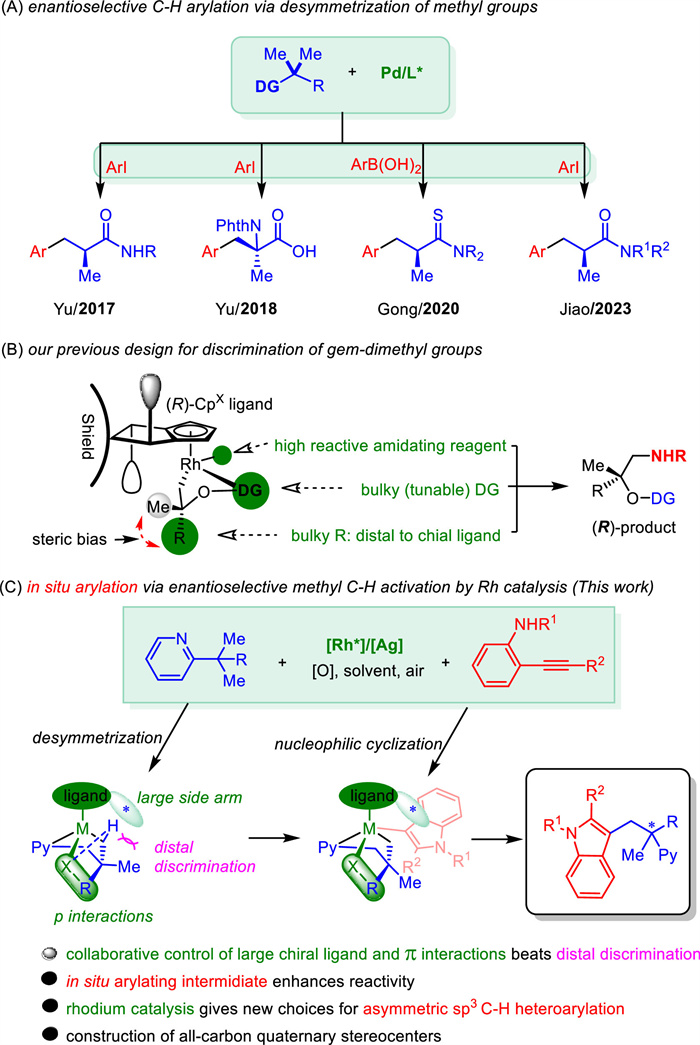
Scheme 1.
Enantioselective intermolecular methyl C(sp3)−H arylations.
Rhodium-catalyzed enantioselective in situ C(sp3)−H heteroarylation by a desymmetrization approach
Yujia Shi , Yan Qiao , Pengfei Xie , Miaomiao Tian , Xingwei Li , Junbiao Chang , Bingxian Liu

Asymmetric C(sp3)-H arylations have made significant progress in the past decade, enabling chemo- and enantio-selective C(sp3)-C(sp2) connections in both intermolecular and intramolecular ways [1–8]. This approach helps in developing versatile building blocks with central chiral carbon stereocenters. Moreover, intermolecular C(sp3)-H arylation also adds the introduction of aromatic groups into molecules, modifying their original properties and broadening new possibilities for drug research [9–13]. The key to achieving good stereocontrol lies in the enantioselective C(sp3)-H activation process, which primarily involves recognizing enantiotopic methylene C−H bonds or desymmetrizing alkyl groups to create an organometallic species with chiral information. Although enantioselective methylene C−H arylations have been well studied using Pd [4,8,14–22], Rh [23,24], Ni [25,26], and Cu [27] catalysts, C−H arylations of methyl groups still have some limitations. In this context, Yu et al. recently described a breakthrough in α-chiral center formation by asymmetric β-C(sp3)–H arylation through desymmetrization of two methyl groups even for challenging isopropyl substrates (Scheme 1A) [28]. After that, Yu's group [29,30], Gong's group [31], and Jiao's group [32] independently reported a few Pd-catalyzed asymmetric C−H arylation reactions using the desymmetrition strategy with prefunctionalized arylating reagents (Scheme 1A). However, the process is currently constrained to palladium catalysts and arylating reagents of arylboronic compounds or aryl halides. On the other hand, while chiral rhodium(Ⅲ)-catalyzed C(sp3)–H activation is still under development [33–37], Matsunaga [34,36] and our group [37] have independently reported representative work on enantioselective amidation through desymmetrization of gem-dimethyl groups. In our previous work, we utilized bulky directing groups (DG) and substituents (R group) to induce steric bias and bring gem-dimethyl groups close to the chiral ligand, resulting in the desymmetrization (Scheme 1B). However, discrimination of the unbiased gem-dimethyl groups with small DGs like a pyridine ring is challenging due to the methyl groups' inability to approach the ligand side arm [38]. To overcome this issue, we speculate that a large chiral ligand may offer help to improve stereocontrol. Additionally, the introduction of noncovalent interaction [39–45] would also provide significant assistance. We herein report a rhodium(Ⅲ)-catalyzed in situ asymmetric C(sp3)–H heteroarylation system using a small pyridine as the directing group and o-aminoaryl alkynes [46–50] as arylating reagents (Scheme 1C). In this system, collaborative control of a large silicon substituted chiral ligand and C−H···π, LP···π interactions between aryl rings of the substrate and a preferred additive facilitated the good stereoselectivity, leading to the construction of chiral indoles containing all-carbon quaternary stereocenters.
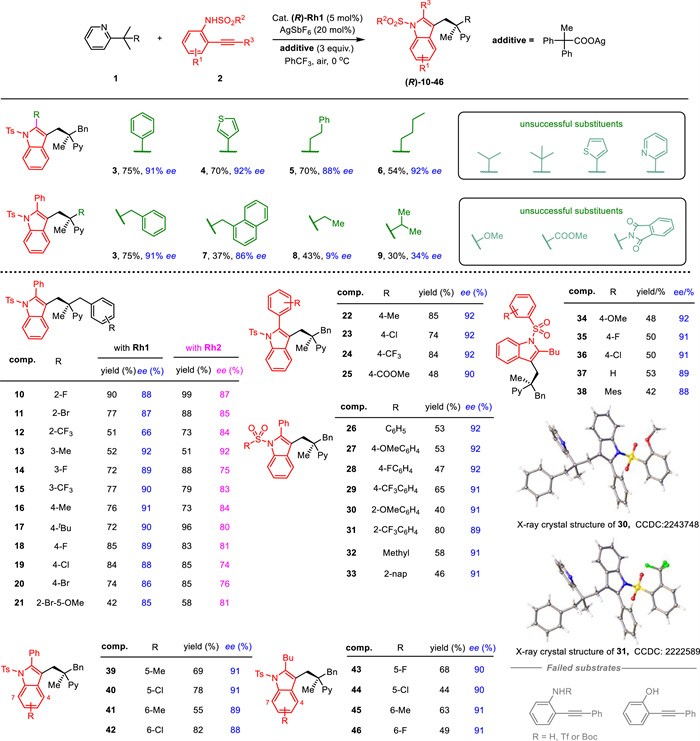
We initiated our investigation by using pyridine derivative 1a and NHTs substituted diarylalkyne 2a as the starting materials in the presence of chiral rhodium catalyst and silver salts [51]. Under the optimal reaction conditions, the model reaction produced the corresponding product 3 in 75% yield and 91% ee, using the chiral rhodium catalyst containing OTDPS (tert-butyldiphenylsilyloxy) substitution (Table 1, entry 1). Reducing the equivalent of the silver carboxylate additive which acts as both the oxidant and the active base for the C−H activation process, the product was obtained with a high ee but a slightly lower yield (entry 2). Other catalysts with different chiral ligands were tested, but only Rh2 with a similar large ligand arm showed increased yield while slightly decreased enantioselectivity (entries 3–5). This indicates that the large steric resistance of the silicon substituents played a vital role in enantioselective control. The change in substrate ratio (1a: 2a) resulted in a lower reaction efficiency while maintaining the enantioselectivity (entry 6, 47% yield, 91% ee). Solvent screening revealed that PhCF3 is optimal for the reaction system (entries 7–10). Lower reaction temperature reduced the reaction efficiency while higher temperature had a negative effect on both selectivity and reaction efficiency (entries 11 and 12). Reducing the reaction concentration had little effect on the reaction while still giving a considerable result (entry 13).
With the optimal conditions in hand, we then investigated the tolerance of functional groups (Scheme 2). We found that phenyl group, heterocyclic group, or alkyl group in the alkynyl unit of o-aminoaryl alkynes all gave moderate to good yields and high enantioselectivity (3–6, 54%−75% yields, 88%−92% ee). However, the presence of bulky isopropyl and tert-butyl substituents inhibited the formation of desired products. Nucleophilic cyclization is impeded by both bulky substituted alkynes and bulky catalysts, and does not occur readily. Likewise, heterocyclic rings with a heteroatom at the ortho-position was also not suitable for this reaction. The heteroatom can act as a coordination site, forming a stable intermediate during nucleophilic cyclization and inhibiting the intended C−H coupling. Interestingly, good enantioselectivity was only achieved when using pyridine substrates that contained an aryl ring substitution (3, 7–9, 30%−75% yields, 9%−91% ee). However, substrates containing an alkyl group (8 and 9) showed low enantioselectivity. The observation could be explained by the possible π interactions between the substrate and carboxylate's aryl rings, considering the phenyl substitution of the silver additive. Other hetero functional groups like OMe, Pht, and ester were found to be unsuitable for the reaction system. These groups could serve as coordination sites that compete with the C−H bond activation process, hindering the delivery of a rhodacycle. We then screened the substrate scopes in detail. Pyridine derivatives with different substitutions in the phenyl ring were tested using Rh1 and Rh2 as catalysts. Better enantioselectivity was achieved in the presence of Rh1 and better efficiency was obtained in the presence of Rh2 in most cases (10–21, 42%−90% yields and 66%−92% ee for Rh1; 51%−99% yields and 74%−92% ee for Rh2). The occupations at 2-, 3-, 4-postion of the phenyl ring by halogen, alkyl group, trifluoromethyl and methoxy all allowed the coupling to give the C(sp3)–H heteroarylation products. The introduction of various functionalities in the phenyl rings of o-aminoaryl alkynes did not affect enantioselectivity and provided the corresponding products with moderate to good yields (22–46, 40%−85% yields, 88%−92% ee). The absolute configuration of compounds 30 and 31 was determined by X-ray single crystal diffraction (CCDC: 2243748 and 2222589). 2-(Phenylethynyl)phenol that can undergo nucleophilic cyclization to afford benzofurans was also tested, but no desired product was obtained.
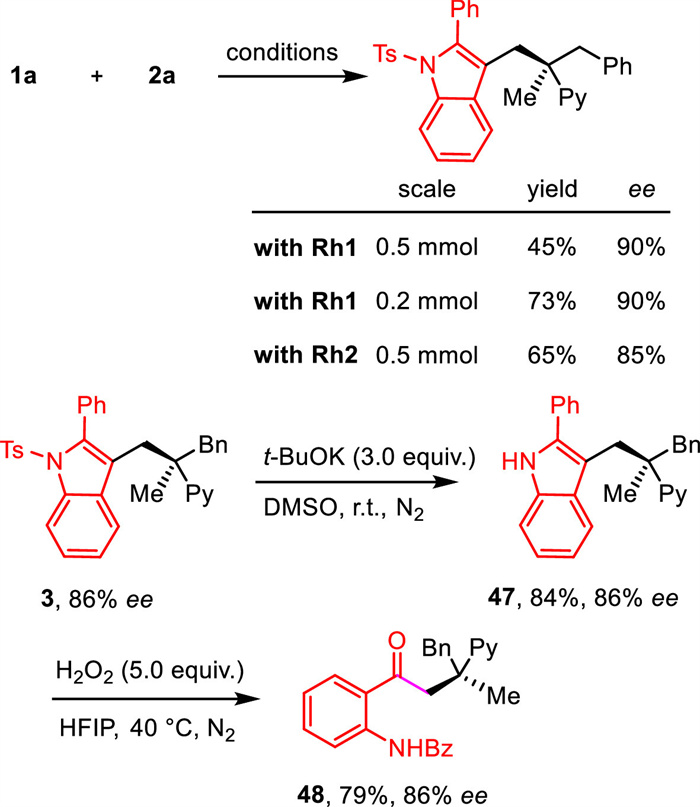
The scale-up synthesis of compound 3 was carried out using both catalyst Rh1 and Rh2 (Scheme 3). The results showed a slight decrease in reaction efficiency and enantioselectivity, though still acceptable. However, when the reaction was performed on a 0.2 mmol scale, compound 3 was generated in 73% yield and 90% ee. The Ts group in compound 3 can be easily eliminated with tBuOK in DMSO, affording the free indole product 47 with a good yield. Subsequent oxidation of compound 47 led to the formation of a ring-opening product 48, which introduced a carbonyl group and an amide group.
To gain a better understanding of the reaction mechanism, some experiments were carried out (Scheme 4). The H/D exchange experiment in the absence of D2O gave both the final product and the recovered pyridine substrate without any deuteration, indicating the irreversibility of the C(sp3)−H activation process (Scheme 4A, ⅰ). The kinetic isotope effect experiment by comparing the initial rates of two parallel reactions resulted in a KIE value of 4.7, confirming the involvement of the C(sp3)−H activation process in the rate-determining step (Scheme 4A, ⅱ). Control experiments were performed to clarify the reaction pathway. The reaction system was easy to generate a Ts-indole compound as a byproduct catalyzed by the silver ion (Scheme 4B, ⅲ). The Ts-indole byproduct can also produce the corresponding product via a potential C−H/C−H coupling progress. Thus, the Ts-indole was synthesized and applied to the system under the standard conditions, showing lower reaction efficiency and enantioselectivity (Scheme 4B, ⅳ). To further rule out the possibility of this intermediate, a competition experiment was conducted between deuterated o-aminoaryl alkyne and the Ts-indole, resulting in the deuterated product as the main product (Scheme 4B, ⅴ, 3:3-D5 = 1:4). The results indicate that the pathway through the Ts-indole intermediate is not as effective. A better approach is to form the Rh-aryl bond through in situ nucleophilic cyclization, which improves the reaction efficiency. To confirm the importance of aromatic ring substitution, three more silver carboxylate oxidants with varying amounts of methyl or phenyl substitutions were tested. The result shows that phenyl substitutions in the additives are also crucial to achieve high enantioselectivity (Scheme 4C). Considering that benzene rings both in the pyridine substrates and the additive are necessary to gain high stereoselectivity, a transition state model with π interactions which preferred to deliver the major rhodacycle intermediate and afforded the final (R)−3 product was assumed (Scheme 4D, TS1R).
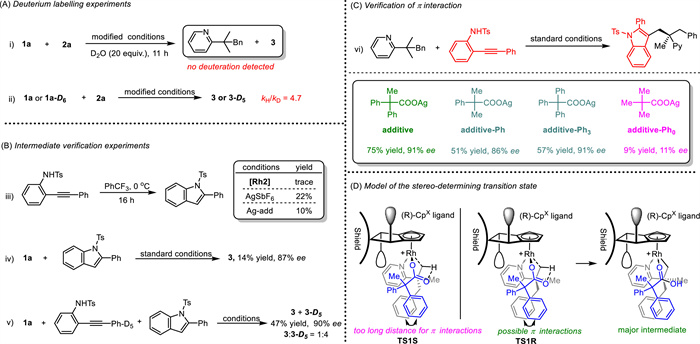
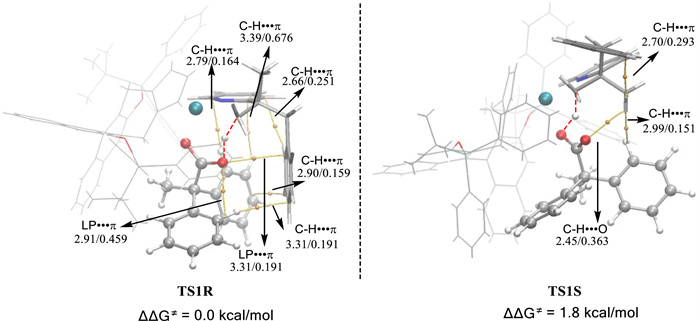
To gain deeper insights into the origin of enantioselectivity, theoretical studies at the DFT levels have been conducted (see Supporting information for details). The calculated results suggest that the C−H activation event is only slightly endergonic (ΔG = 2.8 kcal/mol) with a calculated activation barrier of 19.3 kcal/mol, which seems in line with the 0 ℃ reaction temperature (Fig. S5 and Table S3 in Supporting information). The C−H activation is likely enantio-determining as indicated by the DFT studies. The two transition states of C−H activation that correspond to formation of the R- and S-products have been evaluated. To ensure the lowest energies of these two transition states, eight conformers of TS1R and TS1S were constructed and optimized, and the comparative results are provided in Table S4 (Supporting information), with the most stable ones given in Fig. 1. The TS1R is more stable than TS1S by 1.8 kcal/mol. To rationalize the origin of the stereoselectivity, atom-in-molecule (AIM) analysis [52] was performed on both TS1R and TS1S. The results indicate that in the TS1R the more favorable C−H···π and the LP···π interactions between an enantiotropic phenyl ring of the carboxylate group and the phenyl ring in the pyridine substrate account for the observed stereoselectivity.
Based on the above results and literature reports [47,53–55], a possible reaction pathway has been proposed (Supporting information). The carboxylate assisted C(sp3)−H activation of the pyridine substrate occurred to give a rhodacycle. The transition state with more favorable C−H···π interactions and LP···π interactions eventually delivered the major rhodacyclic intermediate. Subsequently, the intermediate undergoes coordination of an incoming o-aminoaryl alkyne, and a Rh-aryl species was formed by nucleophilic cyclization. The reductive elimination of the Rh(Ⅲ)-aryl species gave the final product and released the rhodium(Ⅰ) catalyst. And the oxidation-induced reductive elimination process via Rh(Ⅳ) intermediate to Rh(Ⅱ) species cannot be ruled out.
In summary, asymmetric synthesis of indole skeleton containing all-carbon quaternary stereocenter was achieved via chiral rhodium catalyzed C(sp3)−H heteroarylation reactions by using an in situ generated arylating reagent. The use of a chiral ligand with a highly hindered silicon substitution and the C−H···π interactions and LP···π interactions between the aryl rings of the carboxylate group and the substrate played crucial roles in achieving stereocontrol. Mechanism studies provided evidence for the proposed catalytic cycle, with in situ nucleophilic cyclization leading to the formation of the Rh-aryl bond and the construction of the final product.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We acknowledge the financial support for this work from the National Key R&D Program of China (No. 2021YFC0864700), the National Natural Science Foundation of China (Nos. 21801066, U1804283 and 82130103), the Central Plains Scholars and Scientists Studio Fund (No. 2018002), the project funded by the Natural Science Foundation of Henan (Nos. 222300420056, 222300420204), and the China Postdoctoral Science Foundation (Nos. 2020M682307, 2021T140183).
Supplementary material associated with this article can be found, in the online version, at doi:
C.G. Newton, S.G. Wang, C.C. Oliveira, N. Cramer, Chem. Rev. 117 (2017) 8908–8976. doi: 10.1021/acs.chemrev.6b00692
J.C.K. Chu, T. Rovis, Angew. Chem. Int. Ed. 57 (2018) 62–101. doi: 10.1002/anie.201703743
K. Yang, M. Song, H. Liu, H. Ge, Chem. Sci. 11 (2020) 12616–12632. doi: 10.1039/d0sc03052j
B. Liu, A.M. Romine, C.Z. Rubel, K.M. Engle, B.F. Shi, Chem. Rev. 121 (2021) 14957–15074. doi: 10.1021/acs.chemrev.1c00519
J.Z. Lv, Y.T. Wei, L.Y. Yang, D.F. Yang, H.L. Li, Catalysts 12 (2022) 537–551. doi: 10.3390/catal12050537
X. Yu, Z.Z. Zhang, J.L. Niu, B.F. Shi, Org. Chem. Front. 9 (2022) 1458–1484. doi: 10.1039/d1qo01884a
B.B. Zhan, L. Jin, B.F. Shi, Trends Chem. 4 (2022) 220–235. doi: 10.1016/j.trechm.2021.12.005
M. Li, J. Wang, Synthesis 54 (2022) 4734–4752. doi: 10.1055/a-1677-5870
E.A. Meyer, R.K. Castellano, F. Diederich, Angew. Chem. Int. Ed. 42 (2003) 1210–1250. doi: 10.1002/anie.200390319
T.J. Ritchie, S.J.F. Macdonald, R.J. Young, S.D. Pickett, Drug Discov. Today 16 (2011) 164–171. doi: 10.1016/j.drudis.2010.11.014
X. Liang, C.J. Lee, J. Zhao, E.J. Toone, P. Zhou, J. Med. Chem. 56 (2013) 6954–6966. doi: 10.1021/jm4007774
S.E. Ward, P. Beswick, Expert Opin. Drug Discov. 9 (2014) 995–1003. doi: 10.1517/17460441.2014.932346
J. Shao, B.P. Kuiper, A.M.W.H. Thunnissen, et al., J. Am. Chem. Soc. 144 (2022) 13815–13822. doi: 10.1021/jacs.2c04986
M. Wasa, K.M. Engle, D.W. Lin, E.J. Yoo, J.Q. Yu, J. Am. Chem. Soc. 133 (2011) 19598–19601. doi: 10.1021/ja207607s
G. Chen, W. Gong, Z. Zhuang, et al., Science 353 (2016) 1023–1027. doi: 10.1126/science.aaf4434
S.Y. Yan, Y.Q. Han, Q.J. Yao, et al., Angew. Chem. Int. Ed. 57 (2018) 9093–9097. doi: 10.1002/anie.201804197
Q.F. Wu, X.B. Wang, P.X. Shen, J.Q. Yu, ACS Catal. 8 (2018) 2577–2581. doi: 10.1021/acscatal.8b00069
H.R. Tong, S. Zheng, X. Li, et al., ACS Catal. 8 (2018) 11502–11512. doi: 10.1021/acscatal.8b03654
S. Jerhaoui, J.P. Djukic, J. Wencel-Delord, F. Colobert, ACS Catal. 9 (2019) 2532–2542. doi: 10.1021/acscatal.8b04946
H.J. Jiang, X.M. Zhong, J. Yu, et al., Angew. Chem. Int. Ed. 58 (2019) 1803–1807. doi: 10.1002/anie.201812426
Y.Q. Han, X. Yang, K.X. Kong, et al., Angew. Chem. Int. Ed. 59 (2020) 20455–20458. doi: 10.1002/anie.202008952
J. Rodrigalvarez, L.A. Reeve, J. Miró, M.J. Gaunt, J. Am. Chem. Soc. 144 (2022) 3939–3948. doi: 10.1021/jacs.1c11921
J.H. Kim, S. Greßies, M. Boultadakis-Arapinis, C. Daniliuc, F. Glorius, ACS Catal. 6 (2016) 7652–7656. doi: 10.1021/acscatal.6b02392
S. Greßies, F.J.R. Klauck, J.H. Kim, C.G. Daniliuc, F. Glorius, Angew. Chem. Int. Ed. 57 (2018) 9950–9954. doi: 10.1002/anie.201805680
X. Cheng, H. Lu, Z. Lu, Nat. Commun. 10 (2019) 3549. doi: 10.1038/s41467-019-11392-6
Y. He, J. Ma, H. Song, et al., Nat. Commun. 13 (2022) 2471. doi: 10.1038/s41467-022-30006-2
W. Zhang, L. Wu, P. Chen, G. Liu, Angew. Chem. Int. Ed. 58 (2019) 6425–6429. doi: 10.1002/anie.201902191
Q.F. Wu, P.X. Shen, J. He, et al., Science 355 (2017) 499–503. doi: 10.1126/science.aal5175
P.X. Shen, L. Hu, Q. Shao, K. Hong, J.Q. Yu, J. Am. Chem. Soc. 140 (2018) 6545–6549. doi: 10.1021/jacs.8b03509
Q. Shao, Q.F. Wu, J. He, J.Q. Yu, J. Am. Chem. Soc. 140 (2018) 5322–5325. doi: 10.1021/jacs.8b01094
H.J. Jiang, X.M. Zhong, Z.Y. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 12774–12778. doi: 10.1002/anie.202004485
C.H. Yuan, X.X. Wang, L. Jiao, Angew. Chem. Int. Ed. 62 (2023) e202300854. doi: 10.1002/anie.202300854
H. Li, R. Gontla, J. Flegel, et al., Angew. Chem. Int. Ed. 58 (2019) 307–311. doi: 10.1002/anie.201811041
S. Fukagawa, M. Kojima, T. Yoshino, S. Matsunaga, Angew. Chem. Int. Ed. 58 (2019) 18154–18158. doi: 10.1002/anie.201911268
L.T. Huang, S. Fukagawa, M. Kojima, T. Yoshino, S. Matsunaga, Org. Lett. 22 (2020) 8256–8260. doi: 10.1021/acs.orglett.0c02872
Y. Kato, L. Lin, M. Kojima, T. Yoshino, S. Matsunaga, ACS Catal. 11 (2021) 4271–4277. doi: 10.1021/acscatal.1c00765
B. Liu, P. Xie, J. Zhao, et al., Angew. Chem. Int. Ed. 60 (2021) 8396–8400. doi: 10.1002/anie.202014080
B.F. Shi, N. Maugel, Y.H. Zhang, J.Q. Yu, Angew. Chem. Int. Ed. 47 (2008) 4882–4886. doi: 10.1002/anie.200801030
S.E. Wheeler, T.J. Seguin, Y. Guan, A.C. Doney, Acc. Chem. Res. 49 (2016) 1061–1069. doi: 10.1021/acs.accounts.6b00096
A.J. Neel, M.J. Hilton, M.S. Sigman, F.D. Toste, Nature 543 (2017) 637–646. doi: 10.1038/nature21701
A. Fanourakis, P.J. Docherty, P. Chuentragool, R.J. Phipps, ACS Catal. 10 (2020) 10672–10714. doi: 10.1021/acscatal.0c02957
C. Li, F. Chen, Q. Mu, C. Xu, Org. Lett. 24 (2022) 8774–8779. doi: 10.1021/acs.orglett.2c03486
M.Y. Jin, Q. Zhen, D. Xiao, et al., Nat. Commun. 13 (2022) 3276. doi: 10.1038/s41467-022-31026-8
J.Y. Li, P.P. Xie, T. Zhou, et al., ACS Catal. 12 (2022) 9083–9091. doi: 10.1021/acscatal.2c00962
D. Baek, H. Ryu, H. Hahm, J. Lee, S. Hong, ACS Catal. 12 (2022) 5559–5564. doi: 10.1021/acscatal.2c00541
Y. Ye, K.P.S. Cheung, L. He, G.C. Tsui, Org. Chem. Front. 5 (2018) 1511–1515. doi: 10.1039/c8qo00191j
M. Tian, D. Bai, G. Zheng, J. Chang, X. Li, J. Am. Chem. Soc. 141 (2019) 9527–9532. doi: 10.1021/jacs.9b04711
R.K. Shukla, A.K. Chaturvedi, C.M.R. Volla, ACS Catal. 11 (2021) 7750–7761. doi: 10.1021/acscatal.1c01234
S. Guo, J. Chen, M. Yi, et al., Org. Chem. Front. 8 (2021) 1783–1788. doi: 10.1039/d0qo01539c
R.R. Xu, X. Bao, Y.W. Huo, et al., Org. Lett. 24 (2022) 6477–6482. doi: 10.1021/acs.orglett.2c02664
R.L. de Carvalho, E.B.T. Diogo, S.L. Homölle, et al., Chem. Soc. Rev. 52 (2023) 6359–6378. doi: 10.1039/d3cs00328k
R.F.W. A Bader, Chem. Rev. 91 (1991) 893–928. doi: 10.1021/cr00005a013
X. Huang, Y. Wang, J. Lan, J. You, Angew. Chem. Int. Ed. 54 (2015) 9404–9408. doi: 10.1002/anie.201504507
J. Mas-Roselló, A.G. Herraiz, B. Audic, A. Laverny, N. Cramer, Angew. Chem. Int. Ed. 60 (2021) 13198–13224. doi: 10.1002/anie.202008166
C.X. Liu, S.Y. Yin, F. Zhao, et al., Chem. Rev. 123 (2023) 10079–10134. doi: 10.1021/acs.chemrev.3c00149

Scheme 2 Substrate scopes. Reaction conditions: 1a (0.05 mmol), 2a (0.1 mmol), Rh1 (5 mol%), AgSbF6 (20 mol%), additive (0.15 mmol), PhCF3 (0.25 mL), 0 ℃, 3 d, under air.

Figure 1 AIM analyses of the transition states TS1R and TS1S (bond distances (Å) and Laplace electron density (eA−3) are shown).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:


 下载:
下载: