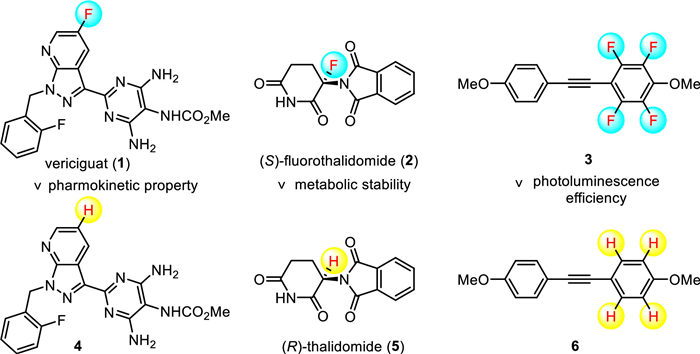
Scheme 1.
Structures of representative drugs, materials and their corresponding analogues.
C-F insertion reaction sheds new light on the construction of fluorinated compounds
Dong-Sheng Deng , Su-Qin Tang , Yong-Tu Yuan , Ding-Xiong Xie , Zhi-Yuan Zhu , Yue-Mei Huang , Yun-Lin Liu

The selective incorporation of fluorine atoms onto organic compounds has emerged as a common and useful strategy to design agrochemicals, pharmaceuticals, medical imaging agents, and functional materials [1–7], since the fluorine substitution can often lead to a dramatic change in physical, chemical, and biological properties [8,9]. For example, vericiguat 1 demonstrates the superior pharmokinetic property [10], chiral fluorinated thalidomide 2 displays the enhanced metabolic stability [11], and fluorinated tolane 3 shows significantly enhanced photoluminescence efficiency [12] when compares with their respective non-fluorinated counterparts 4, 5, and 6 (Scheme 1). Therefore, given the beneficial effects bring about by fluorine atoms, organofluorine molecules have become popular synthetic targets over the years [13–21]. However, how to prepare fluorinated compounds is not as simple as it seems and has been daunting chemists for a long time. This is likely due to the following reasons: (1) The availability of natural fluorine-containing compounds that can potentially be used as starting materials is very limited; (2) the high electronegativity of fluorine atom and the high hydration energy of the fluoride anion makes the C-F bond formation very challenging. Consequently, strategies suitable for the synthesis of chlorinated, brominated, and iodinated compounds cannot be easily extended to fluorinated compounds.
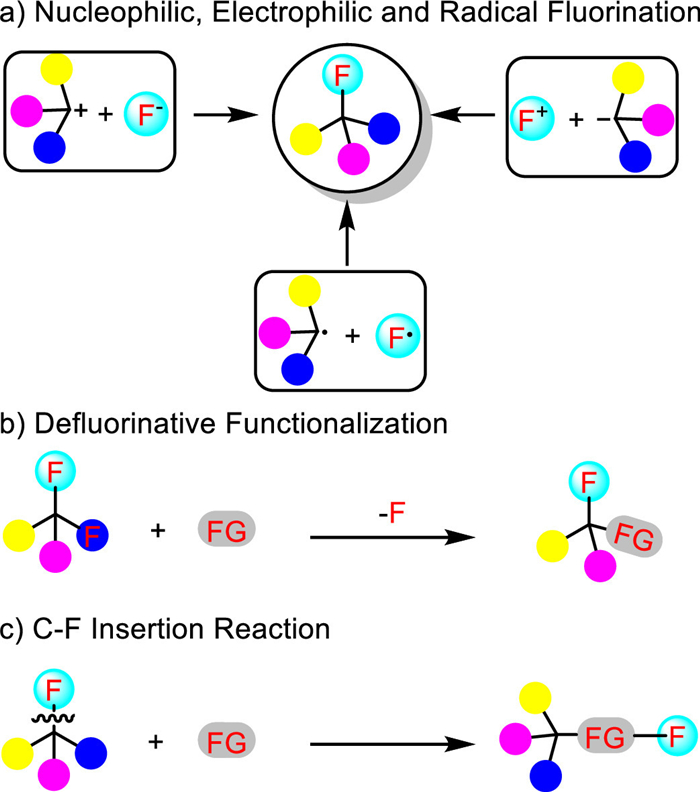
Despite the challenges mentioned above, the wide applications of fluorinated compounds have stimulated extensive research on seeking for efficient strategies for their construction in the past decades. Three commonly used strategies are shown in Scheme 2. These include: (a) Nucleophilic, electrophilic and radical fluorination reaction of non-fluorinated precursors; (b) defluorinative functionalization reaction of poly-fluorinated substances; and (c) C-F bond insertion reaction. Of these strategies developed, the C-F bond insertion reaction which proceeds via the concomitant formation of C-C and C-F bonds is the most desirable as it allows the rapid construction of more complex organofluorine molecules using readily available organofluorine compounds with an atom economy of 100% and without requiring any exogenous fluorinating reagent. However, the merge of C-F bond cleavage and formation in a single transformation is a challenging task because of the high bond dissociation energy of both C-F and metal-F bonds as well as the low reactivity of the fluoride released upon C-F bond cleavage [22]. Considering the advantages of C-F insertion reactions, considerable efforts have been devoted to developing innovative protocols to overcome these challenges, and breakthroughs have been achieved recently. Therefore, this highlight will focus on this emerging field, aiming to inspire and push for new developments. The contents of this highlight will be arranged by the fluorinating agents used.
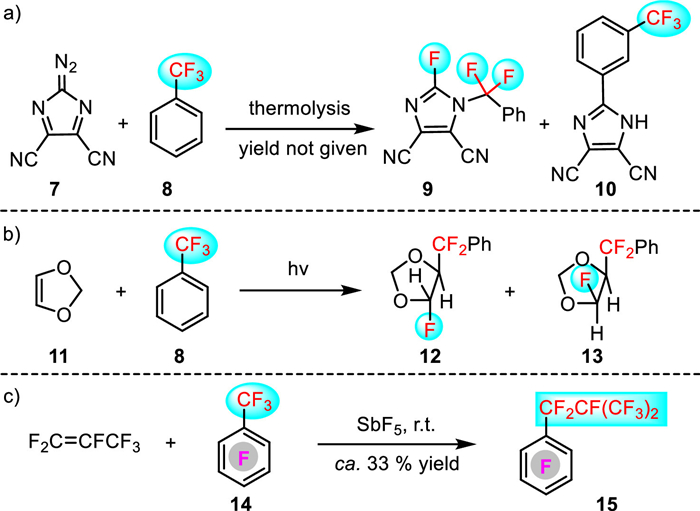
As mentioned above, C-F bond insertion is a highly challenging and complicated process. Therefore, so far, reports of such processes are scarce in the literature. In 1973, Sheppard and Webster found that a C-F bond of trifluoromethylbenzene insertion of a C-N unit with diazonium salt-derived reactive N-heterocyclic carbenes could occur under heating condition (Scheme 3a) [23]. In 1985, Mattay's group reported a photo-mediated insertion of alkenes into C-F bonds of trifluoromethylbenzene to give products 12 and 13 (Scheme 3b) [24]. Later in 2018, a SbF5 promoted insertion of perfluoropropene into C-F bonds of perfluoroarenes 14 was accomplished by the Kapov's group (Scheme 3c) [25]. However, in these early reports, the yields are extremely low as the formation of the desired C-F insertion products was accompanied with substantial amounts of side products.
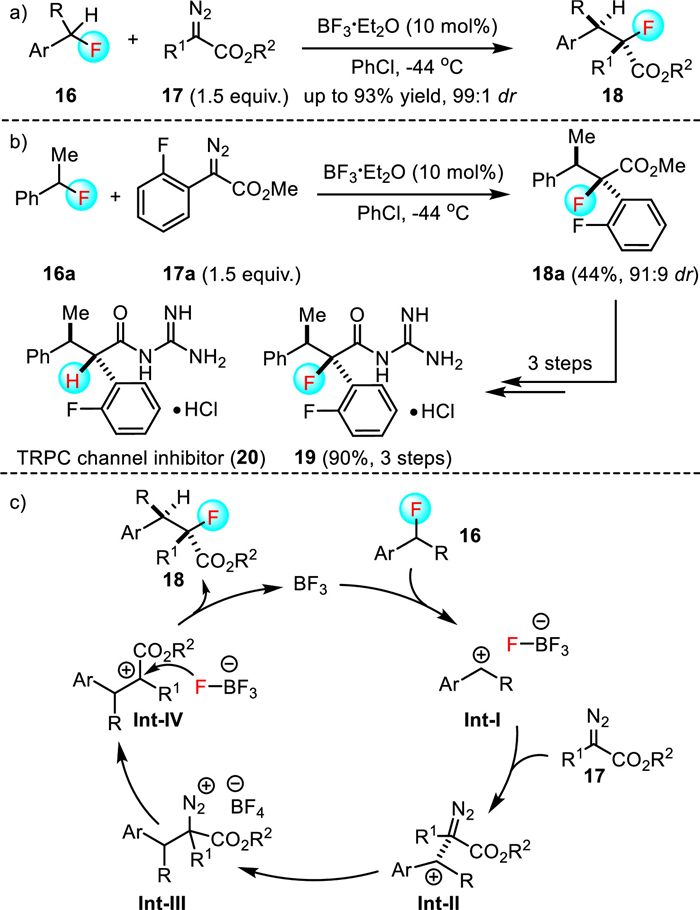
Although the C-F bond insertion reactions described above are not so successful, these pioneering reports demonstrated that it was possible to insert functional groups into a benzylic C-F bond of trifluoro- and perfluoro-arenes. Considering that C-F bond dissociation energy (BDE) increases with increasing fluorine substitution [26], the use of benzylic fluorides with lower BDEs might be amenable to the C-F bond insertion process. In this context, in 2021, Yasuda and Nishimoto reported the first example of a BF3-catalyzed formal insertion of diazo esters 17 into the C-F bonds of benzylic fluorides 16, which furnished the one-carbon elongation products, α-fluoro-α, β-diaryl esters 18, in moderate to good yields and with high diastereoselectivities (Scheme 4) [27]. This catalytic method exhibits high chemoselectivity, broad substrate scope and good functional group tolerance, and can also be applied toward the synthesis of compound 19, a fluorinated analogue of compound 20 that is utilized as a transient receptor and potential canonical (TRPC) channel inhibitor. DFT calculations suggested that the BF3 catalyst initially abstracted F− from benzylic fluoride 16 to generate the benzylic cation and BF4− (Int-Ⅰ). This benzylic cation would thereafter be attacked by diazo esters 17 to form intermediate Int-Ⅱ. N2 Extrusion from Int-Ⅱ afforded contact ion pair Int-Ⅲ. The C-F bond re-formation via the nucleophilic attack of the F atom in BF4− to the carbocation produced the desired α-fluoro-α, β-diaryl esters 18.
Very recently, Hopkinson et al. described a hexafluoroisopropanol (HFIP) mediated C-F insertion of secondary benzyl fluorides 16 with α-fluorinated styrenes 21 to produce gem-difluorinated products 22 in 41%-66% yield (Scheme 5) [28]. In addition, upon increasing the amount of 21 and changing the CH2Cl2: HFIP ratio from to 7:3 to 4:6, the propargylic fluorides 23 can also serve as suitable reaction partners and gave the corresponding products 24 in good yields. However, benzyl fluorides bearing strong electron-withdrawing groups (e.g., NO2, CF3 and CN) at the para-position and primary propargylic fluorides were not compatible with the present reaction conditions likely because of the decreased stability of the in situ formed benzyl or propargyl cation. Interestingly, the authors found that the reactions should be conducted in a PTFE vial rather than in a glass Schlenk tube in that the HF or silicon fluoride species generated from the glass surface caused side reactions. Notably, the use of pure CH2Cl2 totally shut down the reaction. This result confirmed the key role of HFIP as a hydrogen bond donor. Accordingly, the authors proposed that HFIP served as the hydrogen bond donors to activate C-F bonds of benzyl fluorides. This process liberated a benzyl cation Int-Ⅰ and a HFIP stabilized fluoride ion. Subsequent electrophilic addition of the benzyl cation to fluorinated alkenes generate a new cation Int-Ⅱ, which can be trapped by fluoride ion to give the gem-difluorinated products 22.
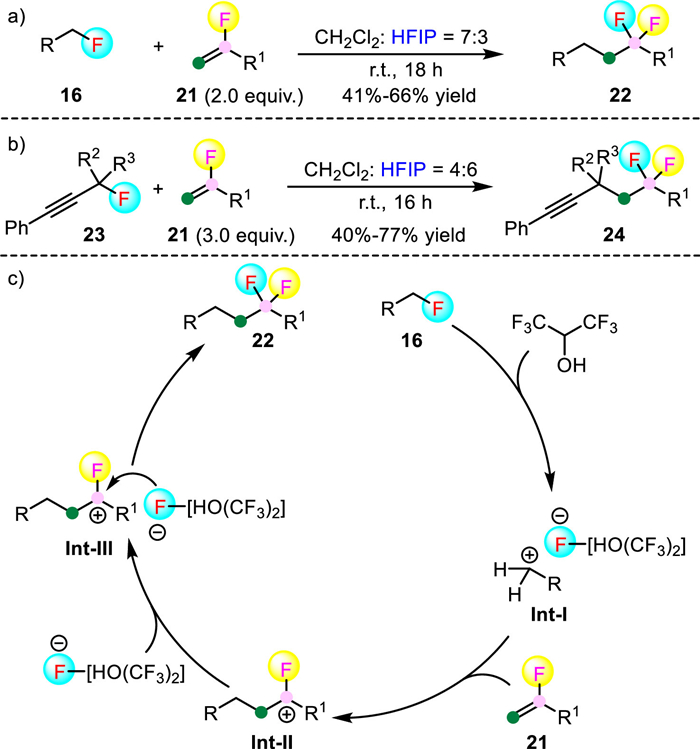
Polyfluorinated arenes constitute a unique class of molecules and play important roles in the fields of pharmaceuticals, pesticides and material science [29–31]. Typically, they can be synthesized by arene fluorination or by partial arene defluorination. However, these classic strategies suffer from lengthy reaction steps or low atom economy [32]. Therefore, the development of novel C-F insertion reactions that occur via mechanistically distinct pathways and also produce complimentary products are of great value. In this context, in 2015, Studer and co-workers disclosed the first light-mediated insertion of aryl and alkyl isonitriles 25 into the C-F bond of polyfluorinated arenes 26, affording various imidoyl fluorides 27 in moderate to excellent yields (Scheme 6a) [33]. The versatile imidoyl fluorides can further be readily transformed into other valuable compounds that are otherwise difficult to prepare using the available methodologies. Unfortunately, high energy and pressure mercury lamp (λexc = 254 nm) was required for the efficient excitation of isonitriles, which resulted in a limited functional group tolerance.
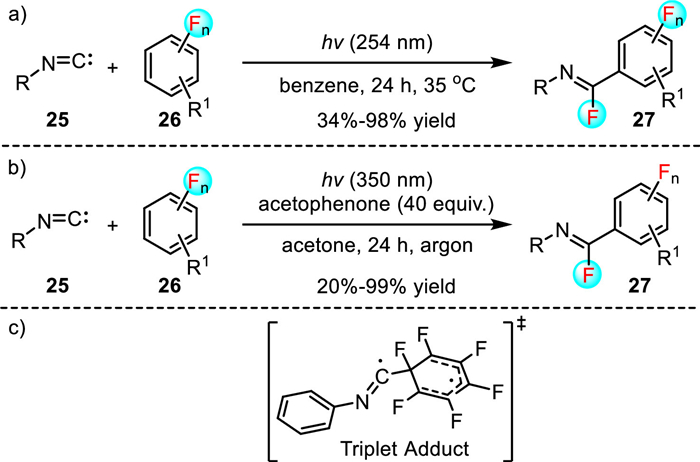
In order to solve this issue, in 2020, the authors reported an alternative photosensitized C-F insertion reaction of polyfluorinated arenes 26 with isonitriles 25 to deliver benzimidoyl fluorides 27 in 20%-99% yields (Scheme 6b) [34]. In this case, acetophenone was proved to be a suitable external photosensitizer, which enabled the reaction to be performed upon irradiation at 350 nm. The mild light warrants the high functional group tolerance of this methodology. For example, this methodology tolerated the halogen functionality (e.g., Cl, Br) in the isonitrile and polyfluorinated arene components, which are challenging substrates in the previous study since 254 nm irradiation caused C-halogen bond homolysis.
Mechanistic studies showed that these C-F insertion reactions proceeded through the triplet state of the isonitriles (Scheme 6c). As for the reaction shown in Scheme 6a, the triplet state was generated via 250 nm excitation, while the acetophenone-sensitized reaction likely proceeded through the Dexter energy transfer at 350 nm (Scheme 6b). However, the mechanism regarding the observed regioselectivity is currently not fully understood, and the precise control of regioselectivity is still an unsolved problem given that the arene contains multiple C-F bonds.
Apart from benzylic and propargylic fluorides, acyl fluorides that display a good balance of highly electrophilic reactivity and stability are also popular substrates for atom-economical C-F insertion reactions as the corresponding fluoroaroylation products are useful for drug discovery [35,36]. In 2020, Tobisu's group reported an elegant phosphine-catalyzed intermolecular acylfluorination of alkyonates 28, which provided rapid and straightforward access to a variety of highly functionalized monofluoroalkene derivatives 30 (Scheme 7) [37]. This transition metal free protocol operates at room temperature, thus permitting a good functional group tolerance. For example, a broad range of acyl fluorides 29 bearing electron-neutral, electron-deficient, or electron-rich groups readily participated in this reaction with (hetero)aryl-substituted alkynoates 28 to produce the corresponding monofluoroalkenes 30 in moderate to excellent yields (30%-94%). However, the alkyl-substituted alkynoates 28 were not compatible, which constituted the limitation of this method. It should be noted that the stereoselectivity of the reaction is substrate dependent. While most products were obtained as an inseparable E and Z isomers (1:1-1:1.6), the use of a 2-pyridyl alkynoate led to excellent Z:E selectivity (96:4 ~ > 99:1). This is possibly due to the stabilizing n-π* interaction between the nitrogen lone pair of pyridyl ring and C=O π* orbital. Based on the control experimental results and density functional theory (DFT) calculations, the authors proposed that the reaction might occur via the addition of tertiary phosphines to alkyonates followed by the nucleophilic substitution with an acyl fluoride to generate the key P(V) intermediate Int-Ⅱ, which was then underwent an unprecedented C-F bond-forming ligand coupling to afford the products 30 and release the catalyst.
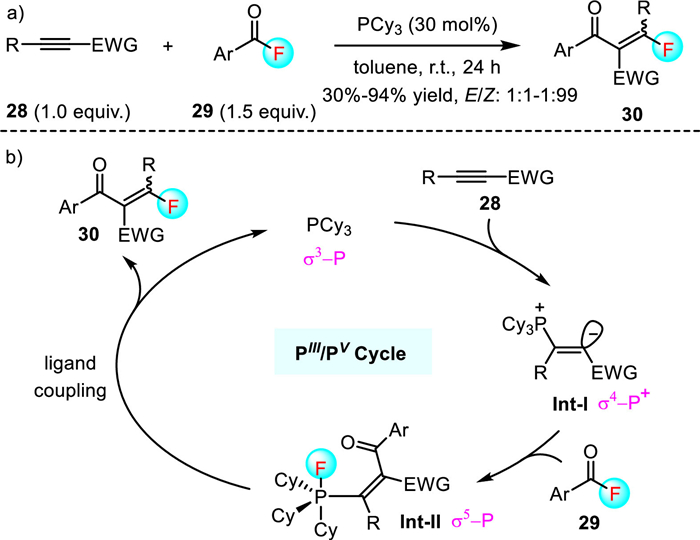
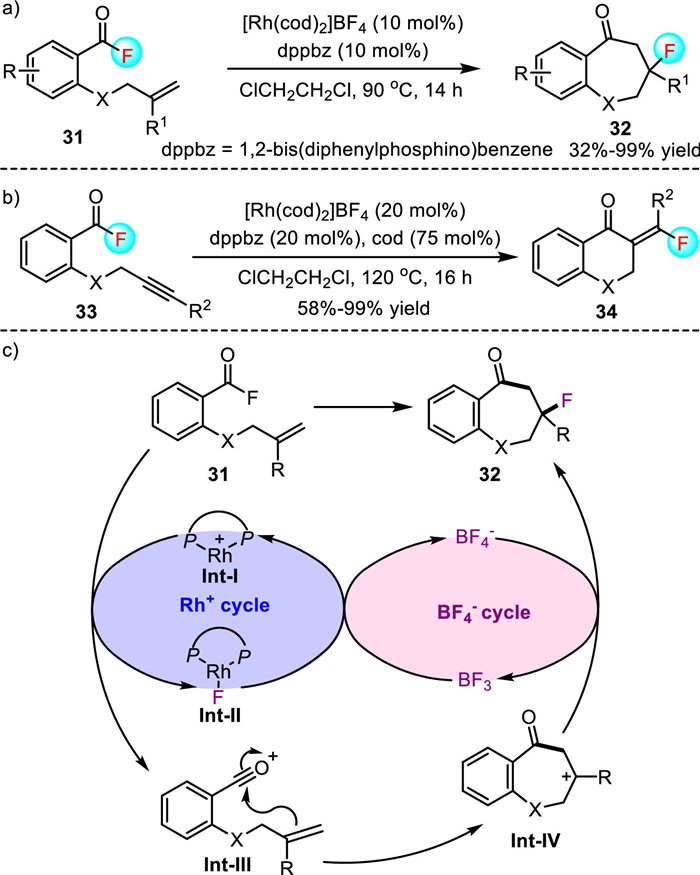
More recently, Tobisu's group further disclosed a [Rh(cod)2]BF4 catalyzed intramolecular carbofluorination reaction of alkene tethered acyl fluorides (Scheme 8) [38]. The reaction allows access to complex tertiary alkyl fluorides 32 bearing a seven-membered heterocycle ring in 32%-99% yield. In addition to alkene tethered acyl fluorides, differently substituted alkyne tethered acyl fluorides 33 were competent substrates, giving the fluorinated 4-chromanone derivatives 34 with high yields. However, although acyl fluorides bearing 1, 1-disubstituted alkenes were found to be well tolerated, substrates with an internal alkene or a mono-substituted alkene moiety failed to participate in this carbofluorination reaction, which constituted the limitations of this approach. Notably, the cationic character of the rhodium catalyst was a key factor for the success of this reaction as the electronically neutral [RhCl(cod)]2 showed no catalytic activity. They proposed that a Lewis acidic rhodium cation initially abstracted a fluoride anion from acyl fluoride to form an acylium cation Int-Ⅲ and a neutral rhodium(Ⅰ)-fluoride complex Int-Ⅱ. The interception of acylium cation Int-Ⅲ with a tethered alkene led to the formation of a tertiary carbocation intermediate Int-Ⅳ. Subsequently, carbocation Int-Ⅳ abstracted a fluoride from BF4− to give product 32 and BF3, which reacted with complex Int-Ⅱ to regenerate cationic rhodium catalyst. In this catalytic cycle, BF4− can be viewed as the fluoride anion shuttle, which acts cooperatively with a rhodium cation to mediate the cleavage and formation of a C-F bond.
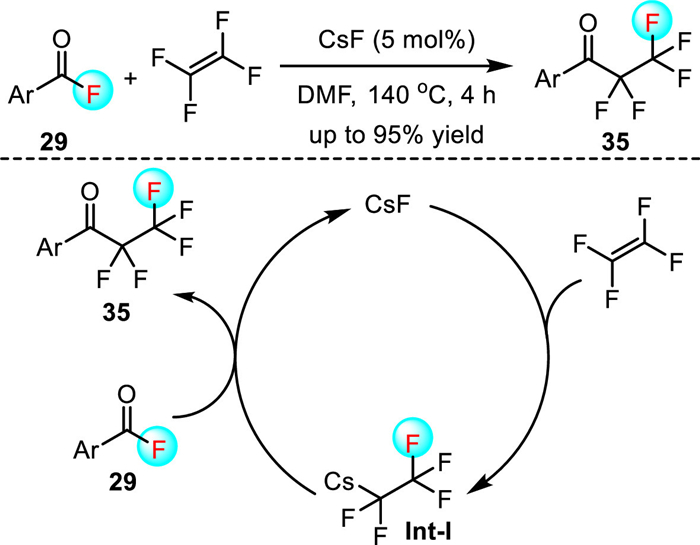
In 2021, Ogoshi and co-workers reported a CsF-catalyzed fluoroacylation of tetrafluoroethylene with acyl fluorides 29 (Scheme 9) [39]. This reaction occurred via the addition of CsF across tetrafluoroethylene to generate intermediate Int-Ⅰ, followed by the reaction with an acyl fluoride 29 to afford the pentafluoroethyl ketones 35. However, the substrate scope of this reaction is largely limited to aromatic acyl fluorides, and high reaction temperature (140 ℃) was required to avoid the formation of side products.
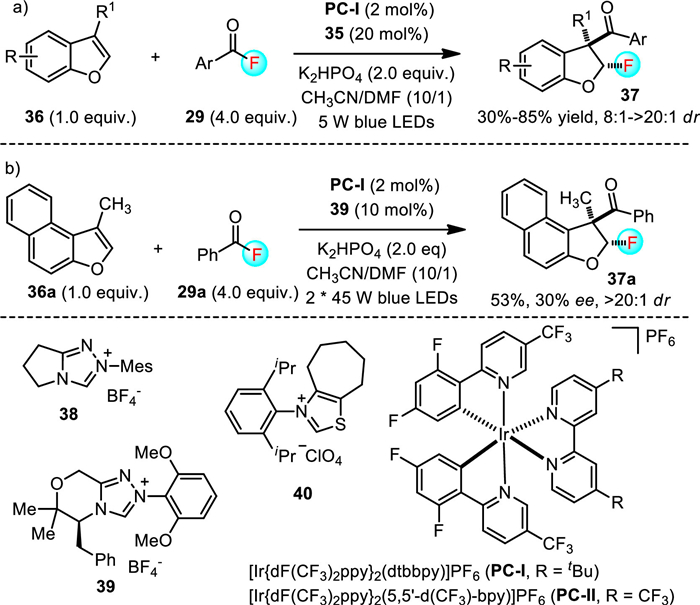
In 2022, Studer and colleagues developed a cooperative N-heterocyclic carbene (NHC)/photoredox catalysis strategy for the 2, 3-fluoroaroylation of benzofurans with aroyl fluorides 29 as the bifunctional reagents (Scheme 10) [40]. In the presence of 2 mol% of photocatalyst PC-1 and 20 mol% of NHC catalyst 38, the dearomatization reactions proceeded smoothly to afford 3-aroyl-2-fluoro-2, 3-dihydrobenzofurans 37 with 30%-85% yields and high diastereoselectivity (8:1 ~ > 20:1 dr). Remarkably, upon running the reaction of 36a and 29a with chiral NHC catalyst 39, the corresponding product 37a could be furnished with 53% yield, 30% ee and > 20:1 dr. In addition, it should be noted that reactions also took place with N-acyl indoles to afford 3-aroyl-2-fluoro-dihydroindoles with moderate yield and excellent diastereoselectivity, which demonstrated the high functional group compatibility of this methodology.
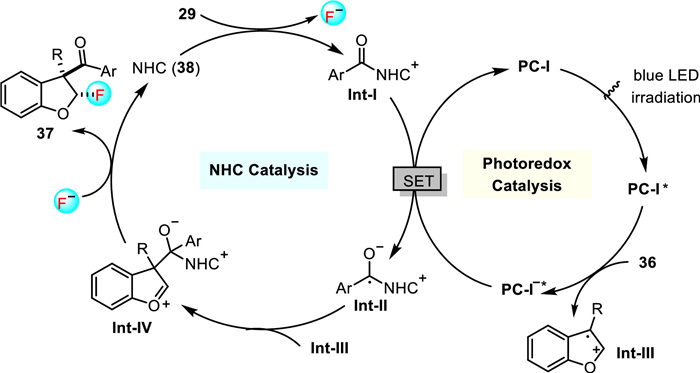
The authors proposed that a photoexcited IrⅢ species (PC-I*) oxidized benzofuran 36 to a radical cation intermediate Int-Ⅲ (Scheme 11). At the meantime, NHC catalyst 38 initially reacted with the acyl fluoride 29 to form an azolium ion intermediate Int-Ⅰ, which was subsequently reduced by Ir(Ⅱ) to give the persistent ketyl radical intermediate Int-Ⅱ. C-C bond coupling of the radical cation Int-Ⅲ and the ketyl radical Int-Ⅱ led to formation of oxocarbenium intermediate Int-Ⅳ, which could be trapped by the fluoride anion trans to the bulky alcoholate moiety to provide the final fluoroacylation products 37 with high diastereoselectivity. This rare radical/radical cation cross-coupling strategy opens up new opportunities to develop reactions that are otherwise difficult to be realized by using other protocols.
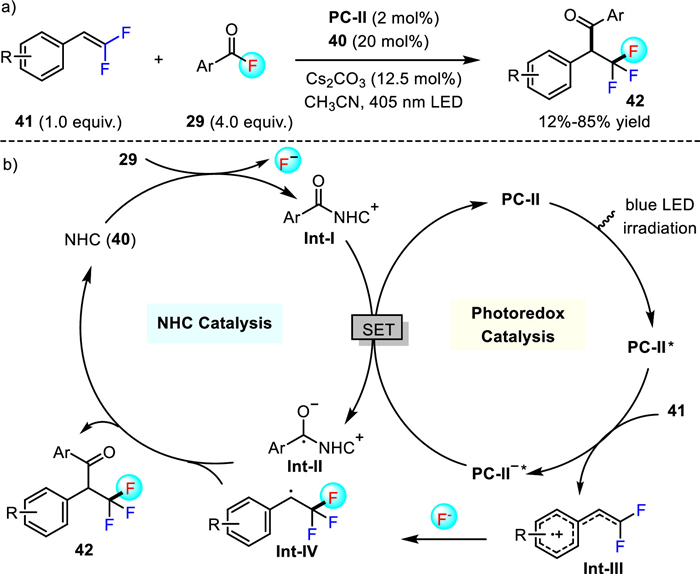
Very recently, Studer et al. further successfully accomplished the fluoroaroylation of gem-difluoroalkenes [41]. The same catalytic strategy using [Ir(dF(CF3)ppy)2(5, 5′-d(CF3)-bpy)]PF6 PC-Ⅱ (2 mol%) as the photoredox catalyst and the triazolium salt 40 as the NHC precatalyst (20 mol%) was adopted to deliver the α-trifluoromethylated ketones 42 with 12%-85% yields (Scheme 12). This method encompasses a broad range of alkenes (e.g., gem-difluoroalkenes, α-fluorostyrenes and simple styrenes) and aroyl fluorides. However, aliphatic acyl fluorides remain problematic substrates. The reaction is postulated to occur in a fashion similar to the mechanism depicted in Scheme 11. In this case, an α-trifluoromethylated benzyl radical species Int-Ⅳ was involved, which finally give rise to the targeted α-CF3-substituted ketones through the cross-coupling with ketyl radical Int-Ⅱ formed by the reaction of aroyl fluoride 29 with NHC catalyst 40 and the subsequent single electron transfer reduction by photocatalyst PC-Ⅱ.
Compared with acyl fluorides, the catalytic C-F bond cleavage of carbamoyl fluorides was much more difficult due to their significantly less electrophilic reactivities [42]. Le and co-workers made a breakthrough in this field. They discovered that BF3·OEt2 could catalyze the carbofluorination reaction of alkyne-tethered carbamoyl fluorides to afford 3-(fluoromethylene)oxindoles 44 and γ-lactams 46 with 45%-99% yield, 94:6 ~ > 95:5 E/Z and 80%-81% yield, > 99:1 Z/E, respectively (Scheme 13) [22]. In particular, this method allowed compounds 47a and 47b, fluorinated derivatives of known protein kinase inhibitors, to be constructed rapidly with moderate yields and excellent stereoselectivities. The mild reaction conditions along with the readily available catalyst make this methodology attractive for drug development.
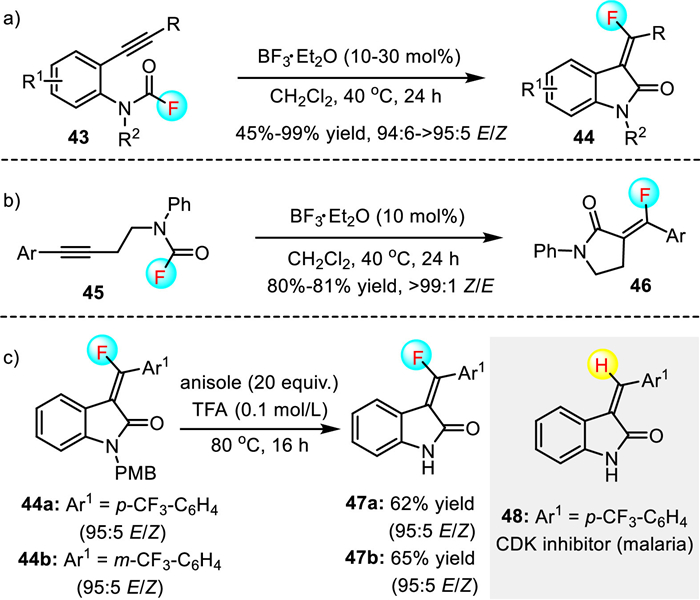
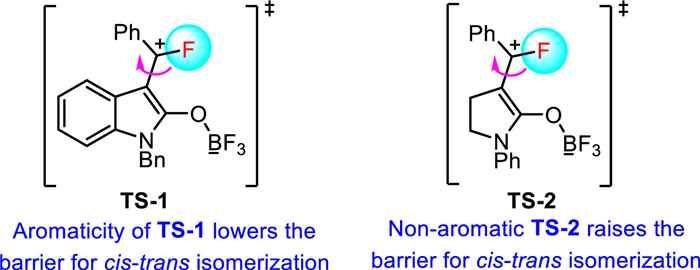
Experimental and computational studies suggested that the fluorocarbamoylation reaction proceeded through a turnover-limiting annulation step, followed by fluoride ion transfer from a BF3-coordinated carbamoyl adduct to forge the C-F bond (Scheme 14). In addition, the calculations also provide insight into the origin of the opposite stereoselectivity observed for fluoromethylene oxindoles 44 and γ-lactams 46. For methylene oxindoles, the transition state TS-1 possesses significant aromatic character, and therefore, easing the barrier for C=C bond isomerization to produce the thermodynamically favored E-isomer as the major product. However, the transition state TS-2 for the isomerization of γ-lactams 46 does not benefit from the aromatic stabilization effect, which leads to a significantly higher barrier for isomerization and the exclusive formation of the Z-γ-lactams.
In recent years, the readily available gem-difluorocyclopropanes have attracted considerable attention partly due to their ability to participate in various ring-opening reactions [42–45]. However, these reactions suffered from low atom economy as the fluorine atom scissored from gem-difluorocyclopropane could not be incorporated into the products. The breakthrough in this field was achieved by Liu et al., they successfully realized an innovative palladium-catalyzed fluorinative bifunctionalization of aziridines and azetidines (Scheme 15) [46]. Under optimum conditions, the regioselective C-C and C-F bond cleavage of gem-difluorocyclopropanes 49 proceeded smoothly to afford various bisfluorinated amines 51 in moderate to excellent yields and with excellent Z/E ratio. This fluorinative bifunctionalization reaction exhibited a broad substrate scope and was easy to scale up. In addition, the products can serve as the important class of building blocks in organic synthesis, as the versatile carbon-carbon double bond permitted various follow-up transformations.
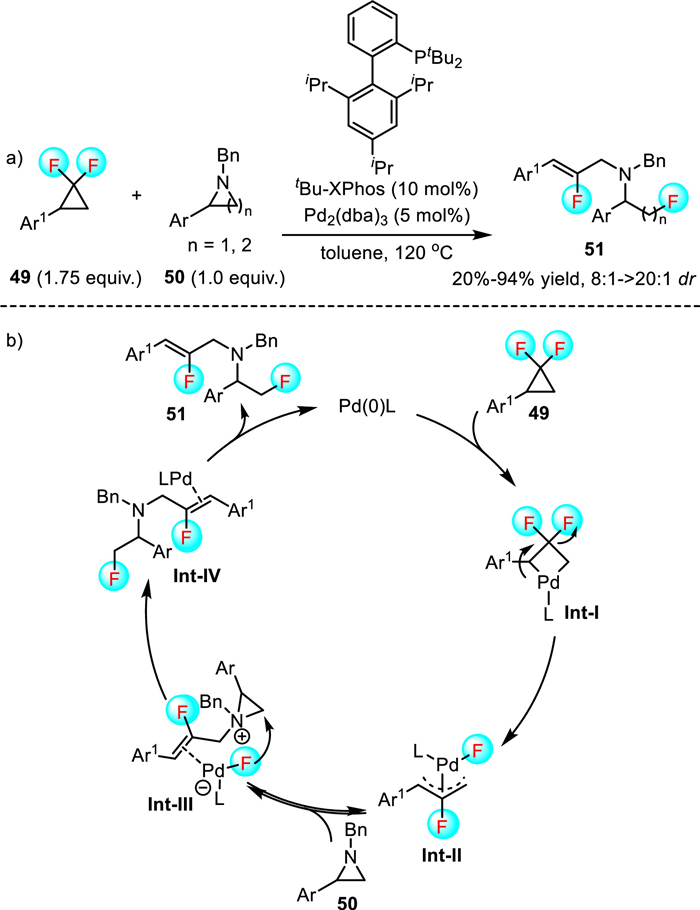
The proposed mechanism began with the oxidative addition of Pd(0)L into gem-difluorocyclopropanes 49 to form the four-membered-ring palladacycle complex Int-Ⅰ. The subsequent β-F elimination generated 2-fluorinated η3-allyl palladium complex Int-Ⅱ. Afterward, cyclic tertiary amines 50 attacked the complex Int-Ⅱ to produce η2-coordinated N-tetrasubstituted allyl ammonium complex Int-Ⅲ. Finally, difluorinated amines 51 were formed from ring opening of aziridinium ion at the electrophilic carbon by fluoride ligand on Pd(0) center. This work represents a notable advance in the C-F insertion chemistry, which opens up opportunities for designing new reactions.
As summarized in this review, C-F insertion reaction represents a reliable and powerful protocol for the rapid and atom-economical synthesis of various versatile fluorinated compounds and some related bioactive natural products and drugs. The past three years have witnessed rapid developments in this field. Nevertheless, despite these excellent achievements, this research area is still in its infancy compared to more established insertion reactions of C-X bonds of the other halogens. This emerging field is still facing some challenges that need to be addressed in the future. First, the substrate scope of fluorinating reagents that can be utilized for C-F insertion reactions is not broad enough so far. The examples described in this highlight clearly shown that catalysts were significant for merging C-F bond cleavage and formation in a single transformation. As a result, the rational design and development of catalytic systems are at the heart of addressing this challenge and will be crucial for advancing this chemistry. Second, catalytic enantioselective C-F insertion reactions remain unknown, albeit this will provide new possibilities for C-F insertion chemistry. Therefore, the development of efficient strategies to achieve this goal could be an important direction for future studies. We hope this highlight allows readers to notice the advantages and drawbacks of current C-F insertion reactions and to identify new activation methods and concepts for addressing the remaining challenges. As this field has recently attracted tremendous research efforts, it can be expected that more and more breakthroughs will appear in the future, and it might become a popular choice for the synthesis of fluorinated compounds.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The Tertiary Education Scientific Research Project of Guangzhou Municipal Education Bureau (No. 202235305) is gratefully acknowledged for financial support.
S. Purser, P.R. Moore, S. Swallow, et al., Chem. Soc. Rev. 37 (2008) 320–330. doi: 10.1039/B610213C
J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432–2506. doi: 10.1021/cr4002879
G. Wang, X. Jin, Y. Luo, Syn. Bio. J. 1 (2020) 358–371. doi: 10.1504/ijitm.2020.110240
M. Inoue, Y. Sumii, N. Shibata, ACS Omega 5 (2020) 10633–10640. doi: 10.1021/acsomega.0c00830
H. Mei, J. Han, S. Fustero, et al., Chem. Eur. J. 25 (2019) 11797–11819. doi: 10.1002/chem.201901840
Y. Zhou, J. Wang, Z. Gu, et al., Chem. Rev. 116 (2016) 422–518. doi: 10.1021/acs.chemrev.5b00392
N.A. Meanwell, J. Med. Chem. 61 (2018) 5822–5880. doi: 10.1021/acs.jmedchem.7b01788
B.E. Smart, J. Fluorine Chem. 109 (2001) 3–11. doi: 10.1016/S0022-1139(01)00375-X
C. Ni, J. Hu, Chem. Soc. Rev. 45 (2016) 5441–5454. doi: 10.1039/C6CS00351F
J. He, Z. Li, G. Dhawan, et al., Chin. Chem. Lett. 34 (2023) 107578. doi: 10.1016/j.cclet.2022.06.001
E. Tokunaga, H. Akiyama, V.A. Soloshonok, et al., PLoS One 12 (2017) e0182152. doi: 10.1371/journal.pone.0182152
M. Morita, S. Yamada, T. Konno, New J. Chem. 44 (2020), 6704–6708. doi: 10.1039/d0nj01268h
J. Hu, W. Zhang, F. Wang, Chem. Commun. 45 (2009) 7465–7478. doi: 10.1039/b916463d
T. Liang, C.N. Neumann, T. Ritter, Angew. Chem. Int. Ed. 52 (2013) 8214–8264. doi: 10.1002/anie.201206566
P.A. Champagne, J. Desroches, J.D. Hamel, et al., Chem. Rev. 115 (2015) 9073–9174. doi: 10.1021/cr500706a
X. Yang, T. Wu, R.J. Phipps, et al., Chem. Rev. 115 (2015) 826–870. doi: 10.1021/cr500277b
S. Fustero, D.M. Sedgwick, R. Román, et al., Chem. Commun. 54 (2018) 9706–9725. doi: 10.1039/c8cc05181j
G. Chandra, D.V. Singh, G.K. Mahato, et al., Chem. Pap. 77 (2023) 4085–4106. doi: 10.1007/s11696-023-02804-5
T.T. Simur, T. Ye, Y.J. Yu, et al. Chin. Chem. Lett. 33 (2022) 1193–1198. doi: 10.1016/j.cclet.2021.08.043
Y.F. Hu, J. Luo, C.X. Lü, Chin. Chem. Lett. 21 (2010) 151–154. doi: 10.1016/j.cclet.2009.10.014
L. Xi, L. Du, Z. Shi, Chin. Chem. Lett. 33 (2022) 4287–4292. doi: 10.1016/j.cclet.2022.01.077
E.A. McKnight, R. Arora, E. Pradhan, et al., J. Am. Chem. Soc. 145 (2023) 11012–11018. doi: 10.1021/jacs.3c03982
W.A. Sheppard, O.W. Webster, J. Am. Chem. Soc. 95 (1973) 2695–2697. doi: 10.1021/ja00789a054
J. Mattay, J. Runsink, T. Rumbach, et al., J. Am. Chem. Soc. 107 (1985) 2558–2560. doi: 10.1021/ja00294a064
T.V. Mezhenkova, V.M. Karpov, Y.V. Zonov, J. Fluorine Chem. 207 (2018) 59–66. doi: 10.1016/j.jfluchem.2017.12.018
J. Wang, Y. Wang, Y. Liang, et al., Angew. Chem. Int. Ed. 62 (2023) e202215062. doi: 10.1002/anie.202215062
F. Wang, Y. Nishimoto, M. Yasuda, J. Am. Chem. Soc. 143 (2021) 20616–20621. doi: 10.1021/jacs.1c10517
A. Garg, N.J. Gerwien, C. Fasting, et al., Angew. Chem. Int. Ed. 62 (2023) e202302860. doi: 10.1002/anie.202302860
W.K. Hagmann, J. Med. Chem. 51 (2008) 4359–4369. doi: 10.1021/jm800219f
P. Jeschke, ChemBioChem 5 (2004) 570–589. doi: 10.1002/cbic.200300833
K. Reichenbächer, H.I. Süss, J. Hulliger, Chem. Soc. Rev. 34 (2005) 22–30. doi: 10.1039/B406892K
J. Weaver, S. Senaweera, Tetrahedron 70 (2014) 7413–7428. doi: 10.1016/j.tet.2014.06.004
A. Dewanji, C. Mück-Lichtenfeld, K. Bergander, et al. Chem. Eur. J. 21 (2015) 12295–12298. doi: 10.1002/chem.201502298
F. Weidlich, N. Esumi, D. Chen, et al. Adv. Syn. Catal. 362 (2020) 376–383. doi: 10.1002/adsc.201901126
Y. Ogiwara, N. Sakai, Angew. Chem. Int. Ed. 59 (2020) 574–594. doi: 10.1002/anie.201902805
E.A. McKnight, D. Cadwallader, C.M. Le, Eur. J. Org. Chem. 2023 e202300017.
H. Fujimoto, T. Kodama, M. Yamanaka, et al., J. Am. Chem. Soc. 142 (2020) 17323–17328. doi: 10.1021/jacs.0c08928
T. Yoshida, M. Ohta, T. Emmei, et al., Angew. Chem. Int. Ed. 62 (2023) e202303657. doi: 10.1002/anie.202303657
N. Ishida, H. Iwamoto, D. Eimi Sunagawa, et al., Synthesis 53 (2021) 3137–3143. doi: 10.1055/s-0040-1705962
X. Yu, Q.Y. Meng, C.G. Daniliuc, et al., J. Am. Chem. Soc. 144 (2022) 7072–7079. doi: 10.1021/jacs.2c01735
X. Yu, A. Maity, A. Studer, Angew. Chem. Int. Ed. 62 (2023) e202310288. doi: 10.1002/anie.202310288
T. Scattolin, S. Bouayad-Gervais, F. Schoenebeck, Nature 573 (2019) 102. doi: 10.1038/s41586-019-1518-3
J. Xu, E.A. Ahmed, B. Xiao, et al. Angew. Chem. Int. Ed. 54 (2015) 8231–8235. doi: 10.1002/anie.201502308
Y. Zhu, Y. Zeng, Z.T. Jiang, et al., Synlett 34 (2023) 1–13. doi: 10.1055/a-1912-3059
L. Lv, H. Qian, Z. Li, ChemCatChem 14 (2022) e202200890. doi: 10.1002/cctc.202200890
D. Li, C. Shen, Z. Si, et al., Angew. Chem. Int. Ed. 62 (2023) e202310283. doi: 10.1002/anie.202310283

Scheme 1 Structures of representative drugs, materials and their corresponding analogues.

Scheme 4 BF3-Catalyzed insertion of diazo esters into C-F bonds of benzylic fluorides.

Scheme 5 Hydrogen-bonding-mediated C-F insertion reactions of benzyl or propargyl fluorides with α-fluorostyrenes.

Scheme 6 Light-mediated insertion of isonitriles into the C-F bond of polyfluorinated arenes.

Scheme 8 [Rh(cod)2]BF4-catalyzed intramolecular carbofluorination of alkenes and alkynes.

Scheme 9 CsF-catalyzed insertion of tetrafluoroethylene into C-F bonds of acyl fluorides.

Scheme 10 Cooperative NHC and photoredox catalysis enabled access to 3-aroyl-2-fluoro-2, 3-dihydrobenzofurans.

Scheme 11 Proposed catalytic cycle for the fluoroaroylation of benzofurans with aroyl fluorides.

Scheme 12 The synthesis of α-CF3-substituted ketones by cooperative NHC and photoredox catalysis.

Scheme 14 The possible transition states and origins of stereoselectivity of the fluorocarbamoylation reaction.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:





 下载:
下载: