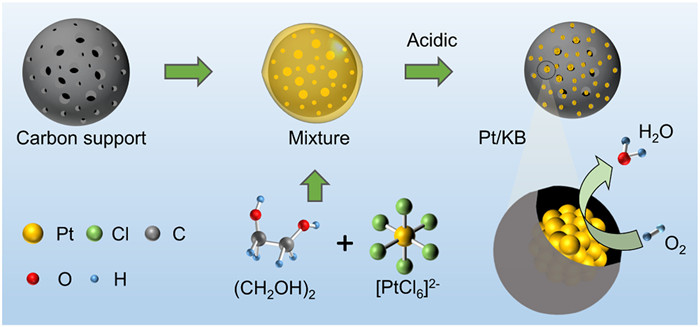
Scheme 1.
Illustration of the synthesis process of Pt/KB catalyst.
Nano-engineered catalysts for high-performance oxygen reduction reaction
Kunsong Hu , Yulong Zhang , Jiayi Zhu , Jinhua Mai , Gang Liu , Manoj Krishna Sugumar , Xinhua Liu , Feng Zhan , Rui Tan

The rapid depletion of traditional fossils and climate-related concerns have raised the need to develop green and sustainable renewables and energy conversion technologies [1,2]. Among various renewables, hydrogen stands out as a clean and sustainable energy carrier, and an important power source for efficient and eco-friendly fuel cells [3,4]. Fuel cells can produce electricity in a low-carbon manner by solely using hydrogen and oxygen [5-8]. Tough promising for advancing carbon neutrality, fuel cells are significantly challenged by the sluggish kinetics of the cathodic oxygen reduction reaction (ORR) [9]. Given this challenge, research efforts have been focused on the development of high-performance ORR catalysts, including noble-metal-based catalysts, e.g., platinum (Pt) [9,10], and non-Pt catalysts, e.g., phosphides [11], sulfides [12], oxides [13,14], nitrides [15] and carbon-based materials [16]. The performance of Pt-based catalysts is much better than the non-Pt alternatives, but their upscaled applications in fuel cells are challenged by massive loadings and high capital costs [17]. In this case, reducing the Pt loading whilst maintaining its high catalytic performance is a practical approach that advances the commercialization of catalysts and efficient fuel cells.
The incorporation of Pt particles within conductive carbon supports has been demonstrated as an effective approach to minimize the Pt usage [18-20]. Conductive carbon materials are capable of providing high surface area with abundant host sites, allowing the dispersion of Pt particles and thus maximizing the performance [21,22]. Several strategies have been proposed to enhance the dispersion of Pt within support materials, including the use of support carriers, e.g., carbon nanotubes and graphene, the addition of surface-active agents [23,24], and carbon coating on the surface of Pt [25]. Specifically, porous carbon supports can reduce activation impedance and oxygen mass transfer resistance, favoring the overall efficiency and performance [26]. However, the lack of ability to remove surface active agents, together with the sophisticated processing procedures and high production costs, has prevented the industrial application of these strategies.
Besides the use of support materials, minimizing the particle size to a nanoscale plays a crucial role in maximizing Pt utilization and catalytic performance [27]. Theoretically, size effects can influence the performance as per the following two reasons: (1) Given the same Pt amount, size-reduced particles can provide increased surface area and more effective catalytic sites [27-29] (2) With the decrease in particle size, the proportion of low-coordination surface sites (corners or edges) increases, altering the electronic structure and enhancing reaction kinetics [29-32]. Alteration in electronic structure can modify the binding energy between active sites and reaction intermediates, allowing the fine-tuning of the catalyst's adsorption strength toward reactants, ultimately resulting in an enhanced reaction activity [33,34]. In light of this, research efforts have been dedicated to developing Pt-based catalysts with smaller particle sizes, aiming for lower Pt usage and superior catalytic performance. However, smaller particles with elevated surface energy tends to form secondary agglomeration or redeposit on the support surface according to Ostwald ripening [27]. To stabilize the surface of Pt nanoparticles (NPs) and avoid agglomeration, functional support materials need to be carefully selected according to the compatibility between embedded particles and support matrix.
In this work, we report a facile structural tuning strategy to encapsulate Pt NPs within the pores of porous carbon support via a one-step microwave-assisted polyol reduction method. We selected commercial Ketjen Black (KB) as the support material due to its large surface area, high porosity, interconnected channels, and excellent conductivity. The substantial surface area and outstanding conductivity of KB provide enhanced space for charge transfer and storage. The high porosity facilitates the deposition of fine Pt NPs by offering a multitude of small pores. The interconnected channels create abundant mass transfer pathways for the ORR. Nano-sized Pt particles (~2.27 nm) were successfully deposited and confined within the abundant pores of carbon support materials. Physio-chemical structures of this composite catalyst were comprehensively studied by various approaches. The effective reaction surface can be maximized within the highly dispersed Pt NPs so as to afford enhanced oxygen reduction performance. Benefiting from this highly dispersed and size-reduced Pt NPs, the electrochemical surface area (ECSA) can be increased to 142.98 m²/gPt, which is 2.25 times higher than that of the commercial counterpart (63.52 m²/gPt). Furthermore, the conductive carbon framework ensures smooth electronic conduction while limiting the overpotential. The size effects and porous carbon support work synergistically to deliver a remarkably catalytic activity with a high half-wave potential (E1/2) of 0.895 V and an improved mass activity (MA) of 0.2289 A/mgPt. Orthogonal experiments were also designed to explore the key process parameters for fabricating Pt/C catalysts. Overall, this one-step approach features a rapid reaction process, short reaction time and environmental sustainability, suggesting significant potential for mass production of high-performing catalysts.
Pt NPs embedded in porous Ketjen Black materials, namely Pt/KB, were synthesized using the rapid microwave-assisted method, as depicted in Scheme 1. Ethylene glycol (EG) serves a dual role as both a reducing agent and a protective/dispersing agent, assisting the dispersion of the catalyst. The reaction mechanism for the reduction of H2PtCl6 by EG can be described as follows [35,36]:
|
|
(1) |
|
|
(2) |
Specifically, EG was initially decomposed into H2O and CH3CHO which serves as a reducing agent to convert Pt4+ ions to Pt NPs. Due to the advantages of rapid reaction processes, short heating times, and energy efficiency, microwave heating was chosen as the external energy source in this study. Microwave radiation allows for uniform internal heating of materials, promoting more even nucleation and faster crystallization. Microwave-assisted rapid synthesis was employed to produce small-sized Pt nanoparticles, subsequently encapsulated within the pores of a porous carbon support. The final outcome was the production of finely dispersed and uniformly porous carbon-supported Pt catalysts. The inner walls within pores immobilize Pt NPs, preserving the high activity of these discrete small-sized particles. Furthermore, the continuous pores in KB also serve as fast transport channels for reactants and reaction products during catalysis, accelerating the reaction rate. Although this approach is very straightforward and facile, a reliable methodology needs to be established by comprehensively investigating the critical process parameters, such as pH, reaction temperature, reaction time and parent solution concentrations. To increase the performance and reproducibility of the fabrication of Pt/KB catalysts, we conducted a six-factor, three-level orthogonal experiment to explore and optimize the process parameters, specific details of which are outlined in Table 1. For simplicity, prototypical samples, named as Pt/KB-4, Pt/KB-10, Pt/KB-14, were selected to elucidate the correlation of structure, property and performance. Additionally, we opted for a commercial catalyst with a 40% Pt loading, denoted as JM 40%, for comparative analysis with our samples. More details about the results of orthogonal experiments are provided in the supporting information and the final section.
Material phases and crystal structures of developed Pt/KB are identified by X-ray diffraction (XRD) as shown in Fig. 1a. The characteristic peaks observed at 39.7° and 46.2° correspond to Pt (111) and Pt (200) crystal facets and align well with the standard Pt characteristic peaks as evidenced by PDF#04–0802. The Pt (111) crystal plane of Pt/KB-4 exhibits a slight leftward shift, and there is limited consensus in the literature regarding the interpretation of this phenomenon [37,38]. Considering the broadening of the Pt (111) peak and the decrease in peak intensity, we attribute this observation to the incomplete development of the crystal lattice, indicative of the presence of ultra-small, not fully matured crystal structures. These indicated crystal facets confirm a typical face-centered cubic (fcc) structure, which can favor the ORR activity. A distinctive C peak (002) is observed in the XRD near 24°, and no other impurity peaks are detected, indicating the effective deposition of Pt onto the carbon support. According to matches the Shirley equation [27], i.e., the inverse correlation between peak width and nanocrystal size, the grain size of the synthesized Pt/KB catalysts is much smaller than that of the commercial samples. Particularly, the Pt/KB-4 sample exhibits the smallest grain size and is anticipated to deliver a high utilization of Pt NPs. To further confirm the loading of Pt, we performed thermogravimetric analysis (TGA) to examine the decomposition behavior of the catalyst at elevated temperatures (Fig. 1b). A noticeable decrease in weight was observed within the temperature ranges of below 150 ℃, 150–350 ℃ and 350–450 ℃. The weight loss below 150 ℃ can be attributed to the evaporation of water within the catalyst. The mass loss between 150 ℃ and 350 ℃ is primarily due to the slow pyrolysis of the carbonaceous support in the catalyst [39]. Subsequently, the combustion of KB at temperatures exceeding 350 ℃ results in a rapid loss in mass. The Pt content indicated by the residual weight ratio in TGA curves can be determined as 27.3%, 28.1% and 27.1% for Pt/KB-4, Pt/KB-10 and Pt/KB-14, respectively. Following this, Pt/KB-4 and Pt/KB-10 were chosen for inductively coupled plasma mass spectrometry (ICP-MS) testing to validate the TGA outcomes. The ICP-MS test results for Pt/KB-4 and Pt/KB-10 were 27.99% and 27.54%, respectively, aligning well with the TGA results. Given the loading of Pt in our developed samples, we select the commercial catalyst with a 40% Pt loading, i.e., JM 40%, for fair comparison.
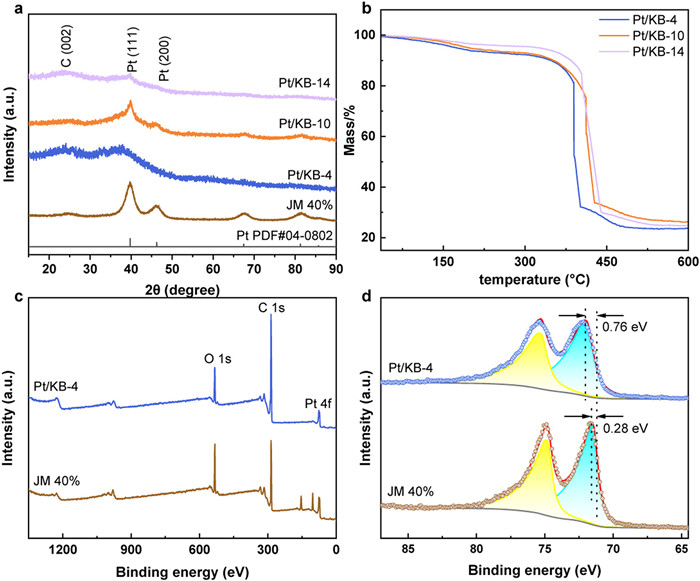
The elemental composition and electronic configuration which play an important role in determining the catalytic performance can be investigated via X-ray photoelectron spectroscopy (XPS). The full-range XPS spectra of the Pt/KB and JM 40% are presented in Fig. 1c and Fig. S1a (Supporting information), respectively. The spectra show the presence of three predominant elemental species, C 1s, O 1s, and Pt 4f. In both samples, Pt is predominantly in its metallic state (Fig. 1d) while Pt 4f7/2 binding energy for the Pt/KB-4 catalyst (71.96 eV) is shifted by 0.76 eV concerning the standard spectrum (71.2 eV). The positive shift in the binding energy indicates the transfer of electrons from Pt to the carbon support, thereby giving rise to a pronounced electronic metal-support interaction (EMSI) [40-43]. The EMSI can modulate the electronic nature of active sites, enhancing the charge transfer at the interface, thereby optimizing catalytic active sites and improving catalytic activity. The optimization of the electronic structure results in a downward shift of the d-band center of Pt, reducing the adsorption of reaction intermediates on the Pt surface and thereby enhancing the catalyst's activity [44-47]. Strong EMSI also contributes to the stabilization of ultrafine Pt nanoparticles, ensuring excellent catalyst durability. Similarly, Pt/KB-10 and Pt/KB-14 exhibit different positive shifts in Pt 4f7/2 binding energy with respect to the standard spectrum (Fig. S1b in Supporting information), suggesting that their d-band centers have also shifted downward. This likely contributes to the higher activity compared to their commercial counterpart.
The microstructure of the catalyst and dispersion of Pt NPs in the carbon support were thoroughly characterized using a transmission electron microscope (TEM) (Figs. 2a-f and Figs. S2a-f in Supporting information). As observed, the selected porous carbon consists of numerous irregular stacked carbon nanospheres and exhibits a loose and porous structure that profoundly provides active sites for the deposition of Pt and facilitates the Pt dispersion. As a result, from Fig. 2a, the Ultrafine Pt NPs are uniformly attached to the carbon support with minimal aggregation. Specifically, most of the Pt NPs of Pt/KB-4 have particle sizes between 1.5 nm and 3 nm, with an average size of 2.27 nm. Likewise, Pt/KB-10 and Pt/KB-14 also exhibit narrow size distributions, with average sizes of 3.44 and 3.28 nm, respectively (Figs. S2a and d). In contrast, the particle size distribution of commercial JM 40% is much larger, ranging from 2 nm to 8 nm with an average size of 4.12 nm (Fig. 2d). High resolution TEM images further Confirm that the commercial JM 40% has large particles (Figs. 2c and f). Insightful TEM observations clearly indicate the successful preparation of ultrafine Pt NPs highly dispersed in the porous carbon support without agglomeration issues.
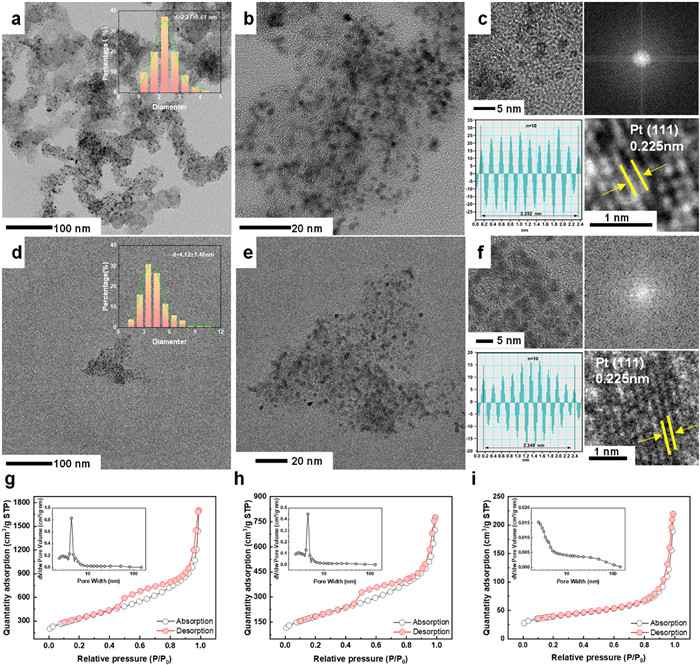
Brunauer-Emmett-Teller (BET) is a straightforward approach to detecting the pore behavior of porous materials and composite materials with impregnated species. As a control, porous KB exhibits characteristic adsorption-desorption hysteresis (Type Ⅳ isotherm), indicating the mixed nature of micro and mesopores (Fig. 2g). Of note, the derived pore size locates in the range of 1–7 nm, which provides ample space for Pt NPs to diffuse and deposit in the nanoscale pores of KB. After loading Pt NPs, the surface area for fresh carbon support (1206.45 m²/g) has significantly reduced to 662.81, 704.73 and 817.78 m²/g for Pt/KB-4, Pt/KB-10 ad Pt/KB-14, respectively (Fig. 2h, Figs. S2g and h in Supporting information). The pore volume of KB is 2.63 cm³/g, and after Pt deposition, the pore volume of Pt/KB-4 decreases to 1.2 cm³/g, Pt/KB-10 to 1.32 cm³/g, and Pt/KB-14 to 1.46 cm³/g. These results strongly confirm that Pt NPs can be highly dispersed in KB and occupy the pore channels while leaving the residual channels for the permeation of reactants, e.g., water and oxygen. Due to the constraining effects of EMSI and mesoporous walls on platinum nanoparticles (Pt NPs), these ultrafine Pt NPs are securely immobilized within the micropores of KB. This likely imparts excellent durability to the prepared catalyst, a proposition subsequently confirmed through durability testing. In comparison, commercial JM 40% only possess a limited surface area of 140.79 m²/g with a small number of micropores (Fig. 2i), suggesting the aggregation of Pt particles with limited reaction area in the commercial samples.
The oxygen reduction performance of our developed catalysts and the commercial counterpart were further evaluated by electrochemical measurements, cyclic voltammetry (CV) and linear sweep voltammetry (LSV) (Fig. 3). Among the CV curves for Pt/KB-4, Pt/KB-10, Pt/KB-14 and the JM 40% commercial samples, Pt/KB-4 exhibits a significantly larger area in the hydrogen adsorption/desorption region (Fig. 3a). As derived from the CV curves, the ECSAs of Pt/KB-4, Pt/KB-10, and Pt/KB-14 are 142.98, 128.55, and 76.99 m2/gPt, respectively, much higher than that of the JM 40% commercial sample (63.52 m2/gPt). Fig. 3b presents the LSV curves for Pt/KB-4, Pt/KB-10, Pt/KB-14, and the JM 40% commercial sample, in which the E1/2 values for Pt/KB-4, Pt/KB-10, and Pt/KB-14 are 0.895, 0.884, and 0.879 V vs. reversible hydrogen electrode (RHE), respectively, all higher than the JM 40% commercial sample (0.878 V vs. RHE). Importantly, our developed catalysts also delivered remarkable MA. MAs of Pt/KB-4, Pt/KB-10, and Pt/KB-14 at 0.9 V vs. RHE are 0.2289, 0.1365 and 0.1327 A/mgPt, higher than that of the JM 40% commercial sample (0.0958 A/mgPt), respectively. These results strongly confirm that the finely dispersed ultrafine Pt NPs embedded in porous KB effectively improve active sites and maximize the usage of Pt NPs to increase the overall performance.
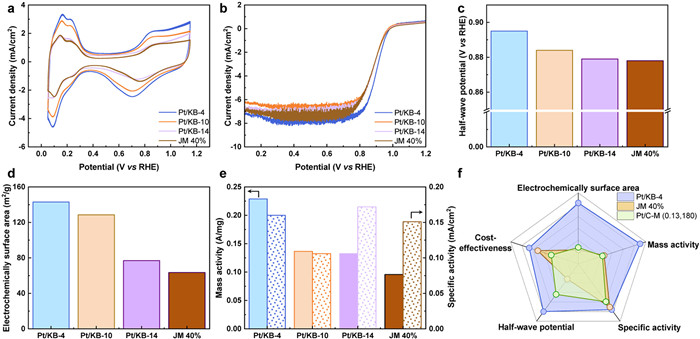
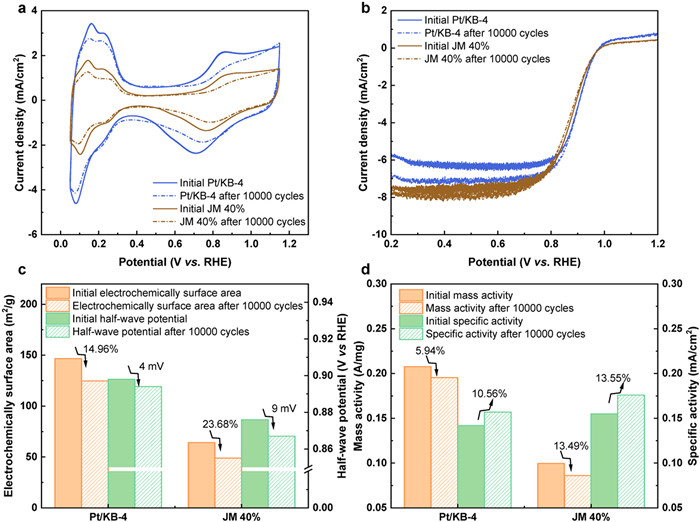
Furthermore, we compared Pt/KB-4 with Pt/C catalysts reported in recent years (Table S1 in Supporting information). A radar plot (Fig. 3f) was used to perform a multifaceted comparison of Pt/KB-4, JM40% commercial sample, and Pt/C-M (0.13, 180) [48]. Compared to the commercial product JM 40% and Pt/C-M (0.13, 180) (60%), Pt/KB-4 (27.3%) features a much lower Pt loading and can be produced from a straightforward approach, microwave-based hydrothermal reaction, hinting at a reduced production cost. Most importantly, despite its lower Pt content, our catalyst outperforms the commercial JM 40% catalyst in terms of overall catalytic performance, such as higher ECSA, E1/2 and MA. To assess the durability disparity between our synthesized catalyst and commercial counterparts, we executed 10,000 accelerated durability tests (ADTs) under identical conditions [5,27,47,49,50]. Comparative analysis of the CV and LSV curves before and after the ADTs cycle was conducted (Figs. 4a and b). The marginal rise in limiting current density could potentially be attributed to the diffusion of oxygen in proximity to the electrode [51]. The slight increase in limiting current density may be attributed to the diffusion of oxygen near the electrode. Subsequently, the ECSA, E1/2, MA, and SA of the catalyst before and after ADTs cycles were calculated for a visual comparison (Figs. 4c and d). After 10,000 ADT cycles, JM 40% exhibited an ECSA loss of 23.68%, while Pt/KB-4 showed a relatively smaller ECSA loss of 14.96% (Fig. 4c). The ECSA loss could be attributed to the aggregation of Pt nanoparticles during Ostwald maturation cycles, leading to their separation from the supporting material [25]. The reduced ECSA loss in Pt/KB-4 compared to JM 40% may be attributed to spatial constraints imposed by KB on ultrafine Pt NPs and the effect of EMSI. Pt/KB-4 demonstrated a negative shift in E1/2 of only 4 mV, whereas JM 40% exhibited a larger negative shift of 9 mV. Furthermore, JM 40% experienced a significant MA decrease of 13.49%, while Pt/KB-4 only showed a 5.94% decrease, indicating excellent long-term stability of Pt/KB-4 for ORR (Fig. 4d). Both Pt/KB-4 and JM 40% showed a certain degree of SA increase, primarily due to the larger decrease in ECSA compared to MA.
Key synthetic factors involved within the synthetic approach were identified meticulously to afford a much reliable and scalable method that is particularly important for industrial manufacturing. Critical synthetic factors include concentrations that determine the formation types of Pt particles [52], pH solutions that corelates to the reduction process during the microwave synthesis [53,54], temperature and reaction time [55-57]. In addition, the concentration of carbon carriers also affect the dispersion of Pt particles and their interaction with the carbon carrier [58]. Carefully tuning these synthetic parameters is important to achieve optimal performance of Pt/C catalysts. Accordingly, a six-factor, three-level orthogonal experiment was designed to investigate the effects of key parameters on the catalyst performance of produced catalysts, in which key factors include Pt concentration (CPt, 1, 2, 3 mgPt/mL), pH of non-reacted solutions (pH1, 10, 11, 12), reaction temperature (T, 140, 160, 180 ℃), reaction time (t, 60, 90, 120 s), carbon slurry concentration (CC, 2, 4, 6 mg/mL), and pH of reacted solutions (pH2, 1, 2, 3). CV and LSV were carried out to evaluate catalytic performance and identify the optimal synthetic factors (Fig. S3 in Supporting information and Table 2).
Influence of Pt concentration (CPt): As recorded in Table S2 (Supporting information), the catalytic performance as indicated by the ECSA, E1/2, and MA reaches their peak values at CPt = 1 (Fig. 5a). In the scenario of using low parent solutions, Pt particles cannot saturate the active sites of catalyst and result in the limited catalytic activity, while in the concentrated reaction solutions, Pt might aggregate and lose catalytic performance.
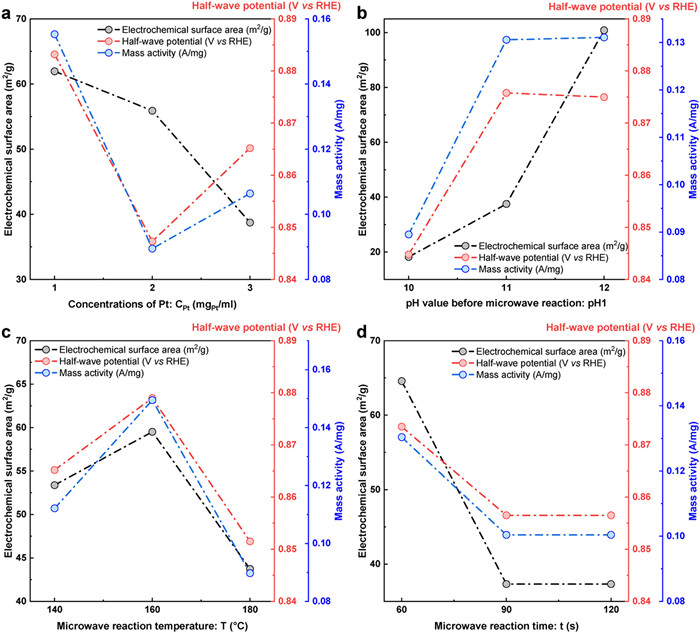
Effects of pH1: pH of parent solutions can not only influences the size of Pt NPs [53], but also determine the reduction process of H2PtCl6 in the microwave process using the EG as reactant. Alkaline condition can provide excessive hydroxide ions to facilitate the decomposition of EG into glycolaldehyde, stabilize and limit the nucleation and growth of Pt NPs. Therefore, ECSA, E1/2 and MA of Pt/KB reasonably reach the highest values when synthesized at the pH1 of 12 (Fig. 5b and Table S3 in Supporting information).
Other factors: Following the same orthogonal table principle, we investigated the effects of other factors (T, t, CC, pH2) on the catalyst performance (Figs. 5c and d, Figs. S4a and b in Supporting information), and the detailed results are listed in Tables S4-S7 (Supporting information). As evidenced in Fig. 5c, modest temperature, i.e., 160 ℃, enables the best catalytic performance because lower temperature cannot induce the completed reaction and higher temperature might accelerate agglomeration of formed NPs. Particularly, at 160 ℃, the reaction can be thoroughly completed within 60 s (Fig. 5d). Regarding the CC and pH2, the influence of these two factors is very limited. Therefore, durable synthetic protocol for the catalysts with optimal performance is the use of 1 mgPt/mL Pt-precursor solutions reacted at 160 ℃ for 60 s in a microwave-based and alkaline condition.
In this work, we successfully developed a facile and rapid microwave-assisted EG reduction method to synthesize a highly active Pt/KB catalyst. Pt NPs with the particle size of ~2.27 nm are uniformly dispersed within the pores of the conductive carbon support. With the largely reduced particle size and the use of continuous electron-conductive framework, Pt/KB catalysts delivered excellent catalytic performance, supported by the high excellent electrochemical specific surface area (142.98 m²/gPt) and improved E1/2 (0.895 V) as well as an enhanced MA of 0.2289 A/mgPt at 0.9 V vs. RHE. In addition to the catalytic performance, this catalyst also possesses a low loading of Pt and can be easily prepared from a rapid and cost-effective protocol. Importantly, to confirm the durability and reproducibility of our approach, we screen out the critical synthetic factors for producing the optimal catalyst materials, which are important for future research work and industrial manufacture. Furthermore, our approach is not limited to the use of a single metal. Various metal combinations can be applied to afford alloy catalyst that can further reduce the capital cost while maximizing the performance.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors acknowledge the support from Warwick Manufacturing Group at the University of Warwick, CITIC Dameng Mining Industries Limited -Guangxi University Joint Research Institute of Manganese Resources Utilization and Advanced Materials Technology, Guangxi University-CITIC Dameng Mining Industries Limited Joint base of Postgraduate Cultivation, and State Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures, and National Natural Science Foundation of China (Nos. 11364003 and 52102470), Guangxi Innovation Driven Development Project (Nos. AA17204100 and AA18118052), the Natural Science Foundation of Guangxi Province (No. 2018GXNSFAA138186).
Supplementary material associated with this article can be found, in the online version, at doi:
L. Dai, K. Huang, Y. Xia, Z. Xu, Green Energy Environ. 6 (2021) 193–211. doi: 10.1016/j.gee.2020.09.015
J. Li, L. Zhang, K. Doyle-Davis, R. Li, X. Sun, Carbon Energy 2 (2020) 488–520. doi: 10.1002/cey2.74
H. Wan, W. Ma, K. Zhou, et al., Green Energy Environ. 7 (2022) 205–220. doi: 10.1016/j.gee.2021.04.001
Y. Pan, F.F. Deng, Z. Fang, et al., Chin. Chem. Lett. 32 (2021) 3440–3445. doi: 10.1016/j.cclet.2021.05.067
M.F. Labata, G. Li, J. Ocon, P.Y.A. Chuang, J. Power Sources 487 (2021) 229356. doi: 10.1016/j.jpowsour.2020.229356
Z. Qiao, S. Hwang, X. Li, et al., Energy Environ. Sci. 12 (2019) 2830–2841. doi: 10.1039/c9ee01899a
L. Huang, S. Zaman, X. Tian, et al., Acc. Chem. Res. 54 (2021) 311–322. doi: 10.1021/acs.accounts.0c00488
Y. Wang, Y. Pang, H. Xu, A. Martinez, K.S. Chen, Energy Environ. Sci. 15 (2022) 2288–2328. doi: 10.1039/d2ee00790h
J. Liu, T. Zhang, G.I.N. Waterhouse, J. Mater. Chem. A 8 (2020) 23142–23161. doi: 10.1039/d0ta09092a
K. Jiao, J. Xuan, Q. Du, et al., Nature 595 (2021) 361–369. doi: 10.1038/s41586-021-03482-7
H. Zeng, X. Liu, F. Chen, et al., ACS Appl. Mater. Interfaces 12 (2020) 52549–52559. doi: 10.1021/acsami.0c13597
C. Zhang, R. Lu, C. Liu, et al., Adv. Sci. 9 (2022) 2104768. doi: 10.1002/advs.202104768
Q. Ji, L. Bi, J. Zhang, H. Cao, X.S. Zhao, Energy Environ. Sci. 13 (2020) 1408–1428. doi: 10.1039/d0ee00092b
X.T. Wang, T. Ouyang, L. Wang, J.H. Zhong, Z.Q. Liu, Angew. Chem. Int. Ed. 59 (2020) 6492–6499. doi: 10.1002/anie.202000690
D. Banham, J. Zou, S. Mukerjee, et al., J. Power Sources 490 (2021) 229515. doi: 10.1016/j.jpowsour.2021.229515
W. Li, C. Han, K. Zhang, S. Chou, S. Dou, J. Mater. Chem. A 9 (2021) 6671–6693. doi: 10.1039/d1ta00203a
X. Duan, F. Cao, R. Ding, et al., Adv. Energy Mater. 12 (2022) 2103144. doi: 10.1002/aenm.202103144
Q. Ma, H. Jin, J Zhu, et al., Adv. Sci. 8 (2021) 2102209. doi: 10.1002/advs.202102209
Q. Chen, X. Tan, Y. Liu, et al., J. Mater. Chem. A 8 (2020) 5773–5811. doi: 10.1039/c9ta11618d
C. Zhang, Z. Feng, Y. Lei, et al., J. Colloid Interface Sci. 628 (2022) 174–188. doi: 10.4103/gmit.gmit_19_22
J. Cheng, C. Lyu, G. Dong, et al., Electrochim. Acta 454 (2023) 142364. doi: 10.1016/j.electacta.2023.142364
K. Paperzh, A. Alekseenko, O. Safronenko, et al., Colloid Interface Sci. Commun. 45 (2021) 100517. doi: 10.1016/j.colcom.2021.100517
F. Sun, R. Su, Y. Zhou, et al., ACS Appl. Mater. Interfaces 14 (2022) 41079–41085. doi: 10.1021/acsami.2c11910
H. Zhong, L. Alberto Estudillo-Wong, Y. Gao, Y. Feng, N. Alonso-Vante, J. Energy Chem. 59 (2021) 615–625. doi: 10.1016/j.jechem.2020.11.033
D. Liu, J. Zhang, D. Liu, et al., J. Phys. Chem. Lett. 13 (2022) 2019–2026. doi: 10.1021/acs.jpclett.1c04005
M. Xie, T. Chu, X. Wang, et al., Int. J. Hydrogen Energy 47 (2022) 28074–28085. doi: 10.1016/j.ijhydene.2022.06.131
A.M. Jauhar, Z. Ma, M. Xiao, et al., J. Power Sources 473 (2020) 228607. doi: 10.1016/j.jpowsour.2020.228607
D.D. Qin, Y. Tang, G. Ma, et al., Int. J. Hydrogen Energy 46 (2021) 25771–25781. doi: 10.1016/j.ijhydene.2021.05.096
W. Zhang, J. Li, Z. Wei, Small 19 (2023) 2300112. doi: 10.1002/smll.202300112
T. Lu, H. Wang, Nano Res. 15 (2022) 9764–9778. doi: 10.1007/s12274-022-4157-1
Z. Ma, H. Tian, G. Meng, et al., Sci. China Mater. 63 (2020) 2517–2529. doi: 10.1007/s40843-020-1449-2
B. Garlyyev, K. Kratzl, M. Rück, et al., Angew. Chem. Int. Ed. 58 (2019) 9596–9600. doi: 10.1002/anie.201904492
Y. Wang, X. Zheng, D. Wang, Nano Res. 15 (2021) 1730–1752.
Q. Wang, M. Ming, S. Niu, et al., Adv. Energy Mater. 8 (2018) 1801698. doi: 10.1002/aenm.201801698
X. Li, W.X. Chen, J. Zhao, W. Xing, Z.D. Xu, Carbon 43 (2005) 2168–2174. doi: 10.1016/j.carbon.2005.03.030
R. Zeng, K. Wang, W. Shao, et al., Chin. J. Catal. 41 (2020) 820–829. doi: 10.1016/S1872-2067(19)63456-X
E. Antolini, F. Cardellini, E. Giacometti, G. Squadrito, JMatS 37 (2002) 133–139.
J. Salgado, E. Antolini, E. Gonzalez, J. Power Sources 141 (2005) 13–18. doi: 10.1016/j.jpowsour.2004.08.048
Y. Li, X. Zhu, Y. Chen, S. Zhang, J. Li, J. Liu, J. Energy Chem. 47 (2020) 138–145. doi: 10.1016/j.jechem.2019.12.004
J. Yang, W. Li, D. Wang, Y. Li, Adv. Mater. 32 (2020) 2003300. doi: 10.1002/adma.202003300
K. Sang, J. Zuo, X. Zhang, et al., Green Energy Environ. 8 (2023) 619–625. doi: 10.1016/j.gee.2022.12.006
G. Ren, M. Shi, S. Liu, et al., Chem. Eng. J. 454 (2023) 140158. doi: 10.1016/j.cej.2022.140158
R.G. Rao, R. Blume, M.T. Greiner, et al., ACS Catal. 12 (2022) 7344–7356. doi: 10.1021/acscatal.2c01063
B.A. Lu, L.F. Shen, J. Liu, et al., ACS Catal. 11 (2020) 355–363.
Z. Mao, C. Ding, X. Liu, et al., ACS Catal. 12 (2022) 8848–8856. doi: 10.1021/acscatal.2c01052
E. Pajootan, S. Omanovic, S. Coulombe, Chem. Eng. J. 426 (2021) 131706. doi: 10.1016/j.cej.2021.131706
J. Kong, Y.H. Qin, T.L. Wang, C.W. Wang, Int. J. Hydrogen Energy 45 (2020) 1991–1997. doi: 10.1016/j.ijhydene.2019.11.016
J. Bai, S. Ke, J. Song, et al., ACS Appl. Mater. Interfaces 14 (2022) 5287–5297. doi: 10.1021/acsami.1c20823
G.T. Song, Y. Wang, Y. Qi, W.M. Li, L.X. Zhang, Rare Met. 39 (2019) 784–791. doi: 10.1145/3343031.3350996
D. Liu, S. Gao, J. Xu, et al., Appl. Surf. Sci. 604 (2022) 154466. doi: 10.1016/j.apsusc.2022.154466
D. Nechiyil, M.S. Garapati, R.C. Shende, et al., J. Colloid Interface Sci. 561 (2020) 439–448. doi: 10.1016/j.jcis.2019.11.015
R. Sharma, S. Gyergyek, S.M. Andersen, ACS Appl. Energy Mater. 5 (2022) 705–716. doi: 10.1021/acsaem.1c03189
J. Quinson, M. Inaba, S. Neumann, et al., ACS Catal. 8 (2018) 6627–6635. doi: 10.1021/acscatal.8b00694
Q. Dong, Z. Mo, H. Wang, et al., ACS Sustain. Chem. Eng. 8 (2020) 6979–6989. doi: 10.1021/acssuschemeng.0c00132
X. Wang, J. Zheng, R. Fu, J. Ma, Chin. J. Catal. 32 (2011) 599–605. doi: 10.1016/S1872-2067(10)60213-6
S. Wu, Y. Liu, Y. Ren, Q. Wei, Y. Sun, Nano Res. 15 (2021) 4886–4892.
Y. Xin, T. Nagata, K. Kato, T. Shirai, ACS Appl. Nano Mater. 5 (2022) 4305–4315. doi: 10.1021/acsanm.2c00236
F. Wang, X. Liu, B. Jiang, et al., J. Colloid Interface Sci. 635 (2023) 514–523. doi: 10.1016/j.jcis.2022.12.160

Figure 1 Physio-chemical characterization. (a) XRD spectra for Pt/KB-4, Pt/KB-10, Pt/KB-14, and JM 40%. (b) TGA curves for Pt/KB-4, Pt/KB-10, and Pt/KB-14. (c) XPS survey spectra and (d) Pt 4f spectra of Pt/KB-4 and JM 40%.

Figure 2 View of nano-sized particles and their porosity evolution. TEM images of (a-c) Pt/KB-4 and (d-f) JM 40%. Inset images in (a) and (d): Particle size distribution. BET nitrogen adsorption-desorption isotherms of (g) Ketjen Black EC-600JD, (h) Pt/KB-4 and (i) JM 40%. Inset images in (g-i): the Barrett-Joyner-Halenda (BJH) pore size distribution.

Figure 3 Catalytic performance. (a) CV curves for Pt/KB-4, Pt/KB-10, Pt/KB-14 and JM 40% in nitrogen-saturated 0.1 mol/L HClO4. (b) LSV curves measured for Pt/KB-4, Pt/KB-10, Pt/KB-14 and JM 40% in oxygen-saturated 0.1 mol/L HClO4. (c) E1/2 of Pt/KB-4, Pt/KB-10, Pt/KB-14 and JM 40%. (d) ECSA of Pt/KB-4, Pt/KB-10, Pt/KB-14 and JM 40%. (e) Pt/KB-4, Pt/KB-10, Pt/KB-14 and JM 40% MA and specific activity (SA) at 0.9 V vs. RHE. (f) Comparison of radargrams for Pt/KB-4, JM 40% and Pt/C-M (0.13, 180).

Figure 4 Catalytic performance before and after 10,000 ADTs cycles. (a) CV curves for Pt/KB-4 and JM 40% in nitrogen-saturated 0.1 mol/L HClO4. (b) LSV curves measured for Pt/KB-4 and JM 40% in oxygen-saturated 0.1 mol/L HClO4. (c) ECSA and E1/2 of Pt/KB-4 and JM 40%. (d) Pt/KB-4 and JM 40% MA and SA at 0.9 V vs. RHE.

Figure 5 Performance versus factor plot. (a) CPt versus ECSA, MA and E1/2 curves. (b) pH1 versus ECSA, MA and E1/2 curves. (c) T versus ECSA, MA and E1/2 curves. (d) t versus ECSA, MA and E1/2 curves.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:



 下载:
下载:
 下载:
下载: