—MnO2 cathode returns to the initial state. Such a repeated appearance and disappearance of new phases of ZnSO4Zn(OH)2·4H2O, MnOOH and demonstrate that the K—MnO2 cathode undergoes a reversible electrochemical reaction during discharge/charge processes. Throughout the charging and discharging process, indicating the presence of a reversible electrochemical reaction.
Upon conducting XPS, TEM, and ex-situ XRD analyses before and following full discharge, we were able to propose a redox mechanism (Eqs. 5–8) and theoretical equations were proposed, assuming all distinct processes to be complete (Fig. 5h).
In cathode:
|
|
(5) |
|
|
(6) |
|
|
(7) |
In anode:
|
|
(8) |
Completing a discharge by exchanging one faraday (1 F), one hydrogen ion (H+) is embedded, as indicated in Eq. 5. This is embedded H+ (Eq. 5) then leads to a rise in local pH at the electrode surface, causing ZnSO4Zn(OH)2·4H2O precipitation according to Eq. 5. The opposite process occurs during charging. This reveals a complex interplay of chemical processes and structural changes within the MnO2 lattice during charge-discharge cycles.
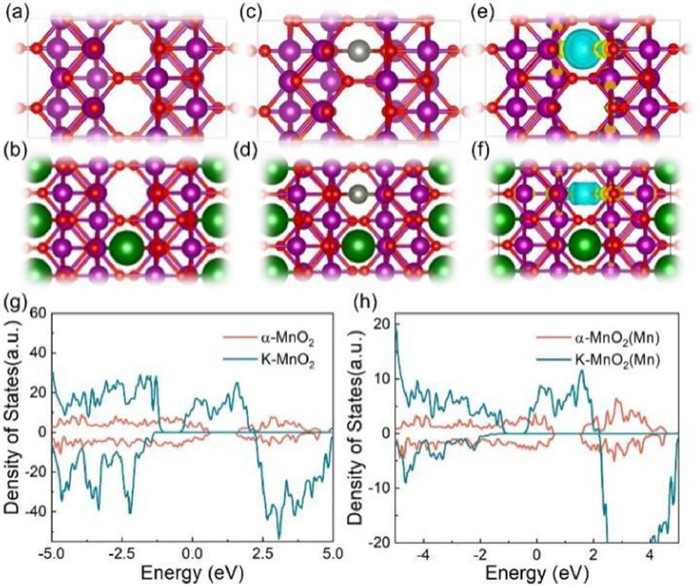
The plausibility of the binding state and Zn2+ storage characteristics arising from K+ doping into tunneled MnO2 was systematically examined through density functional theory (DFT) calculations. The simulation models were constructed based on the crystal structure of α-MnO2, revealing that K+ doping introduced [2 × 2] tunnel structure (Figs. 6a and b) [23]. After the adsorption of zinc ions on the electrode surface (Figs. 6c and d), we calculated the charge density distribution during Zn2+ storage, meticulously analyzing the electronic interactions of Zn2+ in K—MnO2. In Figs. 6e and f, the yellow and blue regions signify charge accumulation and depletion, respectively. Notably, the blue region surrounding Zn2+ in K—MnO2 is comparatively smaller than in α-MnO2, a reduced electron transfer between Zn2+ and K—MnO2. Figs. 6g and h displays the total density of states (TDOS) and partial density of states (PDOS) of K—MnO2 and α-MnO2. The introduction of K+ into the α-MnO2 structure induces a substantial reduction in the band gap, resulting in additional density of states near the Fermi energy level [51]. This outcome leads to an enhancement in the intrinsic conductivity of α-MnO2. As a consequence, K—MnO2 exhibits superior conductivity compared to α-MnO2, thereby facilitating rapid electron transfer [52].
A facile hydrothermal process for potassium ion doping into the α-MnO2 lattice is proposed to produce K—MnO2 nanoscrolls, which can be used as cathode materials in aqueous zinc ion batteries. Results indicate that K doping is highly effective in regulating the type of metal bonding, as well as the average charge distribution of covalent bonding with oxygen atoms in manganese dioxide. Specifically, the Mn—O and Mn—K bonds exhibited by K—MnO2 demonstrate excellent Zn2+ adsorption, thereby facilitating the embedding of Zn2+. The doping-induced oxygen defects have led to the formation of nanoscroll structures, increasing active sites for electrochemical reactions and optimizing the electrical conductivity of α-MnO2. The inclusion of K+ preserves the backbone structure, thus enhancing electrochemical performance during extended cycles. The Zn||K—MnO2 cells display reversible specific capacity and cycling stability, especially where a capacity of about 300.2 mAh/g after 100 cycles at 1 C has been realized. The reaction mechanism was investigated, verifying the continuous insertion/extraction process of H+ and Zn2+ during the electrochemical reaction. These findings indicate the feasibility of a potassium doping strategy for optimizing manganese-based materials in aqueous zinc-ion batteries, yielding new insights for related research.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by Beijing Synchrotron Radiation 4B9A and 1W2A Work Station in China, National Natural Science Foundation of China (No. 52250710161).
Supplementary material associated with this article can be found, in the online version, at doi:
X. Liu, T. Ji, H. Guo, et al., Electrochem. Energy Rev. 5 (2021) 401–433. doi: 10.3390/met11030401
X. Liu, T. Wang, T. Ji, et al., J. Mater. Chem. A 10 (2022) 8031–8046. doi: 10.1039/d1ta10588d
H. Yang, T. Zhang, D. Chen, et al., Adv. Mater. 35 (2023) e2300053. doi: 10.1002/adma.202300053
A. Zhang, R. Zhao, Y. Wang, et al., Energy Storage Mater. 16 (2023) 3240–3301. doi: 10.1039/d3ee01344h
X. Zhao, X. Liang, Y. Li, et al., Energy Storage Mater. 42 (2021) 533–569. doi: 10.1016/j.ensm.2021.07.044
G. Li, L. Sun, S. Zhang, et al., Adv. Funct. Mater. (2023) 2301291.
W. Qiu, H. Xiao, H. Feng, et al., Chem. Eng. J. 422 (2021) 129890. doi: 10.1016/j.cej.2021.129890
S. Kim, B.R. Koo, Y.R. Jo, et al., J. Mater. Chem. A 9 (2021) 17211–17222. doi: 10.1039/d1ta04051k
B. Yang, D. Li, S. Wang, et al., ACS Appl. Mater. 14 (2022) 18476–18485. doi: 10.1021/acsami.2c01362
S. Liu, R. Zhang, J. Mao, et al., Sci. Adv. 8 (2022) eabn5097. doi: 10.1126/sciadv.abn5097
D.L. Chao, W.H. Zhou, F.X. Xie, et al., Sci. Adv. 6 (2020) eaba4098. doi: 10.1126/sciadv.aba4098
Y. Liu, X. Wu, Chin. Chem. Lett. 33 (2022) 1236–1244. doi: 10.1016/j.cclet.2021.08.081
B. Tang, L. Shan, S. Liang, et al., Energy Environ. Sci. 12 (2019) 3288–3304. doi: 10.1039/c9ee02526j
Y. Xu, G. Zhang, J. Liu, et al., Energy Environ. Mater. 6 (2023) e12575. doi: 10.1002/eem2.12575
J. Lei, L. Jiang, Y.C. Lu, Chem. Phys. Rev. 4 (2023) 021307. doi: 10.1063/5.0146094
Q. Zhao, A. Song, S. Ding, et al., Adv. Mater. 32 (2020) 2002450. doi: 10.1002/adma.202002450
Wei Liu, Q. Su, R. Zhu, et al., ACS Appl. Mater. 6 (2023) 6689–6699. doi: 10.1021/acsaem.3c00777
Z. Wang, Y. Wang, Y. Lin, et al., ACS Appl. Mater. 14 (2022) 47725–47736. doi: 10.1021/acsami.2c13030
G. Liu, H. Huang, R. Bi, et al., J. Mater. Chem. A 7 (2019) 20806–20812. doi: 10.1039/c9ta08049j
N. Liang, X. Sun, L. Qi, et al., Energy Technol. 10 (2022) 2200502. doi: 10.1002/ente.202200502
F.W. Fenta, B.W. Olbasa, M.-C. Tsai, et al., J. Mater. Chem. A 8 (2020) 17595–17607. doi: 10.1039/d0ta04175k
Z. Qin, Y. Song, D. Yang, et al., ACS Appl. Mater. 14 (2022) 10526–10534. doi: 10.1021/acsami.1c22674
Y. Yuan, R. Sharpe, K. He, et al., Nat. Sustain. 5 (2022) 890–898. doi: 10.1038/s41893-022-00919-3
Q. Chen, X. Lou, Y. Yuan, et al., Adv. Mater. 35 (2023) 2306294. doi: 10.1002/adma.202306294
X. Yang, Y. Ni, Y. Lu, et al., Angew. Chem. Int. Ed. 61 (2022) e202209642. doi: 10.1002/anie.202209642
Y. Jiao, L. Kang, J. Berry-Gair, et al., J. Mater. Chem. A 8 (2020) 22075–22082. doi: 10.1039/d0ta08638j
M. Wang, X. Zheng, X. Zhang, et al., Adv. Energy Mater. 11 (2020) 2002904.
Y. Wang, J. Xie, J. Luo, et al., Small Methods 6 (2022) e2200560. doi: 10.1002/smtd.202200560
C. Xu, B. Li, H. Du, et al., Angew. Chem. Int. Ed. 124 (2012) 957–959. doi: 10.1002/ange.201106307
N. Zhang, F. Cheng, J. Liu, et al., Nat. Commun. 8 (2017) 405. doi: 10.1007/978-3-319-70090-8_42
X. Gao, H. Wu, W. Li, et al., Small 16 (2020) 1905842. doi: 10.1002/smll.201905842
Y. Huang, J. Mou, W. Liu, et al., Nano-Micro Lett. 11 (2019) 49. doi: 10.1007/s40820-019-0278-9
P. Ruan, X. Xu, D. Zheng, et al., ChemSusChem 15 (2022) e202201118. doi: 10.1002/cssc.202201118
D. Chao, C. Ye, F. Xie, et al., Adv. Mater. 32 (2020) 2001894. doi: 10.1002/adma.202001894
T. Xiong, Z.G. Yu, H. Wu, et al., Adv. Energy Mater. 9 (2019) 1803815. doi: 10.1002/aenm.201803815
J. Zhang, W. Li, J. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202215654. doi: 10.1002/anie.202215654
K. Han, F. An, F. Yan, et al., J. Mater. Chem. A 9 (2021) 15637–15647. doi: 10.1039/d1ta03994f
W. Sun, F. Wang, S. Hou, et al., J. Am. Chem. Soc. 139 (2017) 9775–9778. doi: 10.1021/jacs.7b04471
S. Wang, G. Yuan, J. Yang, et al., ChemSusChem 15 (2022) e202200786. doi: 10.1002/cssc.202200786
C. Zhu, G. Fang, S. Liang, et al., Energy Storage Mater. 24 (2020) 394–401. doi: 10.1016/j.ensm.2019.07.030
X. Wu, G. Liu, S. Yang, et al., Chin. Chem. Lett. 34 (2023) 107540. doi: 10.1016/j.cclet.2022.05.054
J. Tianyi, L. Xiaoxu, Z. Jiupeng, et al., Chem. J. Chin. Univ. 41 (2020) 821–828.
S. Fleischmann, J.B. Mitchell, R. Wang, et al., Chem. Rev. 120 (2020) 6738–6782. doi: 10.1021/acs.chemrev.0c00170
C. Choi, D.S. Ashby, D.M. Butts, et al., Nat. Rev. Mater. 5 (2020) 5–19.
X. Liu, T. Wang, T. Zhang, et al., Adv. Energy Mater. 12 (2022) 202202388.
T. Ji, X. Liu, H. Wang, et al., Research 6 (2023) 0092. doi: 10.34133/research.0092
X. Wang, Y. Liu, Z. Wei, et al., Adv. Mater. 34 (2022) 2206812. doi: 10.1002/adma.202206812
M. Zhang, X. Liu, J. Gu, et al., Chin. Chem. Lett. 34 (2023) 108471. doi: 10.1016/j.cclet.2023.108471
C. Huang, Q. Wang, D. Zhang, et al., Nano Res. 15 (2022) 8118–8127. doi: 10.1007/s12274-022-4498-9
Z. You, W. Hua, N. Li, et al., Chin. Chem. Lett. 34 (2023) 107525. doi: 10.1016/j.cclet.2022.05.039
C. Chen, K. Xu, X. Ji, et al., J. Mater. Chem. A 3 (2015) 12461–12467. doi: 10.1039/C5TA01930C
Y. Ma, M. Xu, R. Liu, et al., Energy Storage Mater. 48 (2022) 212–222. doi: 10.1016/j.ensm.2022.03.024

Figure 1 Morphological and structural characterization of K—MnO2. (a) XRD patterns, (b) Raman spectra, (c) SEM image, (d) TEM image, (e) HRTEM image and (f–i) EDS elemental mapping images.

Figure 2 The chemical compositions and electronic band structures α-MnO2 and K—MnO2. (a) Normalized O K-edge XAFS spectra, (b) normalized Mn K-edge XAFS spectra of standard Mn in the pristine state, (c) EXAFS, (d, e) Wavelet transform spectra and (f) K—MnO2 O 1s spectra.

Figure 3 Electrochemical performances of K—MnO2 and α-MnO2. (a) Schematic diagram of electrochemical processes. (b) Cyclic voltammetry curves at 0.2 mV/s. (c) Charge/discharge curves for 0.2 C in 0.8–1.85 V. (d) Charge/discharge curve. (e) Comparison of multiplicative performance for 0.2–0.4 C in 0.8–1.85 V and (f) cycling performance at 1 C.

Figure 4 Electrochemical kinetic analyses of α-MnO2 and K—MnO2. K—MnO2: (a) Cyclic voltammetry curves at different scan rates (0.1–1.0 mV/s). (b) log(i)-log(v) plots of cyclic voltammetry curves at specific peak currents. (c) Corresponding pseudocapacitive contributions. α-MnO2: (d) Cyclic voltammetry curves at different scan rates (0.1–1.0 mV/s). (e) log(i)-log(v) plots of cyclic voltammetry curves at specific peak currents. (f) Corresponding pseudocapacitive contributions. (g) GITT, (h) Zn2+ diffusion coefficients and (i) Nyquist curves.

Figure 5 Storage mechanism of K—MnO2. XPS before and after complete discharge. (a) Mn 2p3/2, (b) K 2p, (c) O 1s, (d) Zn 2p. (e) High-resolution TEM image of K—MnO2 after discharge. (f) EDS elemental mapping image of K—MnO2 after complete discharge. (g) Ex-situ XRD patterns under different charging and discharging states and (h) schematic of the reaction mechanism.

Figure 6 DFT theoretical calculations. (a) Optimized structures of α-MnO2 and (b) K—MnO2 Mn (purple), O (red), K (green), Zn (light grey). (c) Structures of α-MnO2 and (d) K—MnO2 with adsorption of one Zn ion. (e) Electron cloud distribution of adsorbed zinc ions for α-MnO2 and (f) K—MnO2. (g) The total density of states of α-MnO2 and K—MnO2 and (h) manganese density of states of α-MnO2 and K—MnO2.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: