Glutathione depletion based Pt(Ⅳ) hybrid mesoporous organosilica delivery system to conquer cisplatin chemoresistance: A “one stone three birds” strategy
Citation:
Linjie Ju, Zhongxi Huang, Qian Shen, Chan Fu, Shuanghe Li, Wenjie Duan, Chenfeng Xu, Weizhen An, Zhiqiang Zhai, Jifu Wei, Changmin Yu, Guoren Zhou. Glutathione depletion based Pt(Ⅳ) hybrid mesoporous organosilica delivery system to conquer cisplatin chemoresistance: A “one stone three birds” strategy[J]. Chinese Chemical Letters,
2024, 35(10): 109450.
doi:
10.1016/j.cclet.2023.109450
Glutathione depletion based Pt(Ⅳ) hybrid mesoporous organosilica delivery system to conquer cisplatin chemoresistance: A “one stone three birds” strategy
English
Glutathione depletion based Pt(Ⅳ) hybrid mesoporous organosilica delivery system to conquer cisplatin chemoresistance: A “one stone three birds” strategy
Department of Pharmacy, Jiangsu Cancer Hospital, The Affiliated Cancer Hospital of Nanjing Medical University, Jiangsu Institute of Cancer Research, Nanjing 210009, China
b.
Key Laboratory of Flexible Electronics (KLOFE), School of Flexible Electronics (Future Technologies) & Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech), Nanjing 211816, China
c.
Department of Oncology, Jiangsu Cancer Hospital, The Affiliated Cancer Hospital of Nanjing Medical University, Jiangsu Institute of Cancer Research, Nanjing 210009, China
d.
Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, College of Resources and Environmental Sciences, Nanjing Agricultural University, Nanjing 210095, China
Received Date:
21 November 2023 Accepted Date:
20 December 2023 Revised Date:
18 December 2023 Available Online:
15 October 2024
Abstract:
The occurrence of acquired resistance to cisplatin (DDP) that induces the toxic drug effects has always been a huge challenge and urgently needs to be resolved in the cancer treatment. The combination of anticancer drugs with different mechanisms can remarkably improve the chemotherapeutic efficiency. Given that glutathione (GSH) plays as the driving factors in the resistance of DDP, here we have firstly proposed a “three birds, one stone” based nanoplatform to achieve triple synergetic effects simultaneously addressing DDP resistance in non-small cell lung cancer (NSCLC). Specifically, we initially designed and synthesized a DDP prodrug [Pt(Ⅳ)] bridged silsesquioxane precursor (Pt-Si). Then Pt-Si and bis[3-(triethoxysilyl)propyl]diselenide (BTESePD) were integrated into the framework of mesoporous organosilica nanoparticles (MONs) to obtain a nanocarrier MONPt/Se. After loading with norcantharidin (NCTD) and modifying with the aptamer AS1411 based G-quadruplex (Apt), the Apt@NCTD@MONPt/Se exhibit impressive tumor homing capability. Once being endocytosed, (Ⅰ) the diselenide and -O-Pt(Ⅳ)-O- rich scaffold can be reduced by the excessive GSH, followed by (Ⅱ) breaking the redox homeostasis via GSH depletion and precise release of the DDP. Next, the encapsulated NCTD is also released along with the degradation of the nanocarriers thereby (Ⅲ) achieving the GSH depletion and synergistic anti-tumor effect of NCTD and DDP. Taken together, we believe this “one stone, three birds” strategy may be a promising paradigm to conquer drug resistance for clinical care.
Lung cancer often ranks one of the most prevalent malignancies of cancer-related death globally. Non-small cell lung cancer (NSCLC) accounts for 85% of the lung cancer cases, which has caused serious threat to the public [1]. Over the past decades, the main NSCLC treatments in clinic include surgery, radiotherapy, chemotherapy, and immunotherapy [2]. For most NSCLC patients, cisplatin (DDP)-based chemotherapy remains the first-line chemotherapeutic choice [3]. However, the occurrence of acquired resistance to DDP still restrict the application of DDP [4]. Hence, reversing drug resistance and reducing the toxic drug effects of DDP are urgent issues that need to be resolved for the NSCLC treatment [5].
Specifically, over-expressed thiol containing species in NSCLC cells, especially glutathione (GSH), are considered as the major driving factors in the resistance of DDP, which can cause lower cellular uptake and enhanced cell efflux of the drugs [6,7]. To overcome DDP-resistance, octahedral [Pt(Ⅳ)] complexes have been prepared by oxidizing the square planar [Pt(Ⅱ)] with H2O2 [8]. Such [Pt(Ⅳ)] based complexes are particularity suitable as the prodrugs because they are very stable outside the cancer cell and site-specifically reduced to generate corresponding toxic [Pt(Ⅱ)] drugs inside the tumor cells [9]. Additionally, [Pt(Ⅳ)] prodrugs can exhibit improved properties such as lipophilicity and active targeting by various modification of the axial ligands. To date, numerous [Pt(Ⅳ)] based derivatives have been synthesized by decorating the axial ligands of [Pt(Ⅳ)] with different bioactive agents, including approved drugs, enzyme inhibitors, pathway activators or suppressors, epigenetic modifiers, and even antimetabolites [10,11]. However, the therapeutic efficacy of these small molecules remains limited due to their inherent shortcomings such as short half-life, off-target effects and unpredictable toxic side effects [12]. Thus, integration of [Pt(Ⅳ)] with other therapeutic moieties into the nanoplatform will become a preferred alternative strategy to enhance the anticancer outcome of [Pt(Ⅳ)]-prodrugs.
Since Youyou Tu was awarded the 2015 Nobel Prize in physiology or medicine for the discovery of artemisinin, traditional Chinese medicines (TCMs) have attracted widespread concern [13]. Numerous evidences indicate that TCMs possess the advantages of multi-pathway, multi-target, multi-component anti-tumor pharmacological effects and low risk of adverse effects [14]. Norcantharidin (NCTD), a chemically demethylated analog of cantharidine (isolated from TCM Mylabris), has been widely administered for cancer patients in China [15,16]. Recently, it has been discovered that NCTD is a DNA-damaging agent that synergize DDP by specific inhibition of serine/threonine protein phosphatase 2A (PP2A) [17]. Given the above advantages of [Pt(Ⅳ)], we hypothesize that the combination of [Pt(Ⅳ)] and NCTD will enhance the anticancer effect than the monotherapy, especially overcome the drug resistance and reduce the toxic side effects of DDP.
With the advancement of nanotechnology, nanoscale drug delivery systems (DDSs) have sparked a research boom in the field of medical research [18,19]. Among these advanced DDSs, organo-bridged mesoporous organosilica nanoparticles (MONs) have received considerable attention in biomedical applications due to their stable physicochemical properties, good biocompatibility, suitable mesoporous, and large surface area [20]. Nevertheless, the stable -Si-O-Si- structure and saturation state make MONs relatively resistant to degradation in biological environments, thereby limiting their potential application [21–23]. Hence, targeted drug delivery along with controllable drug release has been regarded as a potential therapeutic strategy for efficient cancer treatment [24,25]. Besides, the biodegradability of MONs mainly depends on their mesoporous structures, compositions and surface modification [26]. To tackle the limited biodegradability of MONs to the utmost, the unique metabolites of tumor cells like redox condition have been utilized as the selective “switches” to achieve responsive cleavability and controlled drug release of MONs [27]. Additionally, the materials for constructing the MONs are preferable to be rapidly and completely degraded after the nanocarriers have accomplished the mission of drug delivery, which can reduce the risk of adverse effects induced by the nondegradable components [28,29]. For instance, Shao and co-workers have reported a series of bioinspired diselenide-bridged mesoporous silica nanoplatforms for dual-responsive “cargos” delivery, which have provided many classic paradigms for the subsequent researches [30,31]. However, whether these nanocarriers could improve the outcomes of various anticancer regimens by GSH-depleting strategy are barely investigated.
In this study, we firstly constructed a “three birds, one stone” based nanoplatform to achieve triple synergetic effects simultaneously address DDP resistance in NSCLC. Briefly, a [Pt(Ⅳ)] bridged silsesquioxane precursor namely Pt-Si was initially designed and synthesized. Then Pt-Si and bis[3-(triethoxysilyl)propyl]diselenide (BTESePD) were introduced into the framework of the nanocarriers (denoted as MONPt/Se) to achieve responsive degradation and circumvent the chemoresistance of DDP resistant lung cancer cells. Subsequently, NCTD was loaded into the obtained MONPt/Se to maximize the efficacy and reduce the toxic side effects of DDP. Besides, tumor affinity molecules decoration could effectively guide the nanoparticles to the tumor site. As a stable DNA aptamer in vivo, the AS1411 with natural G-quadruplex structure (Apt) can specifically bind to the nucleolin which is overexpressed on the surface of tumor cells [32,33]. Thus, Apt was modified on the surface of the MONPt/Se to obtain Apt@NCTD@MONPt/Se, thereby facilitating the uptake of our engineered nanocomposite (Scheme 1). Benefiting from the enhanced permeability and retention (EPR) effect and enhanced active targeting ability, the Apt@NCTD@MONPt/Se displays outstanding tumor homing capability. After cancer cell internalization, (Ⅰ) the diselenide and -O-Pt(Ⅳ)-O- rich scaffold can be reduced by the intracellular excessive GSH and induce biodegradation of the nannocarriers, followed by (Ⅱ) breaking the redox homeostasis and precise release of DDP. Meanwhile, the encapsulated NCTD is also released along with the degradation of the nanocarriers thereby (Ⅲ) achieving the GSH depletion and synergistic anti-tumor effect of NCTD and DDP. Taken together, our current work aimed to develop a novel GSH depletion DDS for enhanced cancer therapy.
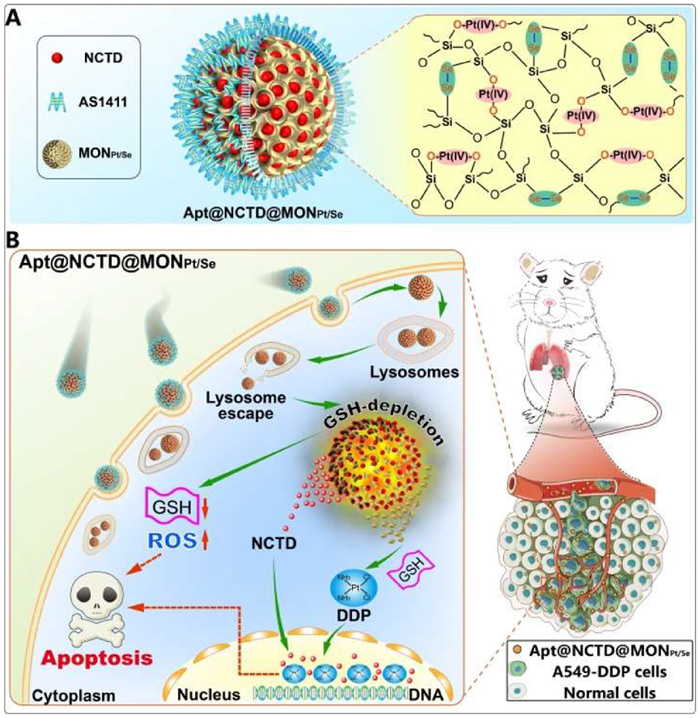
Scheme 1
Scheme 1.
Glutathione depletion based Pt(Ⅳ) hybrid mesoporous organosilica delivery system to conquer cisplatin chemoresistance: a “one stone three birds” strategy. (A) Illustration to show the microstructure of Apt@NCTD@MONPt/Se. (B) Reversing cisplatin resistance through GSH depletion and triple synergetic effects based mesoporous organosilica hybrid nanoplatform.
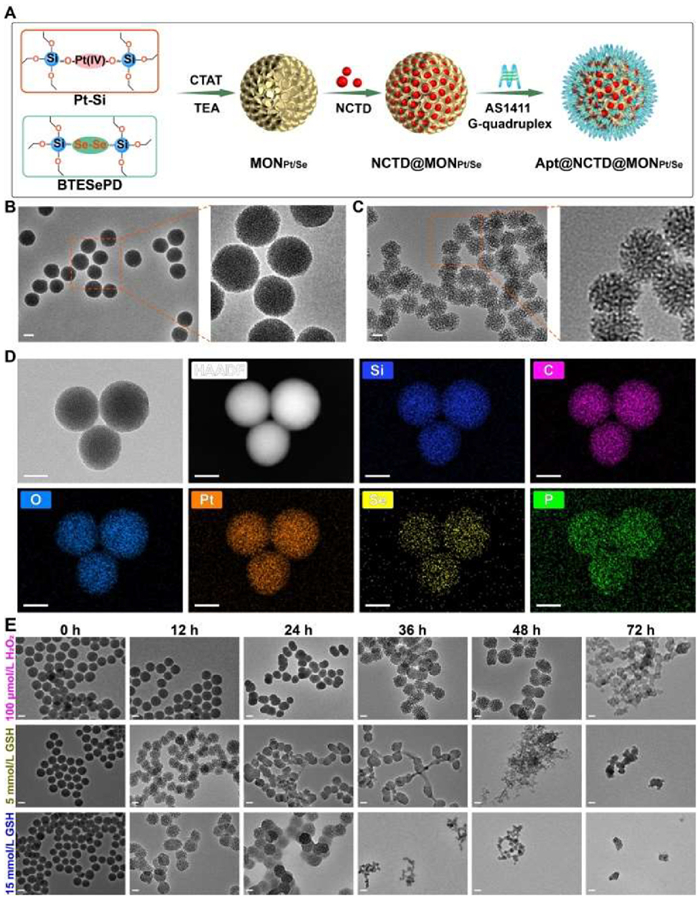
To introduce the -O-[Pt(Ⅳ)]-O- bond-containing organosilica moieties into the framework of MON, a [Pt(Ⅳ)]-based organosilica precursor namely Pt-Si was designed, synthesized and characterized by 1H and 13C NMR spectra (Figs. S1 and S2 in Supporting information). As the diselenide-bond-containing organosilica moieties can endow MONs with better GSH-consuming capability and controlled biodegradability, a diselenide-bond-containing organosilica precursor namely BTESePD was also synthesized and characterized by 1H and 13C NMR spectra (Figs. S3 and S4 in Supporting information). Next, MONPt/Se was prepared through a modified sol–gel method by employing tetraethyl orthosilicate (TEOS), BTESePD and Pt-Si as the co-organosilica precursors, ethyltrimethylammonium tosylate (CTAT) as the structure-directing agent and triethanolamine (TEAH3) as the catalyzer (Fig. 1A) [34]. Meanwhile, the diselenide-bond-bridged MON with the same TEOS/BTESePD ratio of MONPt/Se, termed MONSe, was also constructed in parallel to investigate the GSH depletion advantages of the MONPt/Se. As shown in Figs. 1B and C, transmission electron microscope (TEM) images revealed the uniform spherical structure of MONPt/Se and MONSe with a comparable diameter of ~53 nm and ~58 nm, respectively (Figs. S5 and S6 in Supporting information). NCTD is not only an effective antitumor drug, but also can combine with chemotherapy to improve the treatment effect of various cancers [35]. Thus, NCTD was loaded into the nanocarrier MONPt/Se to obtain NCTD@MONPt/Se along with a loading content of 1.16 × 10−3 mmol/mg (loading efficiency: 19.5%) (Fig. S7 in Supporting information). Next, to endow the engineered MONPt/Se with efficient tumor cell uptake and stability in circulation system, Apt was conjugated on the surface of NCTD@MONPt/Se to form Apt@NCTD@MONPt/Se. TEM confirmed the layer of soft materials (Apt) surrounding the surface of the Apt@NCTD@MONPt/Se (Fig. S8 in Supporting information). Then energy-dispersive X-ray (EDX) spectroscopy analysis indicated that platinum species (namely Pt-Si) have been collocated with carbon, silicon, selenium and oxygen inside the obtained Apt@NCTD@MONPt/Se spheres (Fig. 1D), which is consistent in the inductively coupled plasma optical emission spectrometry (ICP-MS) analysis of platinum density in MONPt/Se (3.92%). Compared with the bare MONPt/Se nanocarriers, dynamic light scattering (DLS) measurements of NCTD@MONPt/Se and Apt@NCTD@MONPt/Se showed evidently increased hydrodynamic diameters, corresponding separately to 112.7 ± 6.3 and 167.7 ± 8.1 nm (Fig. S9 in Supporting information).
Figure 1
Figure 1.
Preparation and characterization of Apt@NCTD@MONPt/Se. (A) Schematic illustration to show the preparation of Apt@NCTD@MONPt/Se. (B) TEM image of MONPt/Se. (C) TEM image of MONSe. (D) HAADF-STEM image of Apt@NCTD@MONPt/Se and corresponding element mapping images of Si, C, O, Pt, Se and P. (D) TEM images of MONPt/Se during biodegradation under various conditions (100 µmol/L H2O2, 5 mmol/L GSH and 15 mmol/L GSH) at different intervals. Scale bar: 50 nm.
Meanwhile, their zeta potentials also changed to −19.4 ± 3.2 and −30.8 ± 4.3 mV (Fig. S10 in Supporting information). Next, Fourier transform infrared spectroscopy was employed to characterize the surface functional groups of Apt@NCTD@MONPt/Se. As showed in Fig. S11 (Supporting information), the broad absorption peak at 3000–3500 cm−1 confirmed the presence of -Si-O-H, -C-H and N—H bonds, which indicated that the surface of Apt@NCTD@MONPt/Se was rich in amide groups. Meanwhile, the absorption peaks at 1660, 880–100 and 630 cm−1 corresponded to the stretching vibrations of C=C, C=N, C= S and -Si-O-Si- bonds. Together, these results indicate that NCTD@MONPt/Se NPs were successfully camouflaged with Apt.
To verify the biodegradable and GSH-depletion capability of our engineered nanocarriers, MONPt/Se was incubated with the media mimicking tumor intracellular oxidative (H2O2, 100 µmol/L), reductive (GSH, 5 and 15 mmol/L) and acidic (pH 6.5) conditions. TEM images (Fig. 1E) indicated that MONPt/Se began to collapse under reduced conditions after 24 h incubation. After 48 h exposure, the framework of the MONPt/Se was almost decomposed by the excessive GSH, which exhibited favorable bioreduced responsiveness and GSH depletion for augmented tumor theranostic. Compared with the reductive stimuli, the structure of MONPt/Se remained stable under 24 h high reactive oxygen species (ROS) level stimulation. Moreover, the basic framework of MONPt/Se still existed after 72 h ROS exposure. Obviously, such slow and weaken degradation is helpful to enhance the intracellular ROS generation, thereby improving the sensitivity of cancer cells to the released DDP. Moreover, there were no obvious changes of MONPt/Se under the acidic conditions at any time points (Fig. S12 in Supporting information). Thus, these results have demonstrated two potential advantages of MONPt/Se over the classic MON noncarriers. On one hand, the GSH-responsiveness has endowed the Pt(Ⅳ) bridged MONPt/Se nanocarrier with “on-demand” degradation for controllable and smart drug release. On the other hand, such GSH-depletion-based degradation undoubtedly restores the chemosensitivity by enhancing the ROS level within the chemoresistance cancer cells.
Because the controlled release of loaded cargos at tumor regions is a key index of the biodegradable nanoparticles for efficient oncotherapy [36], red fluorescent dye Rhodamine B (RB) was selected as the model molecule to prepare Apt@RB@MONPt/Se (RB loading efficiency: 18.2%, Fig. S13 in Supporting information) to investigate the drug loading and releasing properties of the MONPt/Se. The drug-releasing profiles of the RB@MONPt/Se and Apt@RB@MONPt/Se were measured in oxidative (H2O2, 100 µmol/L), reductive (GSH, 5 and 15 mmol/L) and acidic (pH 6.5) conditions, respectively. As shown in Fig. S14 (Supporting information), RB@MONPt/Se only released the loaded RB with 81.74% (H2O2, 100 µmol/L), 77.14% (pH 7.4), and 77.15% (pH 6.5) under the oxidative, normal and acidic conditions after 72 h incubation. In stark contrast, RB@MONPt/Se exhibited rapid RB release in reductive conditions with approximate 98.05% (GSH, 5 mmol/L) and 98.68% (GSH, 15 mmol/L) after 72 h. However, Apt@RB@MONPt/Se showed attenuated and slow RB release at any conditions (Fig. S15 in Supporting information), which were attributed to the degradation of the diselenide- and -O-Pt(Ⅳ)-O- bonds in the high reduction environment and outstanding encapsulation of Apt. Similarly, the cumulative Pt release result also suggested that approximately 96.4% and 84.1% of the total Pt in RB@MONPt/Se and Apt@RB@MONPt/Se was rapidly released under the high reductive conditions (GSH, 15 mmol/L) (Figs. S16 and S17 in Supporting information). However, less than 6% of Pt was released when RB@MONPt/Se and Apt@RB@MONPt/Se were incubated in other conditions without GSH. Given above, these results demonstrate that our GSH-consuming based nanoplatform can efficiently impede the GSH detoxification based chemoresistance and specifically deliver the “cargos” to the tumor tissues.
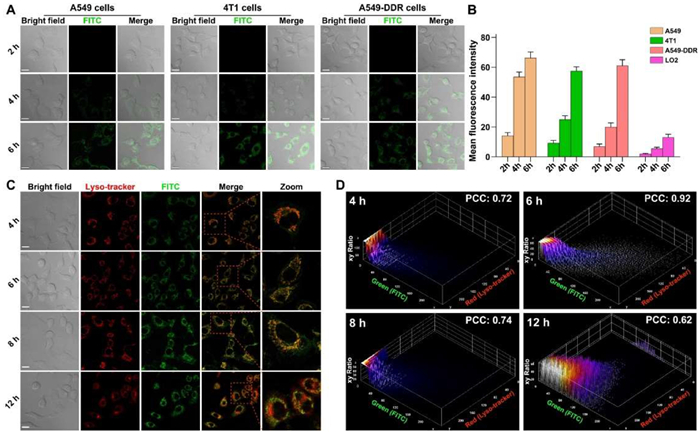
Conventionally, many of the designed nanoplatform can bypass the drug resistance by the virtue of nanometer effect, which may be not sufficient to restore the tumoricidal agents against the resistant malignancies [37]. Effective cancer-targeted delivery of the loaded drugs is one of the pivots for the anticancer activity of the engineered nanoparticles. Thus, human normal liver cell LO2 and three cancer cell lines including mouse breast cancer cell 4T1, human lung cancer cell A549 and human lung cisplatin resistance cancer cell A549-DDP were selected to verify the targeting ability of Apt@NCTD@MONPt/Se. Meanwhile, Apt@MONPt/Se was labeled with fluorescein isothiocyanate isomer (FITC, a green fluorescent dye) to prepare Apt@FITCMONPt/Se. After incubating with the Apt@FITCMONPt/Se for 4 h (Fig. 2A), cancer cells like A549, A549-DDP and 4T1 exhibited nearly equal intracellular green fluorescence intensity (Fig. 2B). Compared to the tumor cells, the human normal liver cell LO2 group showed much weaker intensity of the green fluorescence (Fig. 2B and Fig. S18 in Supporting information). Additionally, the green fluorescence intensity in the cancer cells remained higher than that of the LO2 cells at any subsequent time point, indicating that the Apt on the surface of the Apt@FITCMONPt/Se remarkably increased the targeting capability of the nanoplatform (Figs. 2A and B).
Figure 2
Figure 2.
Cellular uptake and distribution of Apt@NCTD@MONPt/Se. (A) Confocal microscopy images and (B) mean fluorescence intensities of A549, 4T1 and A549-DDP cells incubated with FITC labeled Apt@MONPt/Se (Apt@FITCMONPt/Se) for 2, 4 and 6 h (n = 5). (C) Colocalization of the Apt@FITCMONPt/Se (green) and LysoTracker (deep red) in A549-DDP cells by CLSM. (D) Evolution with time of the Pearson's correlation coefficients (PCC) between the signals from the Apt@FITCMONPt/Se and LysoTracker. Scale bar: 20 µm.
Besides, the escape from endosomes is very important to avoid premature payload release in endo/lysosomal compartments and achieve efficient cargo delivery to the cytoplasm of cancer cells. Thus, the intracellular distribution of Apt@NCTD@MONPt/Se within A549-DDP cells was investigated by employing the commercial lysotracker-Red (denoted as lyso-tracker). After 4 h incubation (Fig. 2C), the green fluorescence of Apt@FITCMONPt/Se overlapped well with the red fluorescence signal of lyso-tracker with an obviously high Pearson's correlation coefficient of 0.92 (Fig. 2D), indicating that considerable Apt@FITCMONPt/Se nanoparticles were captured by the lysosomes. With prolonged incubation time, the green fluorescence of Apt@FITCMONPt/Se began to separate from the red signals of lysosomes with the continuously declining Pearson's coefficient to 0.62 at the 12 h time point. Together, these results demonstrate that the Apt@NCTD@MONPt/Se could effectively enter the cancer cells and escape from the lysosomes to circumvent DDP resistance.
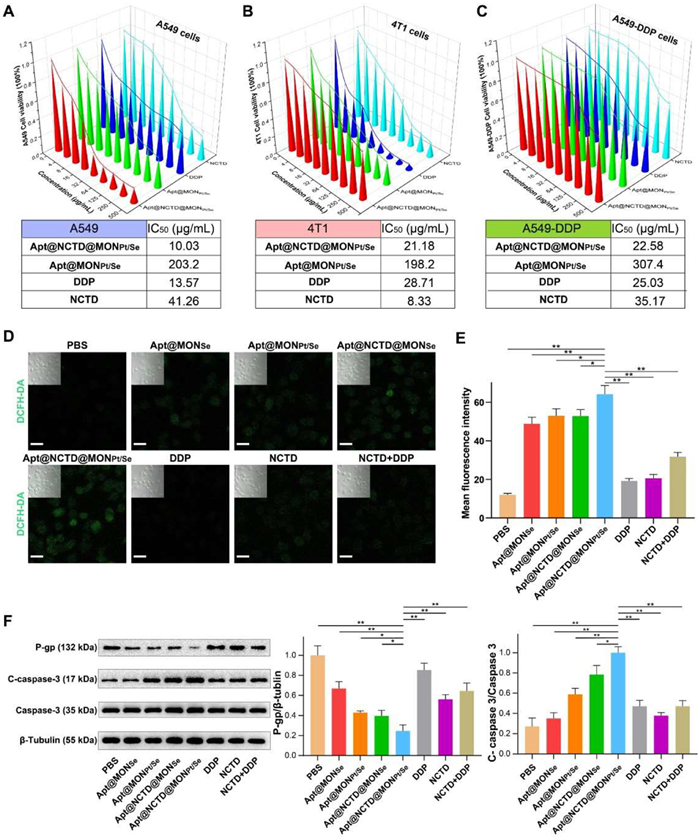
According to the above favorable results, our engineered Apt@NCTD@MONPt/Se exhibited great potential for enhanced targeting and anti-tumor effect. Thus, methyl thiazolyltetrazolium (MTT) assay was preformed to evaluate the therapeutic cell-killing effect of the Apt@NCTD@MONPt/Se on A549, 4T1, A549-DDP, and LO2 cells. As expected, all preparations exhibited dose-dependent inhibitory effects on the viability of the cancer cells. Of note, the cytotoxicity of the treatments was consistently increased for all the cancer cells in the following order: DDP < NCTD < Apt@MONPt/Se < Apt@NCTD@MONPt/Se (Figs. 3A–C). Moreover, each of the above nano preparations (Apt@MONPt/Se and Apt@NCTD@MONPt/Se) had no obvious cytotoxicity on the normal LO2 cell line, indicating their weak side effects and excellent biocompatibility (Fig. S19 in Supporting information). Next, the apoptosis degree of the Apt@NCTD@MONPt/Se to DDP resistance cancer cell A549-DDP was confirmed by fluorescence activated cell sorting (FACS). As shown in Fig. S20 (Supporting information), DDP and NCTD exhibited slightly anticancer effect on the A549-DDP cancer cells. The combination of DDP and NCTD showed a higher anticancer effect on the A549-DDP cells compared with the single chemotherapy drugs. Moreover, Apt@NCTD@MONPt/Se exhibited the strongest anticancer efficacy on the A549-DDP cells.
Figure 3
Figure 3.
Synergistic cytotoxicity and GSH-depleting capability of Apt@NCTD@MONPt/Se. (A) Cytotoxicity analysis of A549 cells treated with various concentrations and types of drugs (n = 5). (B) Cytotoxicity analysis of 4T1 cells treated with various concentrations and types of drugs (n = 5). (C) Cytotoxicity analysis of A549-DDP cells treated with various concentrations and types of drugs (n = 5). (D) Representative fluorescence images of intracellular ROS within A549-DDP cells after treatment of different concentrations and types of drugs. Scale bar: 20 µm. (E) Quantitative mean fluorescence intensity of (D). (F) Relative expression of chemotherapeutic resistance related protein P-gp and apoptosis marker C-caspase 3 within A549-DDP cells after different treatments for 12 h (n = 3). Data are presented as means ± SD. P < 0.05, **P < 0.01.
After escaping from lysosomes (12 h), ICP-MS was then carried out to precisely assess the intracellular Pt content. As shown in Fig. S21A (Supporting information), administration of Apt@NCTD@MONPt/Se resulted in significant enhancement in Pt internalization than that of free DDP in A549-DDP cells. Notably, despite the majority of Pt remained in the cytoplasm because of inherent big volume of cytoplasm, the intranuclear Pt content was significantly improved in A549-DDP cells after Apt@NCTD@MONPt/Se treatment (Fig. S21B in Supporting information). In conclusion, Apt@NCTD@MONPt/Se possesses superior anticancer toward the A549-DDP cells, these results obviously indicate that Apt@NCTD@MONPt/Se has a great potential for the further clinical application.
Typically, excessive intracellular GSH plays a pivotal role in tumor initiation, progression and chemoresistance [38,39]. Herein, we next investigated whether Apt@NCTD@MONPt/Se could overcome cisplatin resistance in NSCLC by GSH-depletion. Given that GSH functions as the major cellular antioxidant against ROS and breaking the intracellular redox balance can restore or maximize the sensitivity of tumor cells to DDP [40], the generation of the oxidized glutathione (GSSG) and GSH of the A549-DDP were initially measured. As shown in Fig. S22A (Supporting information), Apt@MONSe (only contains diselenide bonds) and Apt@MONPt/Se significantly decreased the GSH level of A549-DDP cells. More inspiring results also could be observed in the Apt@NCTD@MONPt/Se administration group, which were attributed to the abundant diselenide- and -O-Pt(Ⅳ)-O- bonds within the framework of the nanocarriers. Next, the GSSG content was further calculated and the result suggested that Apt@NCTD@MONPt/Se could significantly promote the oxidation of GSH compared with the other treatment, thereby disrupting redox balance and inducing oxidative stress in the A549-DDP cells (Fig. S22B in Supporting information).
Additionally, released DDP and NCTD of Apt@NCTD@MONPt/Se were also able to react with the intracellular ROS to generate more cytotoxic free radicals. A fluorescence probe (DCFH-DA) was then employed to test the ROS production in the A549-DDP cells. Confocal laser microscope (CLSM) images showed that A549-DDP cells treated with Apt@NCTD@MONPt/Se displayed the strongest green fluorescence, intuitively demonstrating the highest ROS generation (Figs. 3D and E). Impressed by the excellent GSH consumption of Apt@NCTD@MONPt/Se, we further explored its underlying mechanisms of action. P-glycoprotein (P-gp, also named multi-drug resistance protein, MDR1) is a major drug efflux pump expressing in the human cerebral microvessels that prevents pathogens, toxins and therapeutic drugs from entering the central nervous system [41]. However, the upregulation of P-gp in NSCLC is one of the main threats which encountered in many chemotherapeutic agents. Immunoblotting confirmed the overexpression of P-gp of A549-DDP cells than the A549 cancer cells (Fig. S23 in Supporting information). Caspase-3, a widely expressed protein of the conserved family, is known for its activated formation (namely cleaved-caspase-3, C-caspase-3) which could exacerbate the apoptosis of cancer cells [42]. Compared with the single administration of DDP or NCTD, the combination of DDP and NCTD (denoted as DDP + NCTD) group exhibited slight upregulation of C-caspase-3 and downregulation of the P-gp, which confirmed that NCTD could attenuate the cisplatin resistance and exert synergistic anti-tumor effect with DDP in A549-DDP cells (Fig. 3F). Moreover, the Apt@NCTD@MONPt/Se showed the most efficient anti-DDP-resistance effect than the other administrations by promoting apoptosis of the A549-DDP cells.
Encouraged by the impressive cancer targeting and antitumor efficacy of Apt@NCTD@MONPt/Sein vitro, we further investigated its antitumor efficacy in vivo. All animal experiments were performed under the guidelines evaluated and approved by the institutional animal care and use committee (IACUC) of Nanjing Medical University, China. Mice bearing cisplatin-resistance tumor were established by subcutaneous (hypodermic) injection of the A549-DDP cells. As mentioned above, Apt@NCTD@MONPt/Se could target the tumor with the guidance of the surface Apt. Once internalized by the cancer cells, the Pt(Ⅳ) species within the framework of the Apt@NCTD@MONPt/Se could be reduced to the Pt(Ⅱ) species (namely DDP). Thus, Pt accumulation in the tumor tissues and major organs was initially investigated by ICP-MS after single administration. As shown in Fig. S24 (Supporting information), a time-dependent biodistribution of Pt appeared in both free DDP and the Apt@NCTD@MONPt/Se treated groups. Moreover, the peak of the Pt signals in the tumor tissues of the Apt@NCTD@MONPt/Se group appeared faster and higher than that of the free DDP group. After reaching the Pt content peak (36 h), the Pt signals of the Apt@NCTD@MONPt/Se group begun to decline and remained higher than that of the free DDP group at any time points. Meanwhile, after treating with Apt@NCTD@MONPt/Se for 36 h, the Pt signals were mainly accumulated in the tumor tissues, which were attributed to the EPR effect and Apt based enhanced cancer cell uptake (Fig. S25 in Supporting information). Furthermore, ICP-MS were employed to measure the accumulation of Pt content from tumor tissues and major organs of the mice after different treatments for 14 days. Consistent with our expectations, the Pt content of the Apt@NCTD@MONPt/Se treated group was dramatically higher than that of the other groups, and the order of the Pt concentration in the tumor tissues was as follows: Apt@NCTD@MONPt/Se > Apt@ MONPt/Se > DDP + NCTD > DDP (Fig. S26 in Supporting information). For the Apt@NCTD@MONPt/Se treated group, the Pt content of the tumor tissues was also obviously higher than that of the major organs (Fig. S27 in Supporting information), which was attributed to its outstanding tumor targeting capacity. According to the GSH-depletion effect in vitro, we assumed that Apt@NCTD@MONPt/Se could exert its anticancer effect by disturbing the intratumoral redox balance. As expected, Apt@MONSe, Apt@MONPt/Se, Apt@NCTD@MONSe and Apt@NCTD@MONPt/Se could remarkably decease the GSH level along with increasing the GSSG content within the tumor tissues (Fig. S28A and B in Supporting information). Collectively, these results reveal that Apt@NCTD@MONPt/Se could effectively break the intratumoral redox balance to facilitate its anti-tumor capacity in vivo.
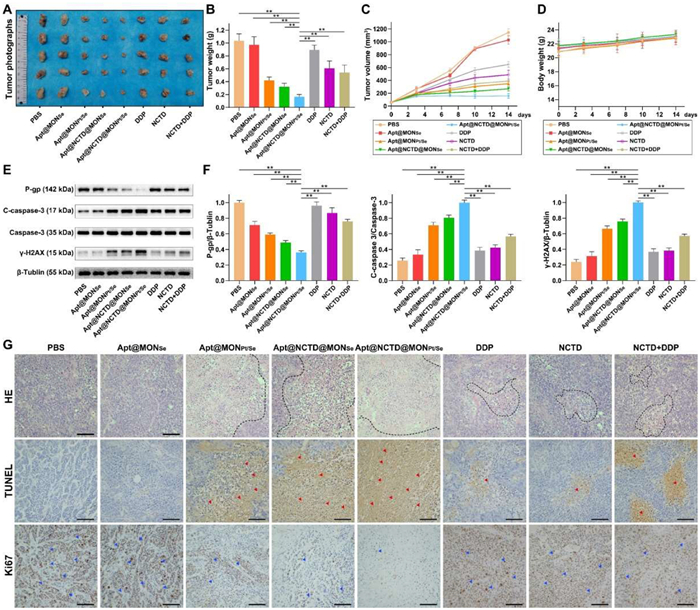
To further investigate the antitumor efficiency of Apt@NCTD@MONPt/Se, cisplatin resistant tumor (A549-DDP) bearing mice were then divided into eight groups with different treatments: (1) phosphate buffer solution treated group (denoted as PBS); (2) Apt@MONSe; (3) Apt@MONPt/Se; (4) Apt@NCTD@MONSe; (5) Apt@NCTD@MONPt/Se; (6) free DDP (denoted as DDP); (7) free NCTD (denoted as NCTD); (8) free NCTD and DDP combination (denoted as NCTD + DDP). The relative volume and weight of tumors were initially monitored to compare the antitumor efficacy of different treatments. As shown in Figs. 4A–C, mice treated with PBS and Apt@MONSe exhibited rapid tumor growth in volume and weight at the end of the treatment. While the DDP group and NCTD group showed a relatively modest therapeutic effect, blaming to the poor tumor target capability and short circulation time of DDP or NCTD. Compared with the DDP group and NCTD group, the combination of NCTD and DDP (NCTD + DDP) showed enhanced anti-tumor effects. However, such improved anti-tumor outcomes remained limited due to the inherent shortcomings of small molecules. Encouragingly, here we found that Apt@NCTD@MONPt/Se treated group showed the highest tumor decrease of 988 mm3 in volume and 0.88 g in weight compared with the PBS treated group in the end without any significant body weight loss (Fig. 4D). These results indicated that our Apt@NCTD@MONPt/Se nanoplatform which integrated NCTD with DDP could improve the antitumor performance. More importantly, the expression of P-gp in the tumor tissues was reduced by Apt@NCTD@MONPt/Se, suggesting that Apt@NCTD@MONPt/Se could effectively conquer the cisplatin resistance in vivo. Furthermore, the strongest upregulation of the γ-H2AX (DNA damage biomarker) and C-caspase-3 in Apt@NCTD@MONPt/Se treated group also confirmed the powerful anti-tumor effect of Apt@NCTD@MONPt/Se (Figs. 4E and F). Together, it was obvious that Apt@NCTD@MONPt/Se could elicit its strongest anti-tumor effect along with alleviating DDP resistance in vivo.
Figure 4
Figure 4.
Antitumor effect of Apt@NCTD@MONPt/Se in xenograft mice. (A) The images of excised tumor tissues and (B) primary tumor weights from each treatment groups (n = 5). (C) Tumor volume curves and (D) body weight changes of the A549-DDP tumor-bearing mice after different treatments for 14 days (n = 5). (E) Relative expression of chemotherapeutic resistance related protein P-gp, apoptosis marker C-caspase 3 and DNA damage biomarker γ-H2AX within the tumor tissues of the A549-DDP tumor-bearing mice after different treatments for 14 days (n = 3). (F) Quantification of the band intensities of (E) (n = 3). (G) Representative H&E staining and immunohistochemical analysis of TUNEL and Ki-67 from tumors treated with different treatments. Scale bar: 100 µm. Data are presented as means ± SD. **P < 0.01.
Next, histological analysis including hematoxylin and eosin (H&E) and immunohistochemistry were conducted to verify the antitumor activity and the biocompatibility of Apt@NCTD@MONPt/Se. H&E staining results suggest that cancer cells of the PBS and Apt@MONSe treated groups displayed large nuclei and spindle shapes, which implied the rapid tumor progression (Fig. 4G). Meanwhile, cancer cells of the free DDP, NCTD and DDP + NCTD treated groups showed mild apoptotic characteristics, such as nuclei shrinkage, fragmentation and even nuclear dissolution, and the DDP + NCTD group was worse. In stark contrast, tumor tissues of the Apt@NCTD@MONPt/Se treated group exhibited the most serious and significant apoptotic characteristics. Similar results also can be observed in the subsequent deoxyuridine triphosphate nick end labeling (TUNEL) assay images (Fig. 4G), which is another indicator of cancer cell apoptosis. Conversely, immunohistochemistry images of the Ki67 (a proliferative signal) suggested that Apt@NCTD@MONPt/Se could effectively inhibit the proliferation of tumor cells. Finally, systemic toxicity and biocompatibility of the Apt@NCTD@MONPt/Se was investigated by H&E assays of main organs (Fig. S29 in Supporting information). Obviously, there were no discernible pathologic lesions appear in all organs (heart, liver, spleen, lung and kidney) of all the treated groups. Collectively, these in vivo results indicate that Apt@NCTD@MONPt/Se possesses the strongest anti-tumor effect, gives the best biocompatibility and has no potential toxicity.
To sum up, our current work has proposed a multifunctional nanoplatform combining Pt(Ⅳ) prodrug and NCTD with GSH-depletion, which can conquer cisplatin resistance and achieve efficient co-delivery of DDP and NCTD. Owing to the particular metabolism of cancer cells, considerable ROS can be produced during their rapid metabolism and proliferation. Thus, elevated levels of GSH could effectively counteract these excessive ROS, and tumor cells are vulnerable to ROS under the situation of GSH deficiency in turn. Besides, intracellular GSH based detoxification plays an important role in the development of DDP-resistance of the NSCLC. Taking advantages of the GSH depletion strategy, a redox-responsive degradable MON nanocarriers rich in diselenide- and -O-Pt(Ⅳ)-O- bonds is developed and loaded with NCTD to achieve enhanced chemotherapy and synergistic antitumor effect. Next, cell-specific aptamer AS1411 was modified on the surface of NCTD@MONPt/Se due to its high specificity and affinity to the cancer cells. In vitro results suggested that intracellular excessive GSH could be effectively consumed by the obtained Apt@NCTD@MONPt/Se and further promote the drug release. Once internalized by the DDP-resistance A549-DDP cancer cells, the released DDP and NCTD could exert a synergistic antitumor effect based on the broken intracellular redox balance. Consistent with the in vitro results, Apt@NCTD@MONPt/Se also effectively inhibited tumor growth by breaking the intracellular redox balance and the synergistic anti-tumor effect of DDP and NCTD in the A549-DDP xenograft-bearing mice. More exciting, this engineered Apt@NCTD@MONPt/Se nanoplatform can achieve directional drug accumulation in vivo, bypasses chemoresistance, possess multifunctionality, and exhibit extremely low toxicity to the normal tissues. Given above, we believe this “one stone three birds” strategy may be a promising paradigm to conquer drug resistance.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Key R&D Program of China (No. 2020YFA0709900), the Excellent Postdoctoral Program of Jiangsu Province (No. 2022ZB803), National Natural Science Foundation of China (No. 82273162), the major science and technology program of Nanjing (No. 202305027), the Open Program of NHC Key Laboratory of Nuclear Medicine and Jiangsu Key Laboratory of Molecular Nuclear Medicine (No. KF202203).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109450.
J. Wang, Q. Zhang, Y. Li, et al., Chin. Chem. Lett. 35 (2023) 108746.
[40]
Y. Xiong, C. Xiao, Z. Li, X. Yang, Chem. Soc. Rev. 50 (2021) 6013–6041. doi: 10.1039/D0CS00718H
[41]
E. Lepeltier, P. Rijo, F. Rizzolio, et al., Drug Resist. Update 52 (2020) 100704. doi: 10.1016/j.drup.2020.100704
[42]
N.B. Hentzen, R. Mogaki, S. Otake, K. Okuro, T. Aida, J. Am. Chem. Soc. 142 (2020) 8080–8084. doi: 10.1021/jacs.0c01823
Scheme 1
Glutathione depletion based Pt(Ⅳ) hybrid mesoporous organosilica delivery system to conquer cisplatin chemoresistance: a “one stone three birds” strategy. (A) Illustration to show the microstructure of Apt@NCTD@MONPt/Se. (B) Reversing cisplatin resistance through GSH depletion and triple synergetic effects based mesoporous organosilica hybrid nanoplatform.
Figure 1
Preparation and characterization of Apt@NCTD@MONPt/Se. (A) Schematic illustration to show the preparation of Apt@NCTD@MONPt/Se. (B) TEM image of MONPt/Se. (C) TEM image of MONSe. (D) HAADF-STEM image of Apt@NCTD@MONPt/Se and corresponding element mapping images of Si, C, O, Pt, Se and P. (D) TEM images of MONPt/Se during biodegradation under various conditions (100 µmol/L H2O2, 5 mmol/L GSH and 15 mmol/L GSH) at different intervals. Scale bar: 50 nm.
Figure 2
Cellular uptake and distribution of Apt@NCTD@MONPt/Se. (A) Confocal microscopy images and (B) mean fluorescence intensities of A549, 4T1 and A549-DDP cells incubated with FITC labeled Apt@MONPt/Se (Apt@FITCMONPt/Se) for 2, 4 and 6 h (n = 5). (C) Colocalization of the Apt@FITCMONPt/Se (green) and LysoTracker (deep red) in A549-DDP cells by CLSM. (D) Evolution with time of the Pearson's correlation coefficients (PCC) between the signals from the Apt@FITCMONPt/Se and LysoTracker. Scale bar: 20 µm.
Figure 3
Synergistic cytotoxicity and GSH-depleting capability of Apt@NCTD@MONPt/Se. (A) Cytotoxicity analysis of A549 cells treated with various concentrations and types of drugs (n = 5). (B) Cytotoxicity analysis of 4T1 cells treated with various concentrations and types of drugs (n = 5). (C) Cytotoxicity analysis of A549-DDP cells treated with various concentrations and types of drugs (n = 5). (D) Representative fluorescence images of intracellular ROS within A549-DDP cells after treatment of different concentrations and types of drugs. Scale bar: 20 µm. (E) Quantitative mean fluorescence intensity of (D). (F) Relative expression of chemotherapeutic resistance related protein P-gp and apoptosis marker C-caspase 3 within A549-DDP cells after different treatments for 12 h (n = 3). Data are presented as means ± SD. P < 0.05, **P < 0.01.
Figure 4
Antitumor effect of Apt@NCTD@MONPt/Se in xenograft mice. (A) The images of excised tumor tissues and (B) primary tumor weights from each treatment groups (n = 5). (C) Tumor volume curves and (D) body weight changes of the A549-DDP tumor-bearing mice after different treatments for 14 days (n = 5). (E) Relative expression of chemotherapeutic resistance related protein P-gp, apoptosis marker C-caspase 3 and DNA damage biomarker γ-H2AX within the tumor tissues of the A549-DDP tumor-bearing mice after different treatments for 14 days (n = 3). (F) Quantification of the band intensities of (E) (n = 3). (G) Representative H&E staining and immunohistochemical analysis of TUNEL and Ki-67 from tumors treated with different treatments. Scale bar: 100 µm. Data are presented as means ± SD. **P < 0.01.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
