Institute of Pharmaceutics, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China
b.
Key Laboratory of Novel Targets and Drug Study for Neural Repair of Zhejiang Province, School of Medicine, Hangzhou City University, Hangzhou 310015, China
c.
Department of Cardiology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
d.
MOE Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology, Department of Chemistry, Tsinghua University, Beijing 100084, China
e.
Emergency Department, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing 100730, China
Received Date:
23 November 2023 Accepted Date:
15 January 2024 Revised Date:
08 January 2024 Available Online:
15 October 2024
Abstract:
Photodynamic therapy (PDT) has garnered significant attention as a promising approach to cancer therapy, harnessing the combined benefits of localized light treatment and the accompanying host immune response. In this study, we engineered an immuno-enhanced PDT nanoplatform, denoted as HM@p-MOF (hybrid membrane@porphyrin-metal organic framework). The core porphyrin-MOF was cloaked with a hybrid membrane derived from B16F10 cancer cells and NK cells, resulting in enhanced stability. In both in vitro and in vivo experiments, our finding demonstrated that the hybrid membrane conferred dual targeting capabilities to the nanoplatform, leveraging the unique properties of the B16F10 membrane and NK membrane to augment immunogenic cell death (ICD) induced by photodynamic effects. Additionally, in conjunction with the immunomodulatory functions of the NK cell membrane, we observed an expansion of in situ immune infiltration leading to a systemic immune response. The HM@p-MOF nanoplatform exhibited the capacity to not only inhibit the growth of mouse melanoma but also suppress metastasis. This innovative HM@p-MOF nanoplatform present a viable strategy to enhance phototherapeutic efficacy for both localized and metastatic tumors. It provides a direction for the fabrication of biomimetic nanomedicines possessing immuno-modulatory function.
The use of immune cells as therapeutic agents in cancer treatment has garnered substantial research and clinical interest due to their potential in tumor targeting, immune stimulation, and tumor elimination [1,2]. Among these immune cells, cytotoxic natural killer (NK) cells, recognized as crucial components of the innate immune system, play a pivotal role in responding to pathogenic conditions and exert protective functions in various inflammatory contexts. They possess the ability to eliminate transformed cells and regulate innate immunopathology through proteins associated with antigen capture/presentation and immunomodulation, thereby contributing to antitumor immunity. The immunomodulatory functions of NK cells are enticing. NK cells can exert influence over other lymphocyte populations and even endothelial cells. NK cells promote the maturation and activation of dendritic cells (DCs) through the secretion of cytokines, including interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α). Furthermore, NK cells interact directly with T and B cells, influencing adaptive immune responses through a combination of cytokines and cell surface receptors [3]. Extensive analyses have revealed that a majority of membrane proteins in human NK cells are linked to immunity, underscoring the immunomodulatory role of the NK cell membrane [4]. Surface proteins on NK cells possess the ability to recognize tumors, polarize macrophages, and activate immune cells in antitumor therapy [5–7]. For example, NKG2D, a prominent activating receptor on NK cell membranes, can engage with major histocompatibility complex (MHC) class I-like ligands, triggering the activation of T cells and macrophages [8]. Although NK cell membranes may not replicate the strength of living cells, they have proven effective due to the presence of membrane proteins [9]. Hence, the NK cell membrane, retaining key biological characteristics of intact cells without being deactivated by the tumor microenvironment (TME), holds promise as a potential immunomodulator.
The primary objectives in anti-tumor studies are to reduce the cancer burden and minimize adverse effects. Traditional treatments, such as targeted therapy and chemotherapy, have long been the mainstays of clinical oncology medical therapy. However, the off-target effects often compromise patient well-being, contribute to overall debilitation, and limit therapeutic effectiveness. To mitigate adverse effects and enhance the efficacy of tumor treatment, researchers have explored various in situ approaches. Photodynamic therapy (PDT) stands out as a minimally invasive treatment that capitalizes on light irradiation directed at a photosensitizer (PS). PDT holds tremendous potential in cancer treatment due to its low toxicity, high precision, and spatiotemporally controlled characteristics [10–12]. Notably, PDT can not only directly eradicate cancer cells by generating reactive oxygen species (ROS) through the PS but also damage tumor blood vessels and induce immunogenic cell death (ICD) [13,14]. ICD results from the death of tumor cells, releasing various damage-associated molecular patterns (DAMPs), including calreticulin (CRT), high mobility group protein box 1 (HMGB1), and adenosine triphosphate (ATP), which activate cytotoxic T cells (CD8+ T) and trigger specific immune responses to further kill the cancer cells [15,16]. Hence, the combination of PDT and locally induced ICD generates an expanded synergistic effect. The effective synergism of PDT and ICD must overcome the challenge of insufficient systemic immune response. Consequently, achieving target accumulation of the PS and incorporating immunoadjuvants become essential for more effective phototherapy aimed at eradicating cancer cells.
Benefiting from nanotechnology-driven membrane engineering, cell membrane camouflage emerges as a pivotal strategy to overcome both biological and physical barriers in vivo, enhancing biocompatibility and enhancing the targeting efficiency of photosensitizers (PS). In contrast to conventional physicochemical modifications, cell membranes, offer diverse advantages as biological source agents, including low toxicity, excellent biocompatibility, inherent tumor-targeting capabilities, and the potential to trigger tumor-specific immune responses owing to the presence of numerous functional proteins on their surfaces. For instance, the erythrocyte membrane facilitates the evasion of camouflaged nanosystems from immune system clearance, primarily attributed to the CD47 protein on its surface [17]. Additionally, cancer cell membranes possess the ability to induce tumor-specific immune responses while recognizing homologous cell lines and tumors [18–20]. Furthermore, to introduce immunomodulatory functionalities, immune cell membranes such as macrophage membranes are harnessed as modulators, enabling the formation of hybrid membranes [21–23]. The feasible fusion of various cell membranes offers the possibility of streamlining the composition of multifunctional nanosystems, addressing the limitations of individual membranes, and conferring them with customizable functions for synergistic cancer therapy [24–27].
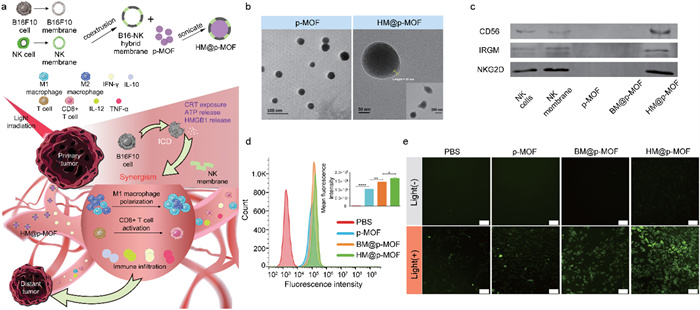
To integrate tumor targeting and immune activation, we conceived a novel nanoplatform named HM@p-MOF (hybrid membrane@porphyrin-metal organic framework). In this platform, the core component comprises supramolecular porphyrin-MOF, synthesized using meso-tetra(4-carboxyphenyl)porphyrin (TCPP) as the photodynamic organic ligand and Zr as the metal cluster. The surface of this core structure is enveloped by a hybrid membrane derived from B16F10 cancer cells and NK cells. The inherent structural advantages of MOFs contribute to enhancing the stability of TCPP. The hybrid membrane is achieved by doping NK cell membrane into B16F10 cell membrane, creating a PS synergistic modification that combines tumor targeting and immune activation. This unique configuration bestows the nanoplatform with superior targeting capabilities, thereby amplifying ICD induced by the photodynamic effects. Additionally, it facilitates the expansion of in situ immune infiltration into a systemic immune response through the immunomodulatory properties of the NK cell membrane. Consequently, this innovative design simultaneously suppresses the growth and metastasis of mouse melanoma (Fig. 1a).
Figure 1
Figure 1.
(a) Schematic illustration of HM@p-MOF nanoplatform for immuno-enhanced tumor phototherapy. Immune activation is induced as a result of augmented immunogenic cell death (ICD) as well as the synergism of NK cell membrane. The light treatment of primary tumor functions effectively in both primary tumor suppression and distant tumor inhibition. TEM image of (b) p-MOF and HM@p-MOF. (c) Western blot images of p-MOF, BM@p-MOF and HM@p-MOF. (d) Flow cytometry analysis of B16F10 cells incubated with p-MOF, BM@p-MOF and HM@p-MOF for 4 h. Fluorescence intensity distribution image and MFI of B16F10 cells of each group (n = 3, P < 0.05, **P < 0.01, **** P < 0.0001). (e) ROS generated by p-MOF, BM@p-MOF and HM@p-MOF under light irradiation detected by DCFH-DA (scale bar = 200 µm).
The photosensitizer TCPP with its highly lipophilic structure exhibits aggregation in aqueous solutions, thus restricting the biological utility. The supramolecular structure of MOF makes TCPP exhibit the capability to circumvent aggregation and self-quenching under physiological conditions. Additionally, the presence of ROS diffusion channels within the supramolecular MOF is able to enhance photodynamic performance [28]. Thus, the supramolecular MOF is constructed as PS instead of free TCPP. The synthesis of porphyrin-MOF followed a solvothermal method as reported previously [29]. Dynamic light scattering (DLS) measurements revealed a hydrodynamic diameter of 57.8 ± 1.2 nm and a zeta potential of 33.1 ± 0.4 mV for the MOF nanoparticles (Figs. S1a and b in Supporting information). PXRD analysis confirmed the MOF's crystal structure similarity to the reported PCN-224 (Figs. S1c in Supporting information). The BET surface area and average pore size of the MOF were determined to be 799.2994 m2/g and 4.7548 nm, respectively (Figs. S1d and e in Supporting information). The structural characteristics of the MOF were validated by FT-IR (-OH (3416 cm−1), C=O (1654 cm−1), Zr-OH (654 cm−1)) (Fig. S1f in Supporting information), while successful organic ligand TCPP insertion was confirmed by UV–vis and fluorescence spectroscopy (Fig. S2 in Supporting information). The absorption spectra of TCPP and MOF contained the porphyrin Soret band at 420 nm and Q band between 500 nm and 700 nm, while the spectrum of MOF was slightly red shifted compared to TCPP, indicating that the TCPP was attached to the Zr6 cluster. The fluorescence emission spectra of TCPP and MOF were quite similar with a peak maximum at 653 nm.
The hybrid membrane was formed through coextrusion of B16F10 cell membrane and primary mouse NK cell membrane at a protein ratio of 10:1. DLS measurements indicated a hydrodynamic diameter of 305.4 ± 4.0 nm and a zeta potential of −19.6 ± 0.2 mV for the hybrid membrane. The morphology of hollow membrane vesicles in the hybrid membrane was visualized using a fluorescence microscope after staining with the cell membrane fluorescence probe DiO (Fig. S3 in Supporting information).
HM@p-MOF was synthesized through ultrasonication in an ice bath. To assess the physicochemical properties of membrane-modified p-MOF, various characterizations were performed using p-MOF, B16F10 cell membrane-camouflaged MOF (BM@p-MOF), and hybrid membrane-camouflaged MOF (HM@p-MOF). UV–vis and fluorescence spectra demonstrated that membrane coating did not affect the optical properties of p-MOF (Fig. S2). Transmission electron microscopy (TEM) analysis (Fig. 1b) revealed that HM@p-MOF featured membrane-wrapped nanoparticles with an outer membrane thickness of approximately 20 nm compared to uncoated p-MOF. The colocalization relationship between DiO-stained hybrid membrane and p-MOF under a fluorescence microscope further confirmed the successful construction of the cell membrane-camouflaged nanoplatform (Fig. S4 in Supporting information). Furthermore, to assess nanoparticle stability after camouflaging, we measured size and zeta potential in both water and a simulated physiological condition (PBS with 10% FBS). Results indicate that cell membrane coating significantly enhanced nanoplatform stability in the simulated physiological environment (Fig. S5 in Supporting information).
SDS-PAGE analysis demonstrated that when NK cell membrane was incorporated into B16F10 cell membrane, both BM@p-MOF and HM@p-MOF retained protein bands consistent with those on the surface of B16F10 membrane (Fig. S6 in Supporting information). Subsequently, western blot assays also confirmed the presence of functional proteins such as CD56, IRGM, and NKG2D in HM@p-MOF (Fig. 1c). These findings further corroborated the successful establishment of the membrane-coated nanoplatform. The retention of B16F10 homologous proteins and functional proteins from the NK cell membrane forms the basis for the biomimetic nanoparticles to fulfill their roles in tumor targeting and immune stimulation.
The essential prerequisite for the photodynamic effect of the nanoplatform is its uptake by tumor cells. B16F10 cells were exposed to PBS, p-MOF, BM@p-MOF, or HM@p-MOF at 37 ℃ for 4 h, and the cellular internalization of the nanoplatforms was assessed based on the red fluorescence of porphyrin. Confocal laser scanning microscopy (CLSM) results (Fig. S7 in Supporting information) demonstrated that both uncoated p-MOF and biomimetic nanoparticles were internalized by B16F10 cells via endocytosis, with the red fluorescence of p-MOF coinciding with the green fluorescence of lysosomes. To further quantify the potential of the hybrid membrane to enhance cellular uptake, flow cytometry was employed to analyze the fluorescence intensity of the cells. Consistent with the CLSM findings, the fluorescence peak position of cells in the nanoparticle-treated groups was notably shifted to the right compared to the PBS group, indicative of successful internalization of these nanoplatforms (Fig. 1d). Importantly, the mean fluorescence intensity (MFI) of p-MOF, BM@p-MOF, and HM@p-MOF (1.02 × 105, 1.43 × 105, and 1.65 × 105, respectively) was significantly higher than that of the PBS group (2.08 × 103), demonstrating a substantial enhancement in cellular uptake. This result suggests that the NK cell membrane-doped B16F10 cell membrane promotes cellular uptake, likely through a dual internalization mechanism involving homologous targeting by the B16F10 cell membrane and ligand-mediated tumor targeting by the NK cell membrane.
MTT assay results revealed that B16F10 cells maintained a high cell survival rate (> 90%) even when exposed to p-MOF, BM@p-MOF, and HM@p-MOF at a high concentration of 100 µg/mL (Fig. S8a in Supporting information), underscoring the low dark cytotoxicity and excellent safety of these nanoplatforms. Under light irradiation, nanoparticles exhibited dose-dependent cytotoxicity, with the survival rate of cells treated with p-MOF, BM@p-MOF, and HM@p-MOF decreasing stepwise at the same concentration, indicative of increased intracellular ROS-induced cell death (Fig. S8b in Supporting information). To evaluate singlet oxygen generation in vitro, a fluorescent probe, DCFH-DA, was employed. Intracellular ROS oxidize the hydrolysate of DCFH-DA into DCF, emitting green fluorescence. As depicted in Fig. 1e, fluorescence signals were detected in p-MOF, BM@p-MOF, and HM@p-MOF-treated cells after light irradiation, compared to groups without light, confirming the light-triggered ROS generation capacity of the nanoplatforms. Notably, B16F10 cells treated with HM@p-MOF exhibited the widest range and highest intensity of fluorescence signals under the same light exposure time. This is possibly attributed to more efficient internalization of HM@p-MOF, leading to higher PS concentration within the cells and enhanced ROS production.
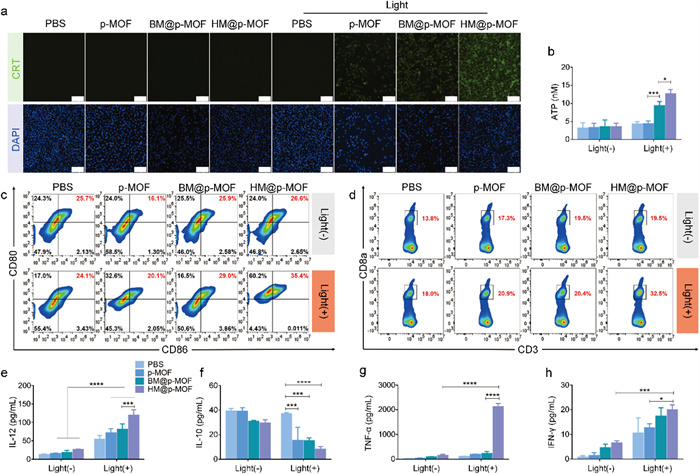
The photodynamic effect can induce ICD of tumor cells, characterized by increased exposure of CRT and ATP release, both of which are crucial signals for antigen uptake and presentation [30]. Fig. 2a demonstrated fluorescence signals exclusively under light irradiation in the nanoparticle-treated group, indicating CRT exposure induced by light-triggered ICD. Furthermore, at the same light exposure duration, the fluorescence intensity of cells treated with p-MOF, BM@p-MOF, and HM@p-MOF significantly increased (Fig. S9 in Supporting information), signifying a positive correlation between CRT exposure and photodynamic cytotoxicity of the nanoplatforms. ATP concentrations in the cell culture supernatants of BM@p-MOF and HM@p-MOF-treated groups subjected to light irradiation were markedly higher than that of the p-MOF group (Fig. 2b), revealing that the hybrid membrane-camouflaged nanoparticle most effectively promoted the production and release of ATP from apoptotic B16F10 cells. The observed high CRT exposure on B16F10 cells and elevated extracellular ATP concentration suggest that the HM@p-MOF system significantly enhances photodynamic-induced ICD effects in vitro.
Figure 2
Figure 2.
(a) CRT exposure in B16F10 cells treated with p-MOF, BM@p-MOF and HM@p-MOF under light irradiation detected by CLSM (scale bar = 200 µm). (b) Extracellular ATP secreted by B16F10 cells treated with p-MOF, BM@p-MOF and HM@p-MOF under light irradiation (n = 3, P < 0.05, ***P < 0.001). (c) Flow cytometric profiles of mature BMDCs (CD80+CD86+) after coculture with p-MOF, BM@p-MOF and HM@p-MOF treated (under light irradiation) culture supernatant from B16F10 cells for 24 h. (d) Flow cytometric profiles of CD8+ T cells (CD3+CD8+) after coculture with different groups of mature BMDCs for 24 h. Cytokines secreted in vitro detected by ELISA: (e) IL-12, (f) IL-10 and (g) TNF-α concentration in BMDCs culture supernatant; (h) IFN-γ concentration in coculture supernatant of BMDCs and spleen lymphocytes (n = 3, P < 0.05, ***P < 0.001, ****P < 0.0001).
HM@p-MOF was confirmed to possess potent phototoxicity, effectively inducing CRT exposure and ATP release from B16F10 cells. Consequently, the culture medium from HM@p-MOF-treated B16F10 cells exposed to light was enriched with a multitude of tumor antigens and immune-activating factors, thus serving as an antigenic solution capable of stimulating DC maturation. Fig. 2c illustrates that the tumor antigen solution from each group without light stimulation failed to induce DC maturation. This outcome underscores the low immunogenicity of culture medium lacking ICD-related immunostimulatory factors and the absence of photodynamic effects. However, with light irradiation, the percentage of mature bone marrow-derived dendritic cells (BMDCs) (CD80+CD86+) in the culture medium from B16F10 cells treated with p-MOF, BM@p-MOF, and HM@p-MOF reached 20.1%, 29.0%, and 35.4%, respectively. This increase suggests that DC maturation was enhanced, likely due to the promotion of ICD. Notably, the higher proportion of mature DCs in the culture medium from HM@p-MOF+light-treated B16F10 cells compared to that in the BM@p-MOF+light group further underscores the superior ICD-inducing effect of the hybrid membrane when compared to the single B16F10 membrane-coated group. This is attributed to enhanced internalization and phototoxicity.
Antitumor immunity, characterized by the generation of cytotoxic T cells (CD8+ T cells), is realized through the antigen presentation process involving DCs and T cells. As the tumor antigen solution generated via HM@p-MOF photodynamic action effectively stimulates BMDC maturation, we further explored the downstream effects on T cells. Fig. 2d reveals a significantly higher proportion of CD8+ T cells cocultured with BMDCs stimulated by the HM@p-MOF group exposed to light, indicating that mature BMDCs in this treatment context possess enhanced antigen presentation efficiency. This subsequently triggers T cell activation and increases the proportion of CD8+ T cells.
Furthermore, cytokine levels in the culture supernatants of BMDCs were assessed using ELISA kits. In Figs. 2e-h, the concentration of interleukin-12 (IL-12) in each nanoplatform group exposed to light was significantly higher than in the corresponding group without light, indicating that photodynamic-induced ICD causes IL-12 release from BMDCs. Among the light-exposed groups, the HM@p-MOF group exhibited significantly higher IL-12 concentrations, suggesting that the HM@p-MOF-induced ICD environment was the most effective in promoting IL-12 secretion from BMDCs. Similar upward trends were observed for TNF-α and IFN-γ, mirroring the IL-12 results. Additionally, IL-10 concentrations were significantly lower in the light-exposed groups compared to the groups without light, reflecting the positive impact of photodynamic-induced ICD on inhibiting IL-10 production by BMDCs and thereby alleviating the inhibitory inflammatory environment. Among the light-exposed groups, the culture supernatant of BMDCs stimulated with the tumor antigen solution from HM@p-MOF exhibited the lowest level of IL-10. These results suggest that the HM@p-MOF-induced immunoenvironment increases the proportion of mature BMDCs by mitigating the inhibitory effect of IL-10 on DC maturation.
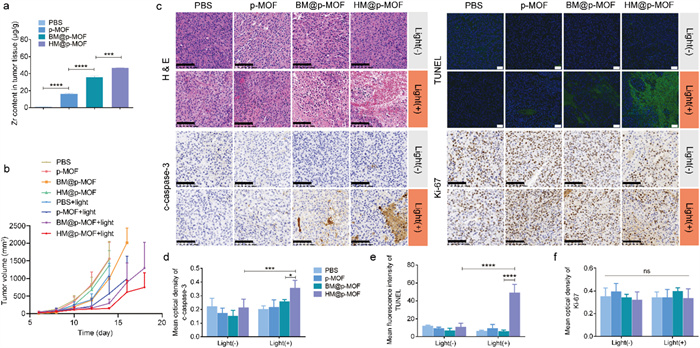
An ideal photosensitizer must exhibit effective in vivo tumor targeting capabilities, enabling the selective destruction of solid tumor tissues upon light excitation while minimizing phototoxicity to normal tissues. We investigated the in vivo distribution of the nanoplatforms using a live fluorescence imaging system based on the inherent red fluorescence of p-MOF. As depicted in Fig. S10 (Supporting information), fluorescence signals were detected at the tumor site 12 h after tail vein injection of BM@p-MOF and HM@p-MOF (indicated by the yellow arrow), diminishing by 24 h. This observation demonstrates that membrane-camouflaged nanoplatforms can successfully reach the tumor site 12 h post-administration, with maximum enrichment at that time. Subsequent dissection of mice at 24 h revealed the main organs (Fig. S11 in Supporting information). The pronounced lung fluorescence in the p-MOF group strongly supports its dispersed and non-targeted in vivo distribution, as well as the lung retention phenomenon associated with p-MOF. Conversely, lung fluorescence in the BM@p-MOF and HM@p-MOF groups was considerably weaker than in the p-MOF group at 24 h post-administration, attributed to the membrane modification, which enhances tumor-targeting capabilities and the in vivo bioavailability of nanoplatforms. To further quantitatively assess the tumor-targeting ability of different nanosystems, we employed ICP-MS to determine the Zr content in tumor tissues 12 h post-administration. The results (Fig. 3a) revealed a significant increase in Zr content per gram of tumor tissue in the p-MOF, BM@p-MOF, and HM@p-MOF groups, confirming the enhanced enrichment of nanoparticles at the tumor site. These results collectively affirm the excellent in vivo tumor-targeting ability of HM@p-MOF. Consequently, we scheduled light irradiation 12 h after tail vein injection for in vivo treatment.
Figure 3
Figure 3.
(a) Zr concentration in tumor tissue dissected from tumor bearing mice at 12 h after i.v. injection of p-MOF, BM@p-MOF and HM@p-MOF detected by ICP-MS (n = 3, ***P < 0.001, ****P < 0.0001). (b) Average tumor volume of the mice after different administrations (n = 5). (c) H & E (scale bar = 100 µm), c-caspase-3 (scale bar = 100 µm), Ki-67 (scale bar = 100 µm) immunohistochemical staining and TUNEL (scale bar = 50 µm) immunofluorescence staining images of tumor sections from the mice after different treatments. Mean optical density of (d) c-caspase-3 and (f) Ki-67 staining. (e) Mean fluorescence intensity of TUNEL staining. P < 0.05, ***P < 0.001, ****P < 0.0001.
We established B16F10 tumor-bearing mice for the antitumor study. Following tail vein administration of the nanoplatforms and subsequent light irradiation, tumor volume measurements demonstrated that the BM@p-MOF+light and HM@p-MOF+light groups effectively suppressed tumor growth, while other groups exhibited negligible differences in tumor growth compared to the PBS group (Fig. 3b and Fig. S12 in Supporting information). Significantly, the average tumor volume in the HM@p-MOF group was lower than that in the BM@p-MOF group at the end of treatment. This difference may be attributed to the immunomodulatory proteins present in the NK membrane, which exerted an immune activation effect and resulted in tumor growth inhibition compared to the single B16F10 membrane-coated nanoparticles. Histological assessments including hematoxylin-eosin (H&E) staining (Fig. 3c), cleaved caspase-3 (Figs. 3c and d), and TdT-mediated dUTP Nick-End Labeling (TUNEL) staining (Figs. 3c and e) of tumor tissue sections revealed the most pronounced apoptosis and necrosis in the tumor tissue of the HM@p-MOF group under light irradiation. However, Ki-67 staining results were similar across all groups, indicating that the main mechanism of tumor growth suppression was not based on proliferation inhibition but rather on apoptosis and necrosis (Figs. 3c and f). Importantly, there was no significant decrease in animal weight during the entire treatment process (Fig. S13 in Supporting information), and H & E staining of major organs indicated negligible tissue damage (Fig. S14 in Supporting information). Collectively, these findings indicate that the HM@p-MOF nanoplatform is safe for in vivo applications and effectively inhibits tumor growth when combined with light irradiation.
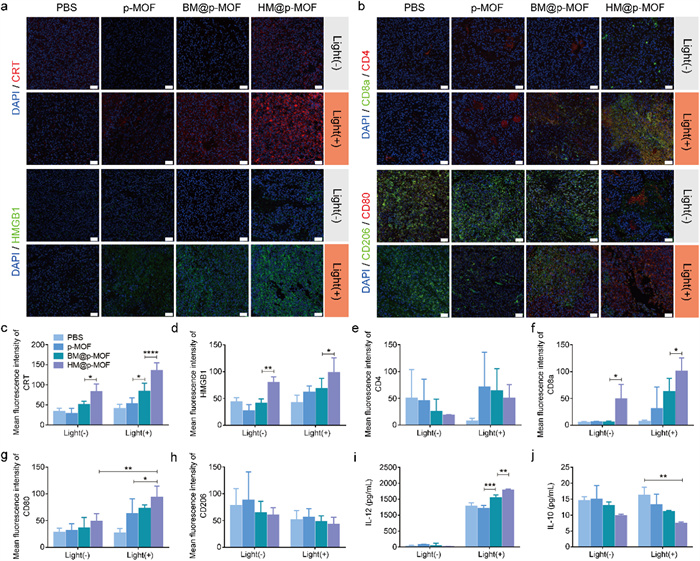
To assess the in vivo induction of ICD by nanoplatforms, we performed immunofluorescence staining of CRT and HMGB1 antibodies on tumor tissue sections post-treatment. In non-irradiated groups, only the HM@p-MOF group exhibited limited CRT and HMGB1 expression (Figs. 4a, c and d), suggesting that NK membrane components may stimulate the immune system in the TME and induce ICD. In the light-irradiated groups, p-MOF, BM@p-MOF, and HM@p-MOF groups all showed increased fluorescence signals compared to the PBS group. Among them, the HM@p-MOF group exhibited the widest range and strongest fluorescence intensity of CRT and HMGB1. Thus, the HM@p-MOF nanoplatform can effectively induce in vivo ICD under light irradiation.
Figure 4
Figure 4.
(a) CRT, HMGB1, (b) CD4+ (red)/CD8+ (green) T cells and M1 (red)/M2 (green) macrophage immunofluorescence staining of tumor sections from tumor bearing mice of different groups at the end of various treatments (scale bar = 50 µm, blue represents nuclei stained by DAPI). Mean fluorescence intensity of (c) CRT and (d) HMGB1 calculated by Image J (*P < 0.05, **P < 0.01, ****P < 0.0001). Mean fluorescence intensity of (e) CD4, (f) CD8a, (g) CD80 and (h) CD206 calculated by Image J (*P < 0.05, **P < 0.01). (i) IL-12 and (j) IL-10 concentration in tumor tissue lysis supernatant from tumor bearing mice of different groups at the end of various treatments detected by ELISA (n = 3, **P < 0.01, ***P < 0.001).
Having demonstrated that the hybrid membrane can stimulate the activation of immune cells in vitro by enhancing the ICD effect, and that HM@p-MOF induces the most robust ICD in vivo, we proceeded to investigate the role of the HM@p-MOF nanoplatform in stimulating immune cells and alleviating the immunosuppressive state of the TME in vivo. Immunofluorescence staining (CD4/CD8a; CD80/CD206) was performed on tumor tissue sections post-treatment to assess the activation of T cells and macrophages in the TME. In Figs. 4b and e-h, only the HM@p-MOF group exhibited expression of CD8a and CD80, indicating that the NK membrane component acted as a modulator to stimulate the activation of cytotoxic T cells (CD8+ T) and the polarization of M1 macrophages (CD80+). Under light irradiation, the fluorescence intensity of CD8a and CD80 in the HM@p-MOF group was significantly higher than in the other groups, indicating a high level of infiltration of CD8+ T cells and M1 macrophages in the TME. Fig. 4i demonstrated that the photodynamic effect could induce the infiltration of IL-12 in the TME. The significantly increased levels of IL-12 in the BM@p-MOF+light group and HM@p-MOF+light group indicated that the infiltration of IL-12 was associated with the enhancement of photodynamic effects induced by membrane-camouflaged nanoparticles. In Fig. 4j, although light treatment had no significant effect on the level of IL-10 in the TME, the concentration of IL-10 in the HM@p-MOF group was lower in both groups with or without light, suggesting that the NK membrane may play a crucial role in inhibiting the secretion of the anti-inflammatory cytokine IL-10 in the TME. These results further illustrate the proinflammatory function of the HM@p-MOF nanoplatform in the tumor microenvironment in vivo.
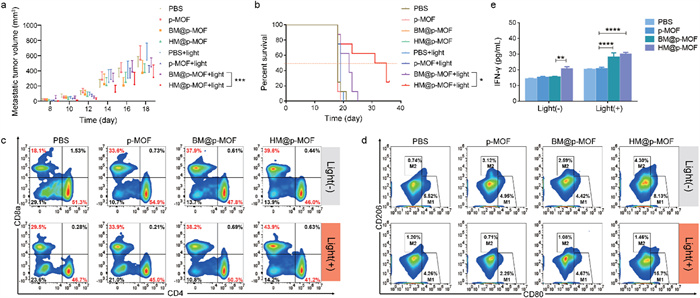
HM@p-MOF has demonstrated potent anti-tumor efficacy in vivo, inducing immune cell infiltration in the tumor microenvironment and regulating cytokine release. It is hypothesized that the therapeutic impact of HM@p-MOF extends beyond the local site, potentially activating a systemic immune response capable of inhibiting the growth of both primary and distant tumors. To investigate this hypothesis, we established a dual-tumor model, treating the primary tumor with intratumoral injections of the nanoplatform and monitoring metastatic tumor size to assess the anti-tumor metastasis effect of HM@p-MOF in vivo. Fig. S15 (Supporting information) illustrates that after three rounds of treatment, there was no significant weight loss in any of the groups, reaffirming the safety of the nanoplatforms in vivo. As shown in Fig. 5a and Fig. S16 (Supporting information), HM@p-MOF effectively inhibited the growth of both primary and metastatic tumors under light irradiation, significantly prolonging the survival time of the treated mice (Fig. 5b).
Figure 5
Figure 5.
(a) Metastatic tumor volume changes and (b) survival curve of bilateral B16F10 tumor model mice from different treatment groups (n = 8, P < 0.05, ***P < 0.001). Systemic immune response induced by nanoplatforms: flow cytometric profiles of (c) CD4+ T cells (CD3+CD4+) and CD8+ T cells (CD3+CD8a+), (d) M1-like macrophages (F4/80+CD80+) and M2-like macrophages (F4/80+CD206+) in spleens of bilateral B16F10 tumor model mice from different treatment groups. (e) IFN-γ concentration in the serum of bilateral B16F10 tumor model mice from different treatment groups detected by ELISA (n = 3, **P < 0.01, ****P < 0.0001).
To further evaluate the impact of HM@p-MOF on systemic immunity, we analyzed T cells and macrophages in the spleen of mice. Fig. 5c demonstrates that in the absence of light irradiation, the proportion of CD8+ T cells (CD3+CD8a+) increased in the p-MOF, BM@p-MOF, and HM@p-MOF groups, suggesting that the B16F10 membrane and NK-doped hybrid membrane themselves stimulated CD8+ T cell activation. After light irradiation, the proportion of CD8+ T cells increased significantly in the p-MOF, BM@p-MOF, and HM@p-MOF groups, indicating that HM@p-MOF further enhanced CD8+ T cell proliferation, likely through its improved ICD effect.
Similarly, Fig. 5d shows that in the absence of light, only the HM@p-MOF group exhibited an increased in M1 macrophages (F4/80+CD80+), indicating that the NK membrane promoted M1 macrophage polarization. Notably, the proportion of M1 macrophages in the p-MOF and BM@p-MOF groups did not increase significantly after light irradiation, suggesting that photodynamic effects alone may have limited impact on M1 macrophage polarization but may reduce the proportion of M2 macrophages (F4/80+CD206+). In the light-irradiated groups, only the HM@p-MOF group demonstrated an increased proportion of M1 macrophages (15.7%). This result indicates that the nanoplatform could enhance spleen M1 macrophage polarization through the combined immunomodulatory function of the NK membrane and the ICD effect induced by photodynamic therapy, thereby eliciting a systemic anti-tumor immune response.
The IFN-γ mediated pathway is one of the mechanisms through which NK cells inhibit tumors, closely related to surface proteins on NK membranes. In Fig. 5e, each administration group showed a significant increase in IFN-γ levels after light stimulation, indicating a photodynamic-induced promotion of IFN-γ secretion. Among them, the serum IFN-γ levels in the BM@p-MOF and HM@p-MOF groups were significantly higher than those in the p-MOF group, reflecting the enhanced photodynamic effect achieved through biomimetic nanoplatforms.
Moreover, in groups without light irradiation, the serum of mice in the HM@p-MOF group had significantly higher IFN-γ content. This highlights the immunostimulatory role of the NK membrane component in the hybrid membrane, which stimulated IFN-γ secretion. Collectively, these findings demonstrate that HM@p-MOF synergizes local photodynamic effects with immunostimulatory effects, resulting in effective inhibition of tumor metastasis.
Different from conventional hybrid membrane, this NK membrane fused hybrid membrane is non-uniform. Taking advantages of NK cell membrane, the HM@p-MOF achieves expanding photodynamic antitumor effect. Moving forward, further advances are needed to address the limitations of the current work. A more in-depth investigation is required to understand how the incorporation of NK cell membrane influences both local and systemic immune responses. This entails mechanistic studies to elucidate the precise role of NK cell membrane in immuno-enhanced phototherapy. Furthermore, the drug-free nanoplatform consisting of MOF materials is considered to exhibit exceptional loading capacity, presenting an opportunity to construct synergistic nanomedicines in combination with other agents such as drugs, proteins, and genes. Lastly, given the tunable nature of NK cell membrane doping ratios, a comprehensive study on different membrane fusion ratios is warranted to strike a balance between efficiency and safety. As such, the asymmetrical hybrid method we employed holds great promise for constructing multifunctional nanoplatforms, providing valuable experimental evidence for the investigation of hybrid membranes.
In conclusion, to enhance the biological applicability of photosensitizers and bolster the effectiveness of PDT, we have introduced an immuno-enhanced phototherapeutic supramolecular platform known as HM@p-MOF. HM@p-MOF employs the photosensitizer TCPP as the organic ligand within the MOF material, and further modifies its surface with a hybrid membrane comprising B16F10 cancer cell membrane and NK cell membrane. HM@p-MOF facilitates the internalization of MOF through hybrid membrane-mediated cell uptake, thereby enhancing the ICD effect and prompting the activation of immune cells in vitro through this reinforced ICD process. In a mice melanoma treatment model, HM@p-MOF demonstrated significant tumor inhibition and ICD induction effects in vivo. It triggered an immune response at the tumor site. In a metastasis model, HM@p-MOF elevated the proportions of CD8+ T cells and M1 macrophages in the mouse spleen, along with increased levels of IFN-γ in mouse serum. This was achieved through the collaborative action of the ICD effect and the NK cell membrane immunomodulatory function, resulting in the inhibition of both primary and metastatic tumors and the extension of the mice's survival. This research offers a novel approach to expand local immune infiltration into systemic antitumor immunity, ultimately suppressing the growth and metastasis of malignant tumors. It underscores the significance of the hybrid membrane camouflaging technique in enhancing immune responses in PDT, and provides a new idea for the biomimetic nanomedicines integrating membrane camouflaging with immuno-modulation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 22175107, 22305140, 51603184, 82270531), Zhejiang Provincial Natural Science Foundation of China (Nos. LY21E030002, LY22H020003), Scientific Research Foundation of Zhejiang University City College (No. J-202104), and Hangzhou Science and Technology Bureau of China (No. 202203B27).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109521.
[1]
Y. Xia, L. Rao, H. Yao, et al., Adv. Mater. 32 (2020) 2002054. doi: 10.1002/adma.202002054
[2]
S. Cheng, C. Xu, Y. Jin, et al., Adv. Sci. 7 (2020) 1903301. doi: 10.1002/advs.201903301
[3]
E. Vivier, E. Tomasello, M. Baratin, et al., Nat. Immunol. 9 (2008) 503–510. doi: 10.1038/ni1582
[4]
G. Deng, Z. Sun, S. Li, et al., ACS Nano 12 (2018) 12096–12108. doi: 10.1021/acsnano.8b05292
[5]
M.J. Smyth, Y. Hayakawa, K. Takeda, et al., Nat. Rev. Cancer 2 (2002) 850–861. doi: 10.1038/nrc928
[6]
D. Piccioli, S. Sbrana, E. Melandri, et al., J. Exp. Med. 195 (2002) 335–341. doi: 10.1084/jem.20010934
[7]
L. Chiossone, P.Y. Dumas, M. Vienne, et al., Nat. Rev. Immunol. 18 (2018) 671–688. doi: 10.1038/s41577-018-0061-z
[8]
A. Diefenbach, A.M. Jamieson, S.D. Liu, et al., Nat. Immunol. 1 (2000) 119–126. doi: 10.1038/77793
[9]
G. Deng, X. Peng, Z. Sun, et al., ACS Nano 14 (2020) 11452–11462. doi: 10.1021/acsnano.0c03824
[10]
D.E.J.G.J. Dolmans, D. Fukumura, R.K. Jain, Nat. Rev. Cancer 3 (2003) 380–387. doi: 10.1038/nrc1071
[11]
L. Benov, Med. Princ. Pract. 24 (2015) 14–28. doi: 10.1159/000362416
J. Park, Q. Jiang, D. Feng, et al., J. Am. Chem. Soc. 138 (2016) 3518–3525. doi: 10.1021/jacs.6b00007
[30]
A.D. Garg, D.V. Krysko, T. Verfaillie, et al., EMBO J. 31 (2012) 1062–1079. doi: 10.1038/emboj.2011.497
Figure 1
(a) Schematic illustration of HM@p-MOF nanoplatform for immuno-enhanced tumor phototherapy. Immune activation is induced as a result of augmented immunogenic cell death (ICD) as well as the synergism of NK cell membrane. The light treatment of primary tumor functions effectively in both primary tumor suppression and distant tumor inhibition. TEM image of (b) p-MOF and HM@p-MOF. (c) Western blot images of p-MOF, BM@p-MOF and HM@p-MOF. (d) Flow cytometry analysis of B16F10 cells incubated with p-MOF, BM@p-MOF and HM@p-MOF for 4 h. Fluorescence intensity distribution image and MFI of B16F10 cells of each group (n = 3, P < 0.05, **P < 0.01, **** P < 0.0001). (e) ROS generated by p-MOF, BM@p-MOF and HM@p-MOF under light irradiation detected by DCFH-DA (scale bar = 200 µm).
Figure 2
(a) CRT exposure in B16F10 cells treated with p-MOF, BM@p-MOF and HM@p-MOF under light irradiation detected by CLSM (scale bar = 200 µm). (b) Extracellular ATP secreted by B16F10 cells treated with p-MOF, BM@p-MOF and HM@p-MOF under light irradiation (n = 3, P < 0.05, ***P < 0.001). (c) Flow cytometric profiles of mature BMDCs (CD80+CD86+) after coculture with p-MOF, BM@p-MOF and HM@p-MOF treated (under light irradiation) culture supernatant from B16F10 cells for 24 h. (d) Flow cytometric profiles of CD8+ T cells (CD3+CD8+) after coculture with different groups of mature BMDCs for 24 h. Cytokines secreted in vitro detected by ELISA: (e) IL-12, (f) IL-10 and (g) TNF-α concentration in BMDCs culture supernatant; (h) IFN-γ concentration in coculture supernatant of BMDCs and spleen lymphocytes (n = 3, P < 0.05, ***P < 0.001, ****P < 0.0001).
Figure 3
(a) Zr concentration in tumor tissue dissected from tumor bearing mice at 12 h after i.v. injection of p-MOF, BM@p-MOF and HM@p-MOF detected by ICP-MS (n = 3, ***P < 0.001, ****P < 0.0001). (b) Average tumor volume of the mice after different administrations (n = 5). (c) H & E (scale bar = 100 µm), c-caspase-3 (scale bar = 100 µm), Ki-67 (scale bar = 100 µm) immunohistochemical staining and TUNEL (scale bar = 50 µm) immunofluorescence staining images of tumor sections from the mice after different treatments. Mean optical density of (d) c-caspase-3 and (f) Ki-67 staining. (e) Mean fluorescence intensity of TUNEL staining. P < 0.05, ***P < 0.001, ****P < 0.0001.
Figure 4
(a) CRT, HMGB1, (b) CD4+ (red)/CD8+ (green) T cells and M1 (red)/M2 (green) macrophage immunofluorescence staining of tumor sections from tumor bearing mice of different groups at the end of various treatments (scale bar = 50 µm, blue represents nuclei stained by DAPI). Mean fluorescence intensity of (c) CRT and (d) HMGB1 calculated by Image J (*P < 0.05, **P < 0.01, ****P < 0.0001). Mean fluorescence intensity of (e) CD4, (f) CD8a, (g) CD80 and (h) CD206 calculated by Image J (*P < 0.05, **P < 0.01). (i) IL-12 and (j) IL-10 concentration in tumor tissue lysis supernatant from tumor bearing mice of different groups at the end of various treatments detected by ELISA (n = 3, **P < 0.01, ***P < 0.001).
Figure 5
(a) Metastatic tumor volume changes and (b) survival curve of bilateral B16F10 tumor model mice from different treatment groups (n = 8, P < 0.05, ***P < 0.001). Systemic immune response induced by nanoplatforms: flow cytometric profiles of (c) CD4+ T cells (CD3+CD4+) and CD8+ T cells (CD3+CD8a+), (d) M1-like macrophages (F4/80+CD80+) and M2-like macrophages (F4/80+CD206+) in spleens of bilateral B16F10 tumor model mice from different treatment groups. (e) IFN-γ concentration in the serum of bilateral B16F10 tumor model mice from different treatment groups detected by ELISA (n = 3, **P < 0.01, ****P < 0.0001).

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
