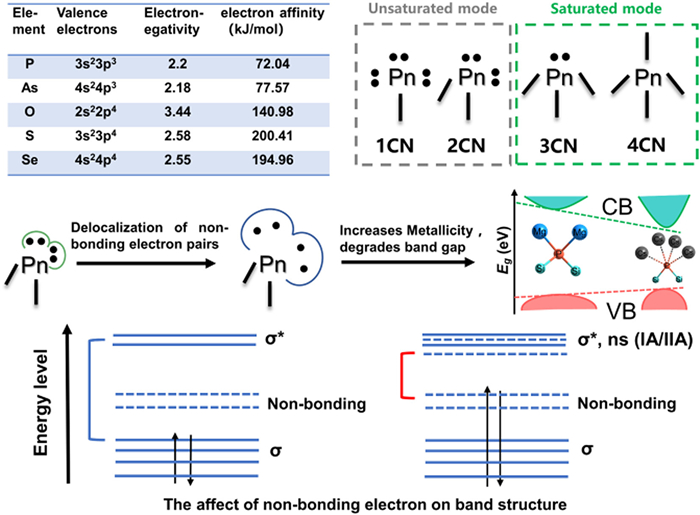
Scheme 1.
The influence of covalent coordination mode of Pn atom on band gap.
Halogen-assisted octet binding electrons construction of pnictogens towards wide-bandgap nonlinear optical pnictides
Lihua Gao , Yinglei Han , Chensheng Lin , Huikang Jiang , Guang Peng , Guangsai Yang , Jindong Chen , Ning Ye

The development of 8–14 µm long-wave infrared (IR) tunable high-power laser is an international cutting-edge research topic [1–3]. The solid-state laser technology with nonlinear optical (NLO) crystals as core components is the main means of realizing long-wave infrared laser outputs with broad tunability, narrow linewidth [4–8]. High-performance infrared NLO crystals require balanced regulation of frequency conversion efficiency, laser damage threshold (LDT), band gap and birefringence. Pnictides generally have a large second harmonic generation (SHG) effect and a wide IR transmission range, which are excellent long-wave IR NLO crystal candidates [9]. Nevertheless, pnictides suffer narrow bandgap (< 2.5 eV for most phosphides and < 1.8 eV for most arsenides) due to relatively weak electronegativity difference between pnictogens and metal component elements. A suitably wide band gap (> 2.33 eV) is vital for IR NLO crystals because the LDT is directly proportional to the band gap, and it determines whether the title crystals can be pumped using mature 1064 nm laser without two-photon absorption. In chalcogenides and oxides, the band gap can be effectively increased by introducing strongly electropositive alkali and alkaline earth metals, to reduce orbital overlap and enhance the ionicity of the system [10–18]. However, our previous work [19] has revealed that this paradigm is not applicable to regulate the band gap of pnictides (Scheme 1). The IA/IIA ions tend to reduce the covalency of pnictogen (Pn) atoms to unsaturated coordination (CN) mode (usually 1CN or 2CN), whereas the contractive electron affinity and electronegativity of Pn atoms are incapable of stabilizing multiple non-bonded electron pairs resulting in their delocalized distribution. Consequently, the metallic interaction occurs between alkali/alkaline-earth metals and neighboring Pn atoms, degrades the band gap. To realized a wide band gap, all composed Pn atoms with at least 3CN are required.
An effective strategy is to explore multiple pnictides diamond-like structure where all pnictogen atoms have 4CN, realizing saturated coordination mode. Under this framework, the comprehensive regulation of key properties can be expected to be achieved through rational chemical element matching, e.g., diamond-like pnictides, MgSiP2, MgSiAs2 and Mg2In3Si2P7, exhibited outstanding optical performances [20–22]. Another alternative strategy is to exclusively incorporate strongly electronegative halogens into pnictides. Since the halogens interact directly with the metal cations and keep a distance from the Pn atoms, the covalent space of the Pn atoms will not be compressed by halogens. For example, Cd3PI3, Cd3AsI3, Cd4As2Cl3, Cd4P2Cl3, and Cd2P3Cl have all component Pn atoms with saturated coordination, exhibiting a large band gap (2.0-2.4 eV) [23,24]. MII-containing halidepnictides (MII = Zn, Cd, Hg, Sn, etc.) are the largest class of halidepnictides, have structural diversity and functional versatility [25–29]. Nevertheless, their nonlinear optical performances are rarely investigated.
Based on these results, two non-centrosymmetric halidepnictides [Cd2Pn]2[CdBr4] (Pn = P and As) were synthesized. As expected, the tetrahedron-coordinated mode, i.e., octet binding valence electron configuration of all Pn atoms were successfully constructed in the anti-tridymite type covalent framework [Cd2Pn]2 under the electrostatic anchor of Br. Simultaneously, synergizing with ionic tetrahedral units [CdBr4], the wide band gap of [Cd2P]2[CdBr4] (2.58 eV) and [Cd2As]2[CdBr4] (1.88 eV) was realized. Remarkably, [Cd2P]2[CdBr4] not only exhibited a widest band gap among Cd-containing pnictides, but also has a SHG effect of 1.2 × AgGaS2, moderate birefringence (0.088@visible light and calcd. 0.043@2050 nm) and a wide IR transmission range (up to ~13.6 µm), indicates it is a potential long-wave IR NLO crystal.
[Cd2P]2[CdBr4] (CPB) and [Cd2As]2[CdBr4] (CAB) were synthesized from a mixture of Cd3Pn2 (3N, Aladdin) and CdBr2 (3N, Adamas) through a mild solid-state reaction (the experimental details are provided in Supporting information). The good fitting of experimental and simulated polycrystalline XRD patterns (Fig. S2 in Supporting information) indicates that the pure phase of title compounds was obtained. The results of Energy-dispersive spectrometry analysis showed that the average atomic ratio of Cd: Pn: Br is 5:2:4, which was consistent with the results of single crystal X-ray diffraction structure solution (Fig. S3 in Supporting information).
CPB crystallized in non-centrosymmetric space group Pna21 with unit cell dimensions of a = 12.1965(10) Å, b = 13.6733(10) Å, c = 7.7112(6) Å, Z = 4 (Table S1 in Supporting information). There are 11 crystallographically independent sites including five Cd, four Br and two P atoms in one asymmetric unit. Its crystal structure is composed of anti-tridymite type [Cd4Pn2]2+ covalent cationic framework and channel-filling [CdBr4]2− tetrahedral anionic isolated units (Fig. 1a). In [Cd4Pn2]2+, the near linear CdP2 units was tetrahedral-connected via P linkers, achieving a three-dimensional framework (Fig. 1b). Since the valence electrons of Cd are 4d105s2, σ bonds with Cd 5s-P 3p orbital hybridization are formed in linear (CdP2) units. Noting that this two-coordinated Cd atom is fairly rare in inorganic compounds. From another perspective, [Cd4Pn2]2+ framework can also be considered to be (Cd4P) anti-tetrahedra via sharing vertex Cd atoms. Six (Cd4P) tetrahedra are interconnected to form a 12-membered ring (Cd18P6) further extended along the c-axis to form a three-dimensional open framework. The Cd-Br bonds in (CdBr4) tetrahedra atoms range from 2.536 Å to 2.715 Å, shorter than CdBr2 (2.76-2.79 Å), Cd4P2Br3 (2.734-3.063 Å) and Cd2P3Br (2.854-3.259 Å). That means there exists certain covalency in Cd-Br bonds. Both (Cd4P) anti-tetrahedra and (CdBr4) tetrahedra have a certain polarity along c-axis, contributing to inherent dipole moment of 15.04 Debye/unit cell. CAB crystallized in a chiral space group P212121, with unit cell dimensions of a = 7.8405(10) Å, b = 13.6415(15) Å, c = 37.217(4) Å, Z = 12. The crystal structure is similar with CPB, consisting of [Cd4As2]2+ framework and [CdBr4]2− units (Fig. S4 in Supporting information). The difference is that the atomic distribution of CAB undergo a symmetry transformation of glide plane (at x, y direction of CPB) to 21 screw axes (at x, z direction of CAB), resulting in the absence of inherent dipole moment.
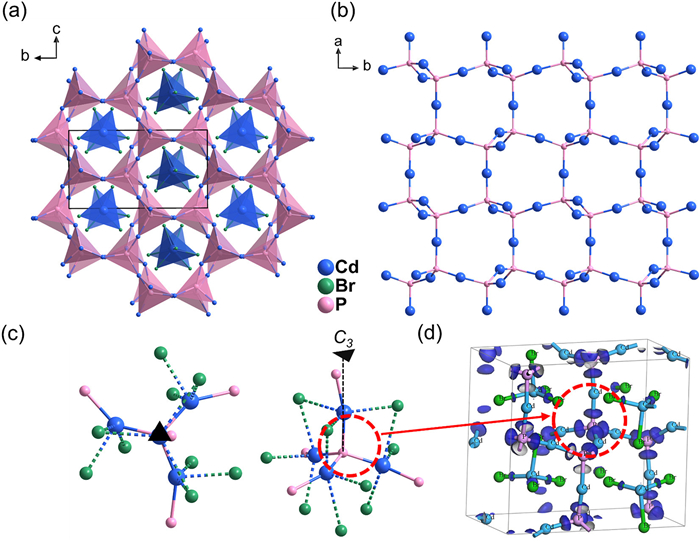
XPS fine specta were performanced to the valence state of component element. As shown in Fig. S8 (Supporting information), the binding energy of Cd 3d5/2 (405.9 eV) and Br 3d5/2 (69 eV) in both CPB and CAB is consistent with those in CdBr2 (406, 69.2 eV), indicates the valence state of Cd and Br is 2+ and 1−, respectively. The binding energy of P 3p3/2 (128.4 eV) and As 3d5/2 (41.1 eV) is consistent with those in InP (128.7 eV) and GaAs (40.9 eV), imply they have 3− valence. The small peaks at high binding energy of P (133.5 eV) and As (44.5 eV) belong to light oxidation phenomenon. Therefore, both CPB and CAB have specific valent electronic formula, i.e., [Cd4Pn2]2+[CdBr4]2−. The Pn atoms are coordinated with four Cd atoms, making the coordination of Pn desired saturation mode, favorable to band gap. To figure out why they can form such complete tetrahedron-coordination mode, we analyzed their nearest and second nearest atomic coordination environments. Due to the structure similarity of CPB and CAB, only the case of CPB is discussed herein. Two independent sites of P atoms, P1 and P2 have proximate nearest and second nearest coordinated conditions. For central P atom, whether the neighbouring Cd atoms or the sub-neighbouring Br and P atoms are trigonometry-arranged along one edge (pseudo threefold axial) of [Cd4P] tetrahedron (Fig. 1c). Due to the electrostatic interaction of Br atoms on Cd, the Cd atoms are anchored in the tetrahedral orientation of P forming four polar covalent bonds, which leads to a stable octet electronic configuration of P atoms. As depicted in the electron density difference (EDD) analysis (Fig. 1d), almost all P atoms have a tetrahedron-rounding electronic structure, which are four bonding electron pairs of P-Cd bonds.
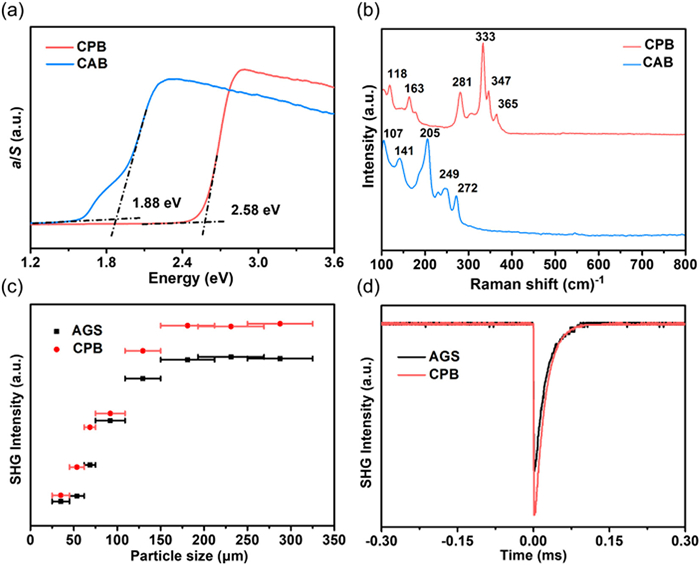
As expected, the measured band gap of CPB and CAB are larger than most phosphides and arsenides like CdSiP2 (~2.2 eV), CdGeAs2 (~0.6 eV) (Table S5 in Supporting information), which are 2.58 and 1.88 eV (Fig. 2a), respectively. In especial, the band gap of CPB is known to be widest among Cd-containing pnictides, indicates that it can be efficiently pumped with mature 1064 nm laser source without two-photon absorption. The mechanism of band gap will be analyzed in first-principles calculation section below. One-order Raman spectra (Fig. 2b) showned that the highest-frequency phonon mode located on 272 cm−1 for CPB and 365 cm−1 for CAB, assigned to the vibration of Cd-P and Cd-As bonds. According to the two-phonon absorption approximation, the IR transmission cutoff can be reckoned as 13.7 µm for CPB and 18.4 µm for CAB, corresponding to shortest the length of Cd-P (2.414 Å) and Cd-As (2.503 Å) bond. The powder SHG responses of CPB and CAB were systematically investigated with AgGaS2 (AGS) as references through the Kurtz and Perry method [30]. CPB exhibited a phase-matching SHG effect of 1.2 × AGS (Figs. 2c and d), attributed to the benign arrangement of the groups. Unfortunately, the SHG response of CAB cannot be observed might because the unfavorable arrangement of the groups counteracts the only second-order susceptibility component χ123 under the restriction of Kleinman's symmetry.
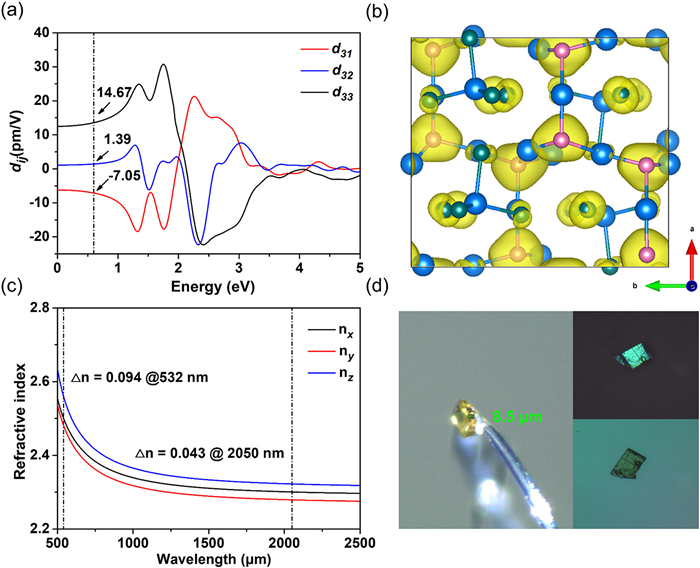
To deeply investigate composition-structure-property relationship, the first-principles calculations were performed. The band structures showed that CPB and CAB have a underestimated direct band gaps of 2.37 and 1.84 eV, respectively (Fig. 3a and Fig. S5a in Supporting information). The valence band maximum (VBM) is dominated by P-3p/As-4p and the conduction band minimum (CBM) is mainly originated from Cd-5s5p (Fig. 3b and Fig. S5b in Supporting information). To understand their wide band gap mechanism, we analyzed the electron density difference (EDD) and electron localization function (ELF) field distribution because the band gap is invariably determined by valence electron behaviors of composed atoms. As shown in Fig. 3c and Fig. S5c (Supporting information), each Cd-Pn bonds in (Cd4Pn) tetrahedra has an isolated EDD maxima shifting to Pn atoms, indicates that the polar covalent bonds are formed between Cd and P atoms. The small-sized EDD maxima of three short Cd-Br bonds in (Cd4Pn) tetrahedra also suggests that there exists certain covalency in them. Due to the formation of octet closed shell of Pn atom via four polar covalent Cd-Pn bonds, the valence electrons of Pn realize tight-binding state with high localization (Fig. 3d and Fig. S5d in Supporting information). Unlike the electronic distribution of weak IA/IIA-Pn interaction where the electron pairs of Pn are nearly free and delocalized, four bonding electron pairs within (Cd4Pn) units all have high electron density and localization. It is known that highly localized valence electrons at VBM can decrease band dispersion (BD). Since VBM was dominantly composed of P-3p/As-4p, these localized valence electrons flatten VBM with BD not exceeding 0.17 eV (Fig. 3a insert). That is remarkable compared to Ba2Si3P6 (BD = 0.4 eV, Eg = 1.88 eV), BaGe2P2 (BD = 0.9 eV, Eg = 1.32 eV), LaSiP3 (BD = 1.2 eV, Eg =1.36 eV) and CaCd2P2 (BD = 1.0 eV, Eg = 1.55 eV), which all have high valence band dispersion, resulting small band gap [31–34]. As a consequence, the covalency and ionicity of system achieve a balance, and the metallicity is maximally reduced, which leads to the effective enhancement of band gaps.

Under the restriction of Kleinman's symmetry, CPB has three independent nonvanishing second-order susceptibility tensor components, namely, d31, d32, and d33, which were 14.67, 1.39, -7.05 pm/V@2050 nm, respectively (Fig. 4a). The largest tensor components, d31 was comparable to that of AgGaS2 (d14 = 13.7 pm/V), accordant with the measured SHG intensity. The sole NLO coefficient of CAB, d14 is only 1.74 pm/V@2050 nm (Fig. S7a in Supporting information), which supports that there is no SHG response observed. To further study structure-NLO performance correlations, the geometrical factor g and structural criterion C of (Cd4Pn) and (CdBr4) tetrahedron groups was calculated based on anionic group theory [35], which can represent arrangement level along largeset NLO susceptibility component (C= ğijk max, ranges from 0 to 1 which represents most unfavorable and most favorable arrangement of groups, respectively.). CPB has C value of 0.62, well consistent with its begign SHG effect and calculated d33. While the C value of CAB is only 0.01, further supports that CAB has a very small calculated d14 and no observed SHG response (Table S7 in Supporting information). The SHG-weighted density of d31 was performed to visualize the SHG contribution of groups or atoms. The plot (Fig. 4b) showed that both (Cd4P) and (CdBr4) groups have an essential contribution to NLO process. The Bader charge analysis uncovered that the concrete SHG contribution of (Cd4P) and (CdBr4) groups 69.6% and 30.4%, respectively. The calculated birefringence of CPB and CAB were 0.043 and 0.045@2050 nm, respectively (Fig. 4c and Fig. S7b in Supporting information). The shortest phase-matching wavelength of CPB is ca. 940 nm, which ensured it could be pumped by a mature 1064 nm laser source. Furthermore, we also measured the birefringence of CPB using a cross-polarizing microscope [36]. The thickness of the measured crystal is 8.5 µm, and the interference color of cross polarized light was the first order blue corresponding to the retardation of 750 nm. Based on the formula R = Δn × d (the detailed physical principle is described in Supporting information), the birefringence Δn was evaluated as 0.088 under visible light, consistent with the calculated value of 0.094@532 nm (Fig. 4d).
In summary, two halidepnictides with wide-band gap, [Cd2P]2[CdBr4] (CPB) and [Cd2As]2[CdBr4] (CAB) were synthesized based on halogen-assisted octet binding electrons construction of pnictogens (Pn). The four valence electrons pairs of Pn with high electron density and localization were formed by four polar covalent Cd-P bonds, creating an extremely flat valence band maximum. Finally, the delicate balance of the covalency and ionicity was successfully realized by the synergy of [Cd4Pn2] and [CdBr4] modules, contributing to the increase of band gap. Remarkably, CPB realizes a widest band gap among Cd-containing pnictides, and also exhibits a SHG effect of 1.2 × AgGaS2, moderate birefringence (0.088@visible light and calcd. 0.043@2050 nm) and a wide IR transmission range (up to ~13.6 µm), indicates it is a promissing NLO crystal. Furthermore, the octet binding electron construction strategy may provide important guidance for designing wide band-gap NLO pnictides.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 22305174, 22375147, 52332001, 51890862, 51902308 and 21921001), the Natural Science Foundation of Fujian Province (No. 2021J05097), and the Natural Science Foundation of Tianjin City (No. 22JCYBJC01380).
Supplementary material associated with this article can be found, in the online version, at doi:
A. Rudenko, P. Rosenow, V. Hasson, et al., Optica 7 (2020) 115–122. doi: 10.1364/optica.382054
D. Woodbury, A. Goffin, R. Schwartz, et al., Phys. Rev. Lett. 125 (2020) 133201. doi: 10.1103/PhysRevLett.125.133201
Z. Chang, L. Fang, V. Fedorov, et al., Adv. Opt. Photon. 14 (2022) 652–782. doi: 10.1364/aop.454797
J. Chen, L. Xiong, L. Chen, et al., J. Am. Chem. Soc. 140 (2018) 14082–14086. doi: 10.1021/jacs.8b10209
C. Qian, X. Duan, B. Yao, et al., Opt. Express 26 (2018) 30195–30201. doi: 10.1364/oe.26.030195
G.Y. Liu, Y. Chen, B.Q. Yao, et al., Opt. Lett. 45 (2020) 2347–2350. doi: 10.1364/ol.389603
F. Ge, X. Han, J. Xu, Laser Photon. Rev. 15 (2021) 2000514. doi: 10.1002/lpor.202000514
P. Liu, W. Li, F. Qi, et al., Appl. Opt. 60 (2021) 10984–10987. doi: 10.1364/ao.446136
C. Wu, X. Jiang, L. Lin, et al., Angew. Chem. Int. Ed. 133 (2021) 22621–22627. doi: 10.1002/ange.202108886
M. Mutailipu, M. Zhang, B. Zhang, et al., Angew. Chem. Int. Ed. 130 (2018) 6203–6207. doi: 10.1002/ange.201802058
Z. Zhang, Y. Wang, B. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 6577–6581. doi: 10.1002/anie.201803392
S. Zhao, X. Yang, Y. Yang, et al., J. Am. Chem. Soc. 140 (2018) 1592–1595. doi: 10.1021/jacs.7b12518
P. Gong, F. Liang, L. Kang, et al., Coordin. Chem. Rev. 380 (2019) 83–102. doi: 10.1016/j.ccr.2018.09.011
B.W. Liu, X.M. Jiang, H.Y. Zeng, et al., J. Am. Chem. Soc. 142 (2020) 10641–10645. doi: 10.1021/jacs.0c04738
A. Abudurusuli, J. Huang, P. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 24131–24136. doi: 10.1002/anie.202107613
X. Hao, M. Luo, C. Lin, et al., Angew. Chem. Int. Ed. 60 (2021) 7621–7625. doi: 10.1002/anie.202016372
H. Chen, M.Y. Ran, S.H. Zhou, et al., Chin. Chem. Lett. 34 (2023) 107838. doi: 10.1016/j.cclet.2022.107838
M. Luo, X. Li, X. Jiang, et al., Chin. Chem. Lett. 35 (2024) 109108. doi: 10.1016/j.cclet.2023.109108
J. Chen, Q. Wu, H. Tian, et al., Adv. Sci. 9 (2022) 2105787. doi: 10.1002/advs.202105787
K.E. Woo, J. Wang, K. Wu, et al., Adv. Funct. Mater. 28 (2018) 1801589. doi: 10.1002/adfm.201801589
J. Chen, H. Chen, F. Xu, et al., J. Am. Chem. Soc. 143 (2021) 10309–10316. doi: 10.1021/jacs.1c03930
J. He, Y. Guan, V. Trinquet, et al., Adv. Opt. Mater. 11 (2023) 2301060. doi: 10.1002/adom.202301060
A. Roy, M. Chhetri, S. Prasad, et al., ACS Appl. Mater. Interfaces 10 (2018) 2526–2536. doi: 10.1021/acsami.7b15992
J. Chen, C. Lin, D. Zhao, et al., Angew. Chem. Int. Ed. 59 (2020) 23549–23553. doi: 10.1002/anie.202010319
X.M. Jiang, G.E. Wang, Z.F. Liu, et al., Inorg. Chem. 52 (2013) 8865–8871. doi: 10.1021/ic401056t
A. Roy, A. Singh, S.A. Aravindh, et al., Angew. Chem. Int. Ed. 131 (2019) 7000–7005. doi: 10.1002/ange.201900936
M.R. Pielmeier, T. Nilges, Angew. Chem. Int. Ed. 60 (2021) 6816–6823. doi: 10.1002/anie.202016257
D.N. Purschke, M.R. Pielmeier, E. Üzer, et al., Adv. Mater. 33 (2021) 2100978. doi: 10.1002/adma.202100978
X.M. Jiang, M.J. Zhang, H.Y. Zeng, et al., J. Am. Chem. Soc. 133 (2011) 3410–3418. doi: 10.1021/ja107921a
S. Kurtz, T. Perry, J. Appl. Phys. 39 (1968) 3798–3813. doi: 10.1063/1.1656857
J. Chen, C. Lin, G. Peng, et al., Chem. Mater. 31 (2019) 10170–10177. doi: 10.1021/acs.chemmater.9b03863
J. Mark, J. Wang, K. Wu, et al., J. Am. Chem. Soc. 141 (2019) 11976–11983. doi: 10.1021/jacs.9b04653
Y. Sun, J. Chen, S. Yang, et al., Adv. Opt. Mater. 9 (2021) 2002176. doi: 10.1002/adom.202002176
Y. Sun, C. Lin, J. Chen, et al., Inorg. Chem. 60 (2021) 7553–7560. doi: 10.1021/acs.inorgchem.1c01052
C. Chen, Y. Wu, R. Li, Int. Rev. Phys. Chem. 8 (1989) 65–91. doi: 10.1080/01442358909353223
C. Wu, X. Jiang, L. Lin, et al., Angew. Chem. Int. Ed. 60 (2021) 22447–22453. doi: 10.1002/anie.202108886

Figure 1 (a) Crystal structure of CPB. (b) [Cd4Pn2]2+ covalent cationic framework. (c) The nearest and second nearest atomic coordination environments of P from two different views. (d) EDD isosurfaces of CPB at η = 0.55.

Figure 2 (a) UV-vis-NIR diffuse reflectance spectra. (b) Raman spectra. (c) Particle size-dependent SHG intensity curves of CPB. (d) SHG signal of CPB with a particle size of 250–325 µm.

Figure 3 (a) Calculated band structure, (b) PDOS, (c) EDD field distribution, (d) ELF field distribution of CPB.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: